
Evaluation of Changes to the Oral Microbiome Based on 16S rRNA Sequencing among Children Treated for Cancer

 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
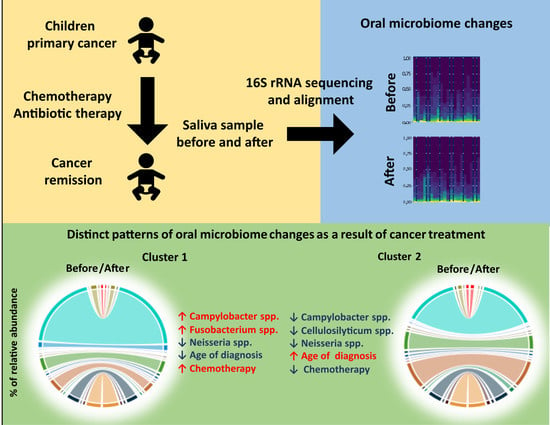
1. Introduction
2. Materials and Methods
2.1. The Cohort
2.2. Saliva Collection and DNA Isolation
2.3. Library Preparation and Sequencing
2.4. NGS Data Processing
2.5. Data Analysis
2.5.1. Alpha and Beta Diversity
2.5.2. Visualization of Data
2.5.3. Statistical Analysis
3. Results
3.1. 16S rRNA Sequencing and Microbiome Abundance Analysis
3.2. Differences in Microbiome in Patients before and after Cancer Treatment
3.3. Patterns of Microbiome Changes during Cancer Treatment Is Associated with Clinical Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greninger, A.L.; Messacar, K.; Dunnebacke, T.; Naccache, S.N.; Federman, S.; Bouquet, J.; Mirsky, D.; Nomura, Y.; Yagi, S.; Glaser, C.; et al. Clinical metagenomic identification of Balamuthia mandrillarisencephalitis and assembly of the draft genome: The continuing case for reference genome sequencing. Genome Med. 2015, 7, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaev, E.A.; Balmasova, I.P.; Mkrtumyan, A.M.; Kostryukova, S.N.; Vakhitova, E.S.; Ilina, E.N.; Tsarev, V.N.; Gabibov, A.G.; Arutyunov, S.D. Metagenomic Analysis of Gingival Sulcus Microbiota and Pathogenesis of Periodontitis Associated with Type 2 Diabetes Mellitus. Bull. Exp. Biol. Med. 2017, 163, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Yi, P.; Yang, J.; Xu, P.; Wang, Y.; Zhang, Z.; Huang, S.; Wang, Z.; Zhang, C. Association of gut microbiota composition and function with a senescence-accelerated mouse model of Alzheimer’s Disease using 16S rRNA gene and metagenomic sequencing analysis. Aging 2018, 10, 4054–4065. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, S.; Kalliala, I.; Nieminen, P.; Salonen, A. Comparative analysis of vaginal microbiota sampling using 16S rRNA gene analysis. PLoS ONE 2017, 12, e0181477. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, F.; Tian, Z. Role of microbiota on lung homeostasis and diseases. Sci. China Life Sci. 2017, 60, 407–1415. [Google Scholar] [CrossRef]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 17, 875. [Google Scholar] [CrossRef] [Green Version]
- Koliarakis, I.; Messaritakis, I.; Nikolouzakis, T.K.; Hamilos, G.; Souglakos, J.; Tsiaoussis, J. Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4146. [Google Scholar] [CrossRef] [Green Version]
- Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648. [Google Scholar]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Fiscella, K.A.; Gill, S.R. Oral microbiome: Possible harbinger for children’s health. Int. J. Oral Sci. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, X.; Yang, X.; Que, J.; Du, Q.; Zhang, Q.; Zou, J. Oral Health, Caries Risk Profiles, and Oral Microbiome of Pediatric Patients with Leukemia Submitted to Chemotherapy. BioMed Res. Int. 2021, 2021, 6637503. [Google Scholar] [CrossRef] [PubMed]
- de Farias Gabriel, A.; Silveira, F.M.; Curra, M.; Schuch, L.F.; Wagner, V.P.; Martins, M.A.T.; da Silveira Matte, U.; Siebert, M.; Botton, M.R.; Brunetto, A.T.; et al. Risk factors associated with the development of oral mucositis in pediatric oncology patients: Systematic review and meta-analysis. Oral Dis. 2021. epub ahead of print. [Google Scholar] [CrossRef]
- Proc, P.; Szczepańska, J.; Skiba, A.; Zubowska, M.; Fendler, W.; Młynarski, W. Dental Anomalies as Late Adverse Effect among Young Children Treated for Cancer. Cancer Res. Treat. 2016, 48, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effinger, K.E.; Migliorati, C.A.; Hudson, M.M.; McMullen, K.P.; Kaste, S.C.; Ruble, K.; Guilcher, G.M.; Shah, A.J.; Castellino, S.M. Oral and dental late effects in survivors of childhood cancer: A Children’s Oncology Group report. Support. Care Cancer 2014, 22, 2009–2019. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A.; Galaxy, T. Manipulation of FASTQ data with Galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. Microbiome Analyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Bearfield, C.; Davenport, E.S.; Sivapathasundaram, V.; Allaker, R.P. Possible association between amniotic fluid micro-organism infection and microflora in the mouth. BJOG Int. J. Obstet. Gynaecol. 2002, 109, 527–533. [Google Scholar] [CrossRef]
- Lif Holgerson, P.; Harnevik, L.; Hernell, O.; Tanner, A.C.; Johansson, I. Mode of birth delivery affects oral microbiota in infants. J. Dent. Res. 2011, 90, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Cephas, K.D.; Kim, J.; Mathai, R.A.; Barry, K.A.; Dowd, S.E.; Meline, B.S.; Swanson, K.S. Comparative analysis of salivary bacterial microbiome diversity in edentulous infants and their mothers or primary care givers using pyrosequencing. PLoS ONE 2011, 6, e23503. [Google Scholar] [CrossRef] [PubMed]
- Munyaka, P.M.; Khafipour, E.; Ghia, J.E. External influence of early childhood establishment of gut microbiota and subsequent health implications. Front. Pediatr. 2014, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.D. Diet and Gut Microbiota in Health and Disease. Intest. Microbiome Funct. Asp. Health Dis. 2017, 88, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Francavilla, R.; Ercolini, D.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Filippis, F.; De Pasquale, I.; Di Cagno, R.; Di Toma, M.; Gozzi, G.; et al. Salivary microbiota and metabolome associated with celiac disease. Appl. Environ. Microbiol. 2014, 80, 3416–3425. [Google Scholar] [CrossRef] [Green Version]
- Docktor, M.J.; Paster, B.J.; Abramowicz, S.; Ingram, J.; Wang, Y.E.; Correll, M.; Jiang, H.; Cotton, S.L.; Kokaras, A.S.; Bousvaros, A. Alterations in diversity of the oral microbiome in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Brinksma, A.; Roodbol, P.F.; Sulkers, E.; Kamps, W.A.; de Bont, E.S.; Boot, A.M.; Burgerhof, J.G.; Tamminga, R.Y.; Tissing, W.J. Changes in nutritional status in childhood cancer patients: A prospective cohort study. Clin. Nutr. 2015, 34, 66–73. [Google Scholar] [CrossRef]
- Aarnoutse, R.; de Vos-Geelen, J.M.P.G.M.; Penders, J.; Boerma, E.G.; Warmerdam, F.A.R.M.; Goorts, B.; Olde Damink, S.W.M.; Soons, Z.; Rensen, S.S.M.; Smidt, M.L. Study protocol on the role of intestinal microbiota in colorectal cancer treatment: A pathway to personalized medicine 2.0. Int. J. Colorectal Dis. 2017, 32, 1077–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Shao, N.; Luo, Y.; Liu, H.; Cai, S.; Dong, X. Transcriptome and Zymogram Analyses Reveal a Cellobiose-Dose Related Reciprocal Regulatory Effect on Cellulase Synthesis in Cellulosilyticum ruminicola H1. Front. Microbiol. 2017, 12, 2497. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.P.; Chang, S.H.; Tang, C.Y.; Liou, M.L.; Tsai, S.J.; Lin, Y.L. Composition Analysis and Feature Selection of the Oral Microbiota Associated with Periodontal Disease. BioMed Res. Int. 2018, 2018, 3130607. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Ren, Z.; Li, A.; Li, J.; Xu, S.; Zhang, H.; Jiang, J.; Yang, J.; Luo, Q.; Zhou, K.; et al. Tongue coating microbiome data distinguish patients with pancreatic head cancer from healthy controls. J. Oral Microbiol. 2019, 11, 1563409. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 15, 925. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir-Kocak, F.; Saygin, H.; Saricaoglu, S.; Cetin, D.; Guven, K.; Spröer, C.; Schumann, P.; Klenk, H.P.; Sahin, N.; Isik, K. Kribbella soli sp. nov., isolated from soil. Antonie Leeuwenhoek 2017, 110, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, C. Campylobacter. Clin. Lab. Med. 2015, 35, 289–298. [Google Scholar] [CrossRef]
- Arane, K.; Goldman, R.D. Fusobacterium infections in children. Can. Fam. Physician 2016, 62, 813–814. [Google Scholar]
- Amitay, E.L.; Werner, S.; Vital, M.; Pieper, D.H.; Höfler, D.; Gierse, I.J.; Butt, J.; Balavarca, Y.; Cuk, K.; Brenner, H. Fusobacterium and colorectal cancer: Causal factor or passenger? Results from a large colorectal cancer screening study. Carcinogenesis 2017, 1, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Gholizadeh, P.; Eslami, H.; Yousefi, M.; Asgharzadeh, M.; Aghazadeh, M.; Kafil, H.S. Role of oral microbiome on oral cancers, a review. Biomed. Pharmacother. 2016, 84, 552–558. [Google Scholar] [CrossRef]
- Takahashi, Y.; Park, J.; Hosomi, K.; Yamada, T.; Kobayashi, A.; Yamaguchi, Y.; Iketani, S.; Kunisawa, J.; Mizuguchi, K.; Maeda, N.; et al. Analysis of oral microbiota in Japanese oral cancer patients using 16S rRNA sequencing. J. Oral Biosci. 2019, 61, 120–128. [Google Scholar] [CrossRef]
- Liu, G.; Tang, C.M.; Exley, R.M. Non-pathogenic Neisseria: Members of an abundant, multi-habitat, diverse genus. Microbiology 2015, 161, 1297–1312. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhao, X.; Zhou, Y.; Wang, J.; Ma, R.; Ren, X.; Wang, H.; Zou, L. Characterization of Oral Microbiome and Exploration of Potential Biomarkers in Patients with Pancreatic Cancer. BioMed Res. Int. 2020, 47, 12498. [Google Scholar] [CrossRef] [PubMed]
- Le, P.T.; Hamasuna, R.; Matsumoto, M.; Furubayashi, K.; Hatanaka, M.; Kawai, S.; Yamaguchi, T.; Uehara, K.; Murakami, N.; Yoshioka, M.; et al. The detection of microorganisms related to urethritis from the oral cavity of male patients with urethritis. J. Infect. Chemother. 2017, 23, 668–673. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | Median (25–75%) or % (n) |
|---|---|
| Age at diagnosis (years) | 13.79 (9.13–16.08) |
| Length of therapy (months) | 10.50 (4.50–24.00) |
| Length of antibiotic therapy (days) | 28.00 (8.00–96.50) |
| Sex (%males) | 65% (13/20) |
| Bacteria | Before Treatment (%) | After Treatment (%) | p-Value |
|---|---|---|---|
| Bifidobacterium | 0.37 (0.28–0.68) | 0.72 (0.37–1.00) | 0.1005 |
| Cellulosilyticum | 0.0021 (0.0007–0.0029) | 0.0011 (0.0007–0.0012) | 0.0438 |
| Kribbella | 0.48 (0.32–0.68) | 0.80 (0.50–1.21) | 0.1454 |
| Prevotella | 12.28 (6.32–17.69) | 9.47 (4.93–12.94) | 0.1259 |
| Tannerella | 0.10 (0.05–0.43) | 0.09 (0.01–0.27) | 0.0366 |
| Bacteria | Before Treatment (Cluster 1) | After Treatment (Cluster 1) | p-Value | Before Treatment (Cluster 2) | After Treatment (Cluster 2) | p-Value |
|---|---|---|---|---|---|---|
| Campylobacter | 0.16 (0.05–0.20) | 0.36 (0.20–0.63) | 0.0128 | 0.66 (0.45–0.76) | 0.38 (0.29–0.44) | 0.1097 |
| Cellulosilyticum | 0.0014 (0.0000–0.0029) | 0.0009 (0.0006–0.0012) | 0.5337 | 0.0029 (0.0021–0.0030) | 0.0011 (0.0009–0.0012) | 0.0506 |
| Fusobacterium | 0.09 (0.01–0.33) | 1.52 (0.15–1.81) | 0.0208 | 0.85 (0.26–1.61) | 0.71 (0.41–0.91) | 0.5940 |
| Gardnerella | 0.01 (0.01–0.03) | 0.06 (0.01–0.14) | 0.1823 | 0.03 (0.01–0.05) | 0.02 (0.01–0.03) | 0.1386 |
| Kribbella | 0.34 (0.27–0.58) | 0.64 (0.45–1.18) | 0.1307 | 0.65 (0.49–0.71) | 1.05 (0.69–1.23) | 0.1731 |
| Megasphaera | 0.12 (0.01–0.42) | 0.08 (0.01–0.22) | 0.0912 | 0.49 (0.13–0.71) | 0.31 (0.16–0.60) | 0.5940 |
| Neisseria | 2.28 (0.08–7.18) | 1.21 (0.06–5.40) | 0.4769 | 1.45 (0.69–2.65) | 0.09 (0.01–0.73) | 0.0284 |
| Cluster 1 | Cluster 2 | ||
|---|---|---|---|
| Clinical Feature | Median (25–75%) or % (n) | Median (25–75%) or % (n) | p-Value |
| Age at diagnosis (years) | 10.25 (6.75–14.00) | 16.16 (13.91–17.66) | 0.0049 |
| Length of anticancer therapy (months) | 19.00 (13.00–32.00) | 4.00 (4.00–7.00) | 0.0038 |
| Length of antibiotic therapy (days) | 71.00 (9.00–108.00) | 26.00 (7.00–35.00) | 0.4467 |
| Sex (% males) | 63.64% (7/11) | 66.67% (6/9) | 0.7415 |
| Steroid therapy (Yes) | 27.27% (3/11) | 66.67% (6/9) | 0.1902 |
| Diagnosis: leukemia/lymphoma | 45.45% (5/11) | 66.67% (6/9) | 0.8812 |
| Diagnosis: CNS tumor | 27.27% (3/11) | 11.11% (1/9) | |
| Diagnosis: soft tissue tumor | 27.27% (3/11) | 22.22% (2/9) | |
| Caries (Yes) | 63.64% (7/11) | 55.56% (5/9) | 0.7136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proc, P.; Szczepańska, J.; Zarzycka, B.; Szybka, M.; Borowiec, M.; Płoszaj, T.; Fendler, W.; Chrzanowski, J.; Zubowska, M.; Stolarska, M.; et al. Evaluation of Changes to the Oral Microbiome Based on 16S rRNA Sequencing among Children Treated for Cancer. Cancers 2022, 14, 7. https://doi.org/10.3390/cancers14010007
Proc P, Szczepańska J, Zarzycka B, Szybka M, Borowiec M, Płoszaj T, Fendler W, Chrzanowski J, Zubowska M, Stolarska M, et al. Evaluation of Changes to the Oral Microbiome Based on 16S rRNA Sequencing among Children Treated for Cancer. Cancers. 2022; 14(1):7. https://doi.org/10.3390/cancers14010007
Chicago/Turabian StyleProc, Patrycja, Joanna Szczepańska, Beata Zarzycka, Małgorzata Szybka, Maciej Borowiec, Tomasz Płoszaj, Wojciech Fendler, Jędrzej Chrzanowski, Małgorzata Zubowska, Małgorzata Stolarska, and et al. 2022. "Evaluation of Changes to the Oral Microbiome Based on 16S rRNA Sequencing among Children Treated for Cancer" Cancers 14, no. 1: 7. https://doi.org/10.3390/cancers14010007
APA StyleProc, P., Szczepańska, J., Zarzycka, B., Szybka, M., Borowiec, M., Płoszaj, T., Fendler, W., Chrzanowski, J., Zubowska, M., Stolarska, M., & Młynarski, W. (2022). Evaluation of Changes to the Oral Microbiome Based on 16S rRNA Sequencing among Children Treated for Cancer. Cancers, 14(1), 7. https://doi.org/10.3390/cancers14010007