
Anti-Angiogenic Properties of Ginsenoside Rg3 Epimers: In Vitro Assessment of Single and Combination Treatments

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
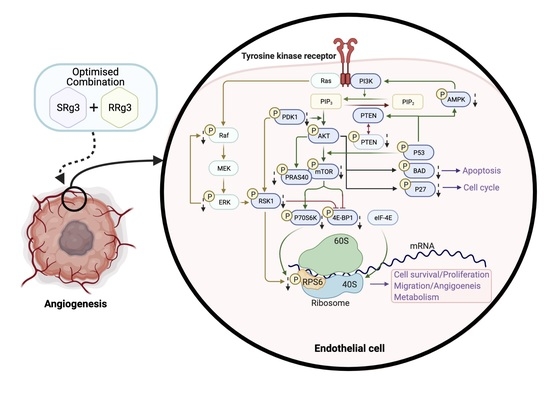
1. Introduction
2. Materials and Methods
2.1. Reagents, Cell Lines, and Cell Culture
2.2. Response Surface Methodology (RSM)
2.3. Proliferation Assay
2.4. Flow Cytometric Analysis of Cell Death
2.5. Flow Cytometric Analysis of Cell Cycles
2.6. Migration Assay
2.7. Loop Formation Assay
2.8. Molecular Docking
2.9. VEGFR2 Specific Interaction
2.10. Quantitative PCR for the Expression of AQP1
2.11. Enzyme-Linked Immunosorbent Assay (ELISA) for the Expression of VEGF-A
2.12. Western Blotting for the Expression of Proteins Involved in Migration and Invasion
2.13. AKT Pathway Phosphorylation Array
2.14. Statistical Analysis
3. Results
3.1. Optimisation of Concentration Combination of SRg3 and RRg3
3.2. Effect of Rg3 on Loop Formation and Migration of Endothelial Cells
3.3. Anti-Proliferative Effects of Rg3 in Endothelial Cells
3.4. The Effect of Rg3 on VEGF, VEGFR2, and Their Interaction
3.5. Effect of Rg3 on the AKT Signalling Pathway and AQP1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ollauri-Ibáñez, C.; Astigarraga, I. Use of antiangiogenic therapies in pediatric solid tumors. Cancers 2021, 13, 253. [Google Scholar] [CrossRef] [PubMed]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular targeting therapy against EGFR family in breast cancer: Progress and future potentials. Cancers 2019, 11, 1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Ganss, R. Modulation of the vascular-immune environment in metastatic cancer. Cancers 2021, 13, 810. [Google Scholar] [CrossRef] [PubMed]
- Taugourdeau-Raymond, S.; Rouby, F.; Default, A.; Jean-Pastor, M.-J. Bevacizumab-induced serious side-effects: A review of the French pharmacovigilance database. Eur. J. Clin. Pharmacol. 2012, 68, 1103–1107. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Haap, M.; Kopp, H.-G.; Lipp, H.-P. Tyrosine kinase inhibitors-a review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Yun, U.-J.; Lee, I.H.; Lee, J.-S.; Shim, J.; Kim, Y.-N. Ginsenoside Rp1, A ginsenoside derivative, augments anti-cancer effects of Actinomycin D via downregulation of an AKT-SIRT1 pathway. Cancers 2020, 12, 605. [Google Scholar] [CrossRef] [Green Version]
- Nakhjavani, M.; Hardingham, J.E.; Palethorpe, H.M.; Tomita, Y.; Smith, E.; Price, T.J.; Townsend, A.R. Ginsenoside Rg3: Potential molecular targets and therapeutic indication in metastatic breast cancer. Medicines 2019, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Nakhjavani, M.; Smith, E.; Townsend, A.R.; Price, T.J.; Hardingham, J.E. Anti-angiogenic properties of ginsenoside Rg3. Molecules 2020, 25, 4905. [Google Scholar] [CrossRef]
- Nakhjavani, M.; Palethorpe, H.M.; Tomita, Y.; Smith, E.; Price, T.J.; Yool, A.J.; Pei, J.V.; Townsend, A.R.; Hardingham, J.E. Stereoselective anti-cancer activities of ginsenoside Rg3 on triple negative breast cancer cell models. Pharmaceuticals 2019, 12, 117. [Google Scholar] [CrossRef] [Green Version]
- Nico, B.; Ribatti, D. Aquaporins in tumor growth and angiogenesis. Cancer Lett. 2010, 294, 135–138. [Google Scholar] [CrossRef]
- Papadopoulos, M.; Saadoun, S.; Verkman, A. Aquaporins and cell migration. Pflügers Arch. Eur. J. Physiol. 2008, 456, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Dorward, H.; Yool, A.J.; Smith, E.; Townsend, A.R.; Price, T.J.; Hardingham, J.E. Role of aquaporin 1 signalling in cancer development and progression. Int. J. Mol. Sci. 2017, 18, 299. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.M.; Lee, J.-H.; Kim, J.-H.; Lee, B.-H.; Yoon, I.-S.; Lee, J.-H.; Kim, D.-H.; Rhim, H.; Kim, Y.; Nah, S.-Y. Stereospecificity of Ginsenoside Rg 3 Action on Ion Channels. Mol. Cells 2004, 18, 383–389. [Google Scholar]
- Kim, J.-H.; Lee, J.-H.; Jeong, S.M.; Lee, B.-H.; Yoon, I.-S.; Lee, J.-H.; Choi, S.-H.; Kim, D.-H.; Park, T.-K.; Kim, B.-K. Stereospecific effects of ginsenoside Rg3 epimers on swine coronary artery contractions. Biol. Pharm. Bull. 2006, 29, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Su, F.; Su, X.; Hu, T.; Hu, S. Stereospecific antioxidant effects of ginsenoside Rg3 on oxidative stress induced by cyclophosphamide in mice. Fitoterapia 2012, 83, 636–642. [Google Scholar] [CrossRef]
- Wei, X.; Chen, J.; Su, F.; Su, X.; Hu, T.; Hu, S. Stereospecificity of ginsenoside Rg3 in promotion of the immune response to ovalbumin in mice. Int. Immunol. 2012, 24, 465–471. [Google Scholar] [CrossRef]
- Wu, R.; Ru, Q.; Chen, L.; Ma, B.; Li, C. Stereospecificity of Ginsenoside Rg3 in the promotion of cellular immunity in hepatoma H22-bearing mice. J. Food Sci. 2014, 79, H1430–H1435. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Choi, W.-I.; Jeon, B.-N.; Choi, K.-C.; Kim, K.; Kim, T.-J.; Ham, J.; Jang, H.J.; Kang, K.S.; Ko, H. Stereospecific effects of ginsenoside 20-Rg3 inhibits TGF-β1-induced epithelial–mesenchymal transition and suppresses lung cancer migration, invasion and anoikis resistance. Toxicology 2014, 322, 23–33. [Google Scholar] [CrossRef]
- Myers, R.H.; Montgomery, D.C.; Anderson-Cook, C.M. Response Surface Methodology: Process and Product Optimization Using Designed Experiments; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Razura-Carmona, F.F.; Pérez-Larios, A.; González-Silva, N.; Herrera-Martínez, M.; Medina-Torres, L.; Sáyago-Ayerdi, S.G.; Sánchez-Burgos, J.A. Mangiferin-loaded polymeric nanoparticles: Optical characterization, effect of anti-topoisomerase I., and cytotoxicity. Cancers 2019, 11, 1965. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.-Y.; Tsou, M.-Y.; Ting, C.-K. Response surface models in the field of anesthesia: A crash course. Acta Anaesthesiol. Taiwanica 2015, 53, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.; Akhter, S.; Ahmad, I.; Hafeez, Z.; Rizvi, M.M.A.; Jain, G.K.; Ahmad, F.J. Improved chemotherapeutic efficacy against resistant human breast cancer cells with co-delivery of Docetaxel and Thymoquinone by Chitosan grafted lipid nanocapsules: Formulation optimization, in vitro and in vivo studies. Colloids Surf. B Biointerfaces 2020, 186, 110603. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Kong, M.; Ayers, G.D.; Lotan, R. Interaction index and different methods for determining drug interaction in combination therapy. J. Biopharm. Stat. 2007, 17, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Malfettone, A.; Silvestris, N.; Paradiso, A.; Mattioli, E.; Simone, G.; Mangia, A. Overexpression of nuclear NHERF1 in advanced colorectal cancer: Association with hypoxic microenvironment and tumor invasive phenotype. Exp. Mol. Pathol. 2012, 92, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Palethorpe, H.; Tomita, Y.; Pei, J.; Townsend, A.; Price, T.; Young, J.; Yool, A.; Hardingham, J. The purified extract from the medicinal plant bacopa monnieri, bacopaside II, inhibits growth of colon cancer cells in vitro by inducing cell cycle arrest and apoptosis. Cells 2018, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Palethorpe, H.M.; Smith, E.; Nakhjavani, M.; Townsend, A.R.; Price, T.J.; Yool, A.J.; Hardingham, J.E. Bumetanide-derived aquaporin 1 inhibitors, AqB013 and AqB050 inhibit tube formation of endothelial cells through induction of apoptosis and impaired migration in vitro. Int. J. Mol. Sci. 2019, 20, 1818. [Google Scholar] [CrossRef] [Green Version]
- Palethorpe, H.; Tomita, Y.; Smith, E.; Pei, J.; Townsend, A.; Price, T.; Young, J.; Yool, A.; Hardingham, J. The aquaporin 1 inhibitor bacopaside II reduces endothelial cell migration and tubulogenesis and induces apoptosis. Int. J. Mol. Sci. 2018, 19, 653. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-s. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thieltges, K.M.; Avramovic, D.; Piscitelli, C.L.; Markovic-Mueller, S.; Binz, H.K.; Ballmer-Hofer, K. Characterization of a drug-targetable allosteric site regulating vascular endothelial growth factor signaling. Angiogenesis 2018, 21, 533–543. [Google Scholar] [CrossRef]
- Brozzo, M.S.; Bjelić, S.; Kisko, K.; Schleier, T.; Leppänen, V.-M.; Alitalo, K.; Winkler, F.K.; Ballmer-Hofer, K. Thermodynamic and structural description of allosterically regulated VEGFR-2 dimerization. Blood 2012, 119, 1781–1788. [Google Scholar] [CrossRef]
- Keung, M.-H.; Chan, L.-S.; Kwok, H.-H.; Wong, R.N.-S.; Yue, P.Y.-K. Role of microRNA-520h in 20 (R)-ginsenoside-Rg3-mediated angiosuppression. J. Ginseng Res. 2016, 40, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-P.; Zhao, F.-L.; Yuan, Y.; Sun, T.-T.; Zhu, L.; Zhang, W.-Y.; Liu, M.-X. Studies on anti-angiogenesis of ginsenoside structure modification HRG in vitro. Biochem. Biophys. Res. Commun. 2017, 492, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Wenthur, C.J.; Gentry, P.R.; Mathews, T.P.; Lindsley, C.W. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, A.K. Use of allosteric targets in the discovery of safer drugs. Med Princ. Pract. 2013, 22, 418–426. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The clinical importance of assessing tumor hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef]
- Luo, D.; Liu, H.; Lin, D.; Lian, K.; Ren, H. The clinicopathologic and prognostic value of hypoxia-inducible factor-2α in cancer patients: A systematic review and meta-analysis. Cancer Epidemiol. Prev. Biomark. 2019, 28, 857–866. [Google Scholar] [CrossRef]
- Ma, Q.; Reiter, R.J.; Chen, Y. Role of melatonin in controlling angiogenesis under physiological and pathological conditions. Angiogenesis 2020, 23, 91–104. [Google Scholar] [CrossRef]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef]
- Carpini, J.D.; Karam, A.K.; Montgomery, L. Vascular endothelial growth factor and its relationship to the prognosis and treatment of breast, ovarian, and cervical cancer. Angiogenesis 2010, 13, 43–58. [Google Scholar] [CrossRef]
- Weigand, M.; Hantel, P.; Kreienberg, R.; Waltenberger, J. Autocrine vascular endothelial growth factor signalling in breast cancer. Evidence from cell lines and primary breast cancer cultures in vitro. Angiogenesis 2005, 8, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Masłowska, K.; Halik, P.K.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The Role of VEGF receptors as molecular target in nuclear medicine for cancer diagnosis and combination therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.C.; Hsu, W.L.; Chang, W.L.; Chou, T.C. Antiangiogenic activity of phthalides-enriched Angelica Sinensis extract by suppressing WSB-1/pVHL/HIF-1α/VEGF signaling in bladder cancer. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Karar, J.; Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Mi, C.; Ma, J.; Wang, K.S.; Zuo, H.X.; Wang, Z.; Li, M.Y.; Piao, L.X.; Xu, G.H.; Li, X.; Quan, Z.S.; et al. Imperatorin suppresses proliferation and angiogenesis of human colon cancer cell by targeting HIF-1α via the mTOR/p70S6K/4E-BP1 and MAPK pathways. J. Ethnopharmacol. 2017, 203, 27–38. [Google Scholar] [CrossRef]
- Saraswati, S.; Kumar, S.; Alhaider, A.A. α-santalol inhibits the angiogenesis and growth of human prostate tumor growth by targeting vascular endothelial growth factor receptor 2-mediated AKT/mTOR/P70S6K signaling pathway. Mol. Cancer 2013, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [Green Version]
- Herbert, S.P.; Costa, G. Sending messages in moving cells: mRNA localization and the regulation of cell migration. Essays. Biochem. 2019, 63, 595–606. [Google Scholar]
- Willett, M.; Brocard, M.; Davide, A.; Morley, S.J. Translation initiation factors and active sites of protein synthesis co-localize at the leading edge of migrating fibroblasts. Biochem. J. 2011, 438, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Petrimpol, M.; Molle, K.D.; Hall, M.N.; Battegay, E.J.; Humar, R. Hypoxia-induced endothelial proliferation requires both mTORC1 and mTORC2. Circ. Res. 2007, 100, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.; Milewski, W.M.; Hara, M.; Steiner, D.F.; Dey, A. GSK-3 inactivation or depletion promotes β-cell replication via down regulation of the CDK inhibitor, p27 (Kip1). Islets 2011, 3, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Hermida, M.A.; Kumar, J.D.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [Green Version]
- Lamanuzzi, A.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Leone, P.; Racanelli, V.; Nico, B.; Ribatti, D.; Ditonno, P.; Prete, M. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget 2018, 9, 20563. [Google Scholar] [CrossRef] [Green Version]
- De Ieso, M.L.; Yool, A.J. Mechanisms of aquaporin-facilitated cancer invasion and metastasis. Front. Chem. 2018, 6, 135. [Google Scholar] [CrossRef] [Green Version]
- Nicchia, G.P.; Stigliano, C.; Sparaneo, A.; Rossi, A.; Frigeri, A.; Svelto, M. Inhibition of aquaporin-1 dependent angiogenesis impairs tumour growth in a mouse model of melanoma. J. Mol. Med. 2013, 91, 613–623. [Google Scholar] [CrossRef]
- Kaneko, K.; Yagui, K.; Tanaka, A.; Yoshihara, K.; Ishikawa, K.; Takahashi, K.; Bujo, H.; Sakurai, K.; Saito, Y. Aquaporin 1 is required for hypoxia-inducible angiogenesis in human retinal vascular endothelial cells. Microvasc. Res. 2008, 75, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Index | Concentration (µM) | ||
|---|---|---|---|---|
| Lowest value (−1) | Centre Value (0) | Highest Value (+1) | ||
| SRg3 | A | 0 | 50 | 100 |
| RRg3 | B | 0 | 25 | 50 |
| Molecule | Binding Score (kJ/mol) (Number of H-Bonds) | ||
|---|---|---|---|
| VEGFR1 | VEGFR2 1 | VEGFR2 2 | |
| SRg3 | 4.8 (0) | −9.0 (8) | −7.2 (3) |
| RRg3 | −7.4 (6) | −8.9 (5) | −7.0 (5) |
| Sorafenib | −4.9 (0) | −9.9 (0) | − |
| Lenvatinib | −8.9 (0) | −9.1 (0) | − |
| Compound | IC50 | EC50 | 95% CI 1 | R Squared |
|---|---|---|---|---|
| VEGF | − | 0.001 | 0.001–0.002 | 0.9781 |
| Bevacizumab | 0.11 | − | 0.08–0.15 | 0.9644 |
| SRg3 | 21.23 | − | 3.25–8008 | 0.3391 |
| RRg3 | 20.67 | − | 15.06–30.82 | 0.6963 |
| SRg3 + VEGF | − | 27.95 | 23.79–32.44 | 0.9670 |
| RRg3 + VEGF | − | 6.52 | 4.84–8.66 | 0.9411 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakhjavani, M.; Smith, E.; Yeo, K.; Palethorpe, H.M.; Tomita, Y.; Price, T.J.; Townsend, A.R.; Hardingham, J.E. Anti-Angiogenic Properties of Ginsenoside Rg3 Epimers: In Vitro Assessment of Single and Combination Treatments. Cancers 2021, 13, 2223. https://doi.org/10.3390/cancers13092223
Nakhjavani M, Smith E, Yeo K, Palethorpe HM, Tomita Y, Price TJ, Townsend AR, Hardingham JE. Anti-Angiogenic Properties of Ginsenoside Rg3 Epimers: In Vitro Assessment of Single and Combination Treatments. Cancers. 2021; 13(9):2223. https://doi.org/10.3390/cancers13092223
Chicago/Turabian StyleNakhjavani, Maryam, Eric Smith, Kenny Yeo, Helen M. Palethorpe, Yoko Tomita, Tim J. Price, Amanda R. Townsend, and Jennifer E. Hardingham. 2021. "Anti-Angiogenic Properties of Ginsenoside Rg3 Epimers: In Vitro Assessment of Single and Combination Treatments" Cancers 13, no. 9: 2223. https://doi.org/10.3390/cancers13092223