
CNA Profiling of Single CTCs in Locally Advanced Esophageal Cancer Patients during Therapy Highlights Unexplored Molecular Pathways

 ,
,  ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients Recruitment and Sample Collection
2.2. Cell Culture
2.3. CTC Enrichment, Grab-All Assay and Single Cell Isolation
2.4. Single CTC WGA and Quality Control (QC)
2.5. Library Preparation and Next Generation Sequencing (NGS)
2.6. CNA Calling from Low-Resolution Whole Genome Sequencing
2.7. Generation of Jaccard Indexes and Statistical Analysis
2.8. Identification of Recurrent Esophageal Cancer Aberrations on Single CTCs
2.9. Enrichment and Network Generation
3. Results
3.1. Grab-All Assay Setup
3.2. Detection Efficiency of Grab-All Assay on Metastatic Esophageal Cancer Patients
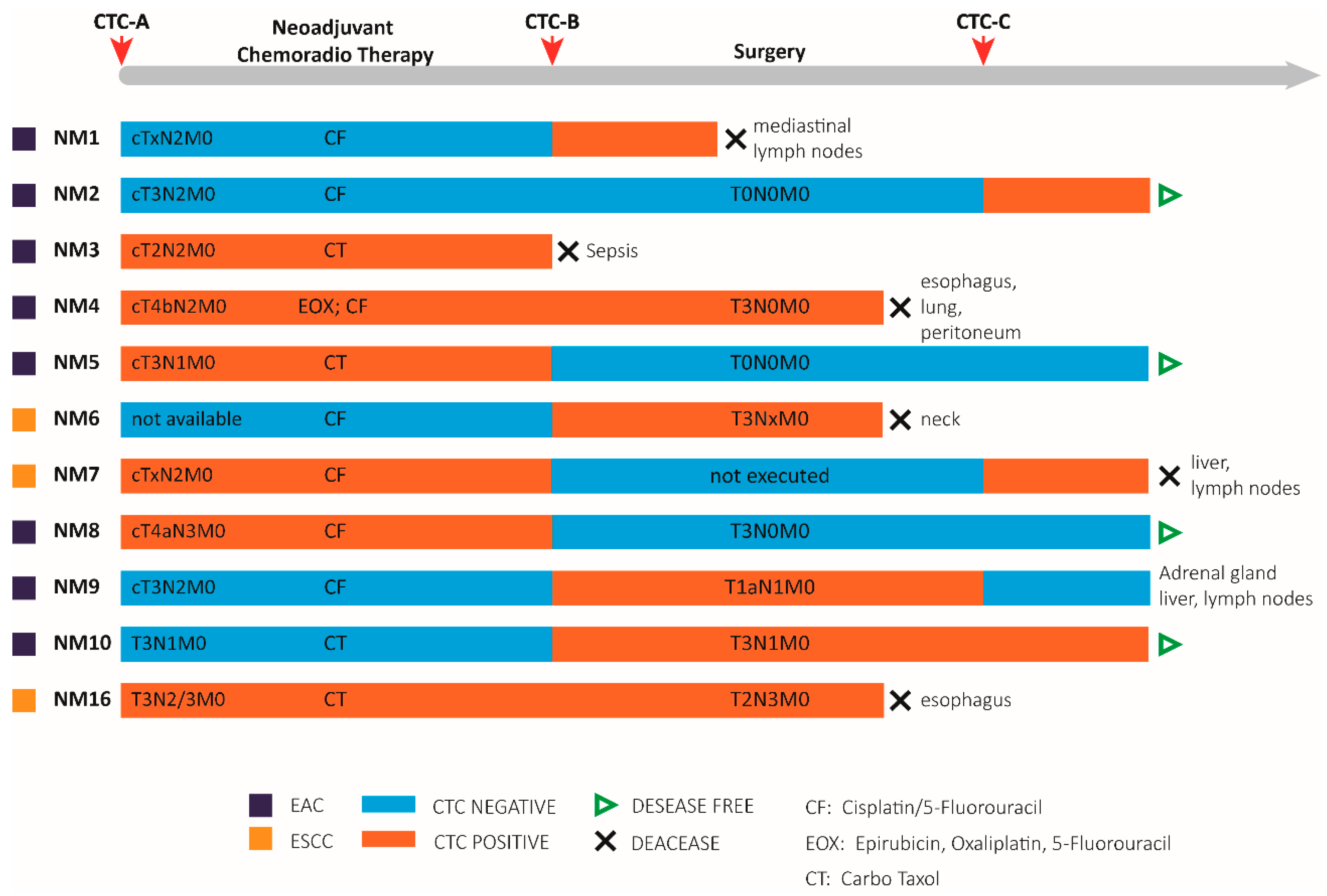
3.3. Patient Characteristics
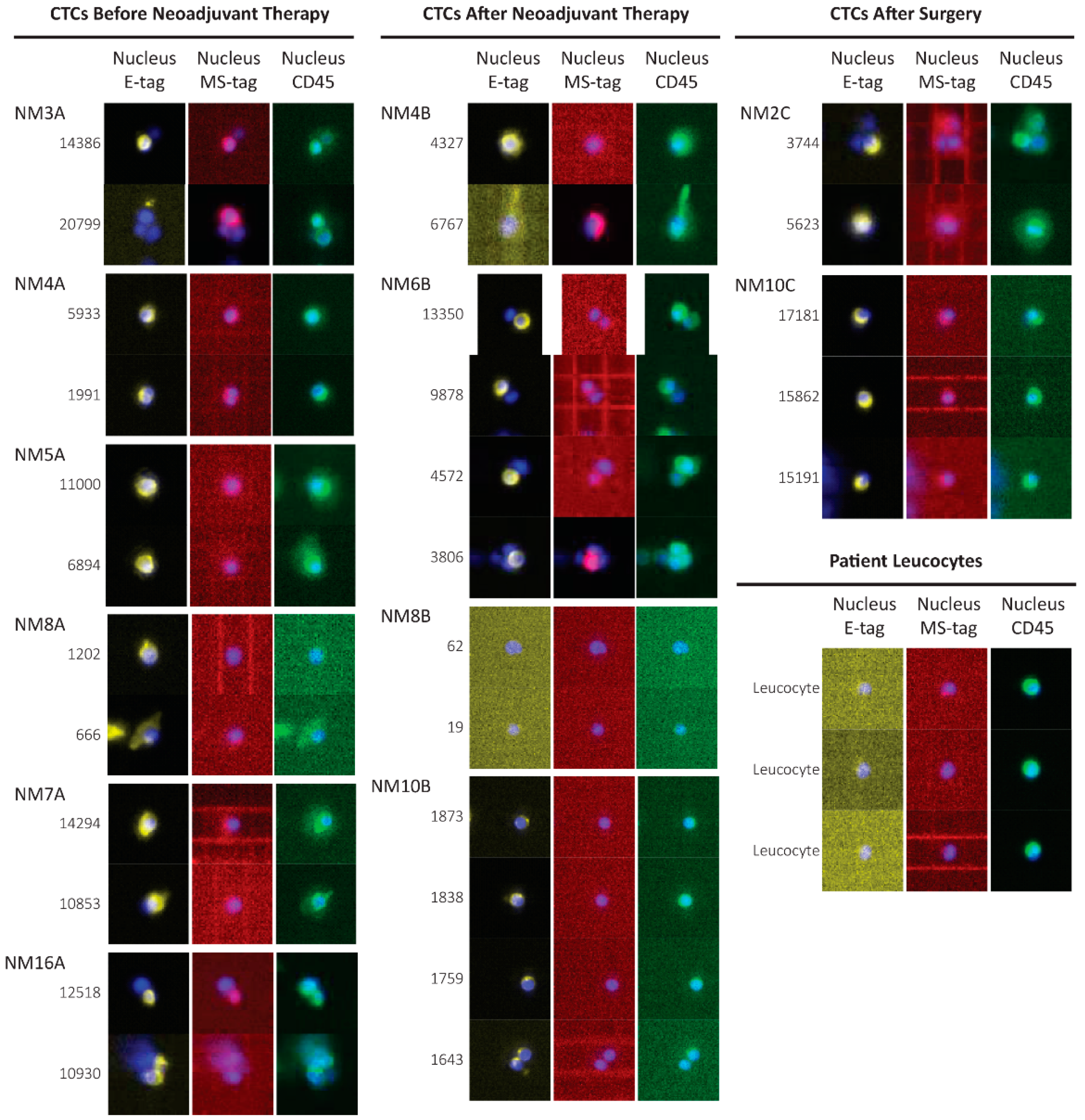
3.4. CTCs Identification in the Blood of Non-Metastatic Patients during Treatment
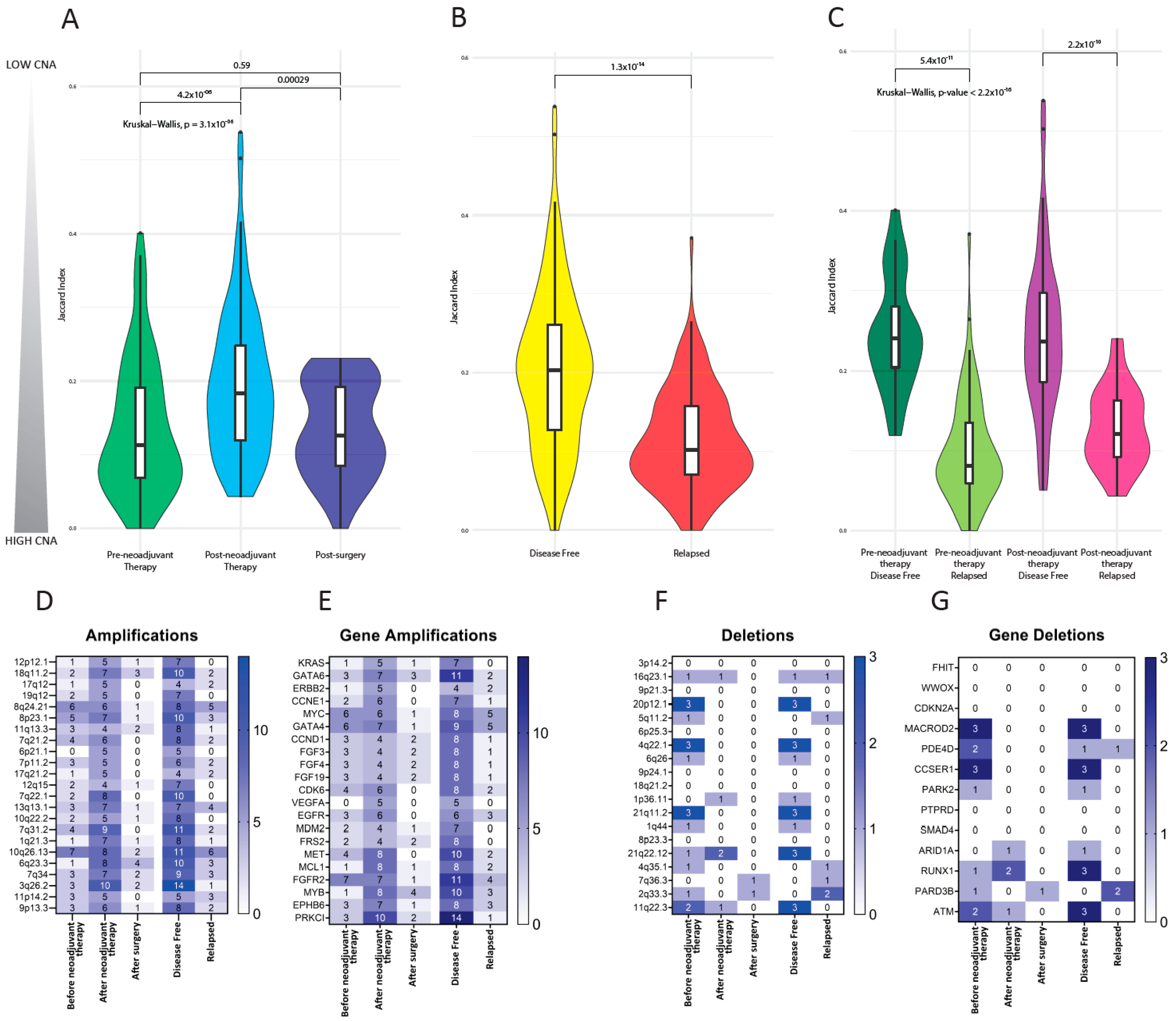
3.5. Copy Number Alterations in CTCs
3.6. Investigation of Previously Reported Esophageal Cancer-Specific Chromosomal Aberration in CTCs
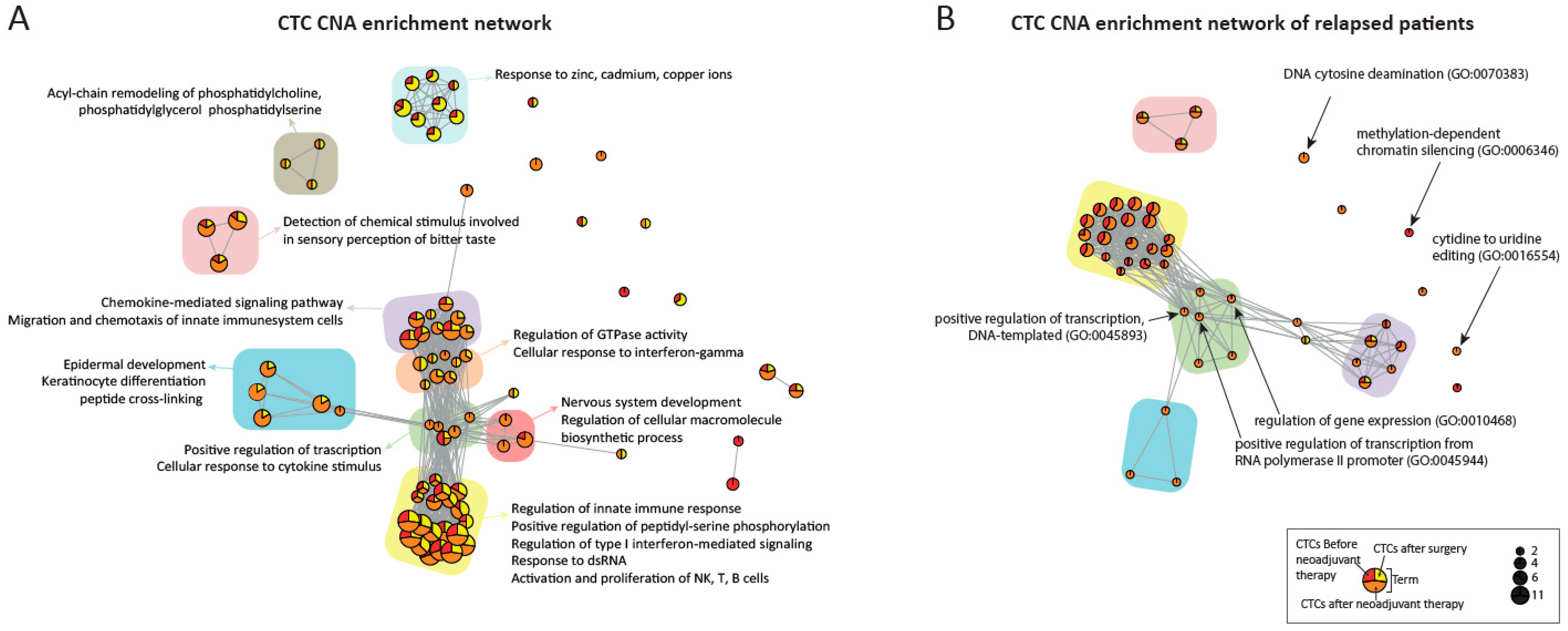
3.7. Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal Cancer. Nat. Rev. Dis. Prim. 2017, 3, 17048. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.; van Lanschot, J.J.B.; Hulshof, M.C.C.M.; van Hagen, P.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.L.; van Laarhoven, H.W.M.; Nieuwenhuijzen, G.A.P.; Hospers, G.A.P.; Bonenkamp, J.J.; et al. Neoadjuvant Chemoradiotherapy plus Surgery versus Surgery Alone for Oesophageal or Junctional Cancer (CROSS): Long-Term Results of a Randomised Controlled Trial. Lancet Oncol. 2015, 16, 1090–1098. [Google Scholar] [CrossRef]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated Genomic Characterization of Oesophageal Carcinoma. Nature 2017, 541, 169–175. [Google Scholar]
- Dulak, A.M.; Schumacher, S.E.; Van Lieshout, J.; Imamura, Y.; Fox, C.; Shim, B.; Ramos, A.H.; Saksena, G.; Baca, S.C.; Baselga, J.; et al. Gastrointestinal Adenocarcinomas of the Esophagus, Stomach, and Colon Exhibit Distinct Patterns of Genome Instability and Oncogenesis. Cancer Res. 2012, 72, 4383–4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhawan, D.; Zack, T.I.; Ren, Y.; Strickland, M.R.; Lamothe, R.; Schumacher, S.E.; Tsherniak, A.; Besche, H.C.; Rosenbluh, J.; Shehata, S.; et al. Cancer Vulnerabilities Unveiled by Genomic Loss. Cell 2012, 150, 842–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic Catastrophes Frequently Arise in Esophageal Adenocarcinoma and Drive Tumorigenesis. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups with Therapeutic Relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-Genome Sequencing Provides New Insights into the Clonal Architecture of Barrett’s Esophagus and Esophageal Adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Noorani, A.; Li, X.; Goddard, M.; Crawte, J.; Alexandrov, L.B.; Secrier, M.; Eldridge, M.D.; Bower, L.; Weaver, J.; Lao-Sirieix, P.; et al. Genomic Evidence Supports a Clonal Diaspora Model for Metastases of Esophageal Adenocarcinoma. Nat. Genet. 2020, 52, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Maltoni, R.; Fici, P.; Amadori, D.; Gallerani, G.; Cocchi, C.; Zoli, M.; Rocca, A.; Cecconetto, L.; Folli, S.; Scarpi, E.; et al. Circulating Tumor Cells in Early Breast Cancer: A Connection with Vascular Invasion. Cancer Lett. 2015, 367, 43–48. [Google Scholar] [CrossRef]
- Reeh, M.; Effenberger, K.E.; Koenig, A.M.; Riethdorf, S.; Eichstädt, D.; Vettorazzi, E.; Uzunoglu, F.G.; Vashist, Y.K.; Izbicki, J.R.; Pantel, K.; et al. Circulating Tumor Cells as a Biomarker for Preoperative Prognostic Staging in Patients with Esophageal Cancer. Ann. Surg. 2015, 261, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Rossi, T.; Gallerani, G.; Angeli, D.; Cocchi, C.; Bandini, E.; Fici, P.; Gaudio, M.; Martinelli, G.; Rocca, A.; Maltoni, R.; et al. Single-Cell NGS-Based Analysis of Copy Number Alterations Reveals New Insights in Circulating Tumor Cells Persistence in Early-Stage Breast Cancer. Cancers 2020, 12, 2490. [Google Scholar] [CrossRef]
- Kuvendjiska, J.; Bronsert, P.; Martini, V.; Lang, S.; Pitman, M.; Hoeppner, J.; Kulemann, B. Non-Metastatic Esophageal Adenocarcinoma: Circulating Tumor Cells in the Course of Multimodal Tumor Treatment. Cancers 2019, 11, 397. [Google Scholar] [CrossRef] [Green Version]
- Woestemeier, A.; Ghadban, T.; Riethdorf, S.; Harms-Effenberger, K.; Konczalla, L.; Uzunoglu, F.G.; Izbicki, J.R.; Pantel, K.; Bockhorn, M.; Reeh, M. Absence of HER2 Expression of Circulating Tumor Cells in Patients with Non-Metastatic Esophageal Cancer. Anticancer Res. 2018, 38, 5665–5669. [Google Scholar] [CrossRef]
- Carter, L.; Rothwell, D.G.; Mesquita, B.; Smowton, C.; Leong, H.S.; Fernandez-Gutierrez, F.; Li, Y.; Burt, D.J.; Antonello, J.; Morrow, C.J.; et al. Molecular Analysis of Circulating Tumor Cells Identifies Distinct Copy-Number Profiles in Patients with Chemosensitive and Chemorefractory Small-Cell Lung Cancer. Nat. Med. 2016, 23, 114–119. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Kraan, J.; Bolt, J.; Van Der Spoel, P.; Elstrodt, F.; Schutte, M.; Martens, J.W.M.; Gratama, J.W.; Sleijfer, S.; Foekens, J.A. Anti-Epithelial Cell Adhesion Molecule Antibodies and the Detection of Circulating Normal-like Breast Tumor Cells. J. Natl. Cancer Inst. 2009, 101, 61–66. [Google Scholar] [CrossRef]
- Gall, T.M.H.; Frampton, A.E. Gene of the Month: E-Cadherin (CDH1). J. Clin. Pathol. 2013, 66, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Portillo, F.; Cano, A. Transcriptional Regulation of Cadherins during Development and Carcinogenesis. Int. J. Dev. Biol. 2004, 48, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Joosse, S.A.; Hannemann, J.; Spötter, J.; Bauche, A.; Andreas, A.; Muller, V.; Pantel, K. Changes in Keratin Expression during Metastatic Progression of Breast Cancer: Impact on the Detection of Circulating Tumor Cells. Clin. Cancer Res. 2012, 18, 993–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Chen, H.C.; Zhang, D.; Fu, J. Twist: A Molecular Target in Cancer Therapeutics. Tumour. Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bennett, S.A.L.; Wang, L. Role of E-Cadherin and Other Cell Adhesion Molecules in Survival and Differentiation of Human Pluripotent Stem Cells. Cell Adh. Migr. 2012, 6, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Fu, Z.; Xu, S.; Xu, Y.; Xu, P. The Prognostic Value of CD44 Expression in Gastric Cancer: A Meta-Analysis. Biomed. Pharmacother. 2014, 68, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Luo, W.; Hu, R.-T.; Zhou, Y.; Qin, S.-Y.; Jiang, H.-X. Meta-Analysis of Prognostic and Clinical Significance of CD44v6 in Esophageal Cancer. Medicine 2015, 94, e1238. [Google Scholar] [CrossRef]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is Required for Two Consecutive Steps in HGF/c-Met Signaling. Genes Dev. 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [Green Version]
- Belthier, G.; Homayed, Z.; Grillet, F.; Duperray, C.; Vendrell, J.; Krol, I.; Bravo, S.; Boyer, J.-C.; Villeronce, O.; Vitre-Boubaker, J.; et al. CD44v6 Defines a New Population of Circulating Tumor Cells Not Expressing EpCAM. Cancers 2021, 13, 4966. [Google Scholar] [CrossRef]
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.W.; Bates, S.E. ABCG2: Determining Its Relevance in Clinical Drug Resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, S.; Okumura, T.; Ito, T.; Kondo, K.; Ortiz, C.; Tanaka, E.; Watanabe, G.; Itami, A.; Sakai, Y.; Shimada, Y. ABCG2 Expression is an Independent Unfavorable Prognostic Factor in Esophageal Squamous Cell Carcinoma. Oncology 2006, 71, 251–258. [Google Scholar] [CrossRef]
- Lordick, F.; Mariette, C.; Haustermans, K.; Obermannová, R.; Arnold, D.; Committee, G. Oesophageal Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up. Ann. Oncol. 2016, 27, v50–v57. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Line, E.C.; Peterson, W.D.; Brenz, R.; Mcgrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; et al. Isolation and Characterization of a Spontaneously Immortalized Isolation and Characterization of a Spontaneously Immortalized Human Breast. Cancer Res. 1990, 50, 6075–6086. [Google Scholar] [PubMed]
- Fici, P.; Gallerani, G.; Morel, A.-P.; Mercatali, L.; Ibrahim, T.; Scarpi, E.; Amadori, D.; Puisieux, A.; Rigaud, M.; Fabbri, F. Splicing Factor Ratio as an Index of Epithelial-Mesenchymal Transition and Tumor Aggressiveness in Breast Cancer. Oncotarget 2016, 8, 2423–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltoni, R.; Gallerani, G.; Fici, P.; Rocca, A.; Fabbri, F. CTCs in Early Breast Cancer: A Path Worth Taking. Cancer Lett. 2016, 376, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and Recovery of Circulating Colon Cancer Cells Using a Dielectrophoresis-Based Device: KRAS Mutation Status in Pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef]
- Ferrarini, A.; Forcato, C.; Buson, G.; Tononi, P.; Del Monaco, V.; Terracciano, M.; Bolognesi, C.; Fontana, F.; Medoro, G.; Neves, R.; et al. A Streamlined Workflow for Single-Cells Genome-Wide Copy-Number Profiling by Low-Pass Sequencing of LM-PCR Whole-Genome Amplification Products. PLoS ONE 2018, 13, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular Profiling of Single Circulating Tumor Cells with Diagnostic Intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and Genetic Profiling of Circulating Tumor Cells in Small-Cell Lung Cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-Cadherin Promotes Motility in Human Breast Cancer Cells Regardless of Their E-Cadherin Expression. J. Cell Biol. 1999, 147, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R.; Badve, S.; Nakshatri, H. CD44+/CD24- Breast Cancer Cells Exhibit Enhanced Invasive Properties: An Early Step Necessary for Metastasis. Breast Cancer Res. 2006, 8, R59. [Google Scholar] [CrossRef] [Green Version]
- Gress, D.M.; Edge, S.B.; Greene, F.L.; Washington, M.K.; Asare, E.A.; Brierley, J.D.; Byrd, D.R.; Compton, C.C.; Jessup, J.M.; Winchester, D.P.; et al. AJCC Cancer Staging Manual; Amin, M.B., Edge, S.B., Greene, F.L., Byrd, D.R., Brookland, R.K., Washington, M.K., Gershenwald, J.E., Compton, C.C., Hess, K.R., Sullivan, D.C., et al., Eds.; Springer International Publishing: Cham, Switzerland, 2017; ISBN 978-3-319-40617-6. [Google Scholar]
- Shinohara, M.; Io, K.; Shindo, K.; Matsui, M.; Sakamoto, T.; Tada, K.; Kobayashi, M.; Kadowaki, N.; Takaori-Kondo, A. APOBEC3B Can Impair Genomic Stability by Inducing Base Substitutions in Genomic DNA in Human Cells. Sci. Rep. 2012, 2, 806. [Google Scholar] [CrossRef] [Green Version]
- Roper, N.; Gao, S.; Maity, T.K.; Banday, A.R.; Zhang, X.; Venugopalan, A.; Cultraro, C.M.; Patidar, R.; Sindiri, S.; Brown, A.-L.; et al. APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep. 2019, 26, 2651–2666.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Wang, W.; Tang, Y.; Zhou, D.; Gao, Y.; Zhang, Q.; Zhou, X.; Zhu, H.; Xing, L.; Yu, J. MRNA and Methylation Profiling of Radioresistant Esophageal Cancer Cells: The Involvement of Sall2 in Acquired Aggressive Phenotypes. J. Cancer 2017, 8, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Lobbardi, R.; Pinder, J.; Martinez-Pastor, B.; Theodorou, M.; Blackburn, J.S.; Abraham, B.J.; Namiki, Y.; Mansour, M.; Abdelfattah, N.S.; Molodtsov, A.; et al. TOX Regulates Growth, DNA Repair, and Genomic Instability in T-Cell Acute Lymphoblastic Leukemia. Cancer Discov. 2017, 7, 1336–1353. [Google Scholar] [CrossRef] [Green Version]
- Meier, T.; Timm, M.; Montani, M.; Wilkens, L. Gene Networks and Transcriptional Regulators Associated with Liver Cancer Development and Progression. BMC Med. Genom. 2021, 14, 41. [Google Scholar] [CrossRef]
- Zhang, L.-W.; Wang, B.; Yang, J.-X.; Yang, H. Circ-PRMT5 Stimulates Migration in Esophageal Cancer by Binding MiR-203. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9965–9972. [Google Scholar]
- Shi, X.; Chen, Z.; Hu, X.; Luo, M.; Sun, Z.; Li, J.; Shi, S.; Feng, X.; Zhou, C.; Li, Z.; et al. AJUBA Promotes the Migration and Invasion of Esophageal Squamous Cell Carcinoma Cells through Upregulation of MMP10 and MMP13 Expression. Oncotarget 2016, 7, 36407–36418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garancher, A.; Lin, C.Y.; Morabito, M.; Richer, W.; Rocques, N.; Larcher, M.; Bihannic, L.; Smith, K.; Miquel, C.; Leboucher, S.; et al. NRL and CRX Define Photoreceptor Identity and Reveal Subgroup-Specific Dependencies in Medulloblastoma. Cancer Cell 2018, 33, 435–449.e6. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, Y.; Wang, J.; Kang, M.; Tang, W.; Chen, S. Assessment of PPARGC1A, PPARGC1B, and PON1 Genetic Polymorphisms in Esophageal Squamous Cell Carcinoma Susceptibility in the Eastern Chinese Han Population: A Case–Control Study Involving 2351 Subjects. DNA Cell Biol. 2020, 39, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Baslan, T.; Kendall, J.; Volyanskyy, K.; McNamara, K.; Cox, H.; D’Italia, S.; Ambrosio, F.; Riggs, M.; Rodgers, L.; Leotta, A.; et al. Novel Insights into Breast Cancer Copy Number Genetic Heterogeneity Revealed by Single-Cell Genome Sequencing. Elife 2020, 9, 1–21. [Google Scholar] [CrossRef]
- Yasui, K.; Imoto, I.; Fukuda, Y.; Pimkhaokham, A.; Yang, Z.-Q.; Naruto, T.; Shimada, Y.; Nakamura, Y.; Inazawa, J. Identification of Target Genes within an Amplicon at 14q12-Q13 in Esophageal Squamous Cell Carcinoma. Genes Chromosom. Cancer 2001, 32, 112–118. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, A.; Zheng, L.; Johnathan, A.-F.; Zhang, J.; Zhang, G. The Biological Significance and Regulatory Mechanism of C-Myc Binding Protein 1 (MBP-1). Int. J. Mol. Sci. 2018, 19, 3868. [Google Scholar] [CrossRef] [Green Version]
- Sawada, G.; Ueo, H.; Matsumura, T.; Uchi, R.; Ishibashi, M.; Mima, K.; Kurashige, J.; Takahashi, Y.; Akiyoshi, S.; Sudo, T.; et al. CHD8 Is an Independent Prognostic Indicator That Regulates Wnt/β-Catenin Signaling and the Cell Cycle in Gastric Cancer. Oncol. Rep. 2013, 30, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Lin, L.; Shen, Z.; Li, Y.; Cao, H.; Peng, L.; Qiu, Y.; Cheng, X.; Meng, M.; Lu, D.; et al. CEBPG Promotes Esophageal Squamous Cell Carcinoma Progression by Enhancing PI3K-AKT Signaling. Am. J. Cancer Res. 2020, 10, 3328–3344. [Google Scholar] [PubMed]
- Diamantopoulou, Z.; Castro-Giner, F.; Aceto, N. Circulating Tumor Cells: Ready for Translation? J. Exp. Med. 2020, 217, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, M.J.D.; Ruurda, J.P.; Lolkema, M.P.; Sitarz, R.; ten Kate, F.J.W.; van Hillegersberg, R. The Role of Biological Markers of Epithelial to Mesenchymal Transition in Oesophageal Adenocarcinoma, an Immunohistochemical Study. J. Clin. Pathol. 2015, 68, 529–535. [Google Scholar] [CrossRef]
- Murugaesu, N.; Wilson, G.A.; Birkbak, N.J.; Watkins, T.B.K.; McGranahan, N.; Kumar, S.; Abbassi-Ghadi, N.; Salm, M.; Mitter, R.; Horswell, S.; et al. Tracking the Genomic Evolution of Esophageal Adenocarcinoma through Neoadjuvant Chemotherapy. Cancer Discov. 2015, 5, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plum, P.S.; Löser, H.; Zander, T.; Essakly, A.; Bruns, C.J.; Hillmer, A.M.; Alakus, H.; Schröder, W.; Büttner, R.; Gebauer, F.; et al. GATA Binding Protein 6 (GATA6) is Co-Amplified with PIK3CA in Patients with Esophageal Adenocarcinoma and Is Linked to Neoadjuvant Therapy. J. Cancer Res. Clin. Oncol. 2021, 147, 1031. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Cozic, N.; de la Fouchardière, C.; Meriaux, E.; Plaza, J.; Mineur, L.; Guimbaud, R.; Samalin, E.; Mary, F.; Lecomte, T.; et al. The Activity of Crizotinib in Chemo-Refractory MET-Amplified Esophageal and Gastric Adenocarcinomas: Results from the AcSé-Crizotinib Program. Target. Oncol. 2021, 16, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Bandla, S.; Pennathur, A.; Luketich, J.D.; Beer, D.G.; Lin, L.; Bass, A.J.; Godfrey, T.E.; Litle, V.R. Comparative Genomics of Esophageal Adenocarcinoma and Squamous Cell Carcinoma. Ann. Thorac. Surg. 2012, 93, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTC Count | |||||
|---|---|---|---|---|---|
| Before Therapy (A) | After Neoadjuvant Therapy (B) | After Surgery (C) | |||
| 20 | 27 | 10 | |||
| Relapsed | Disease Free | Relapsed | Disease Free | Relapsed | Disease Free |
| 15 | 5 | 13 | 14 | 1 | 9 |
| Gene | Term | Percentages of CTC in Relapsed Patients (n = 29) | Percentages of CTC in Disease-Free Patients (n = 28) | Association with EC and Cancer [Reference] |
|---|---|---|---|---|
| APOBEC3 cluster gene | cytidine to uridine editing (GO:0016554) DNA cytosine deamination (GO:0070383) | 24.1% | 7.1% | [43,44] |
| SALL2 | regulation of gene expression (GO:0010468) positive regulation of transcription, DNA-templated (GO:0045893) positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 27.6% | 0% | [45] |
| TOX4 | regulation of gene expression (GO:0010468) positive regulation of transcription, DNA-templated (GO:0045893) | 27.6% | 0% | [46] |
| ZNF219 | regulation of gene expression (GO:0010468) | 27.6% | 0.0% | [47] |
| PRMT5 | regulation of gene expression (GO:0010468) | 27.6% | 3.6% | [48] |
| AJUBA | regulation of gene expression (GO:0010468) | 24.1% | 3.6% | [49] |
| NRL | regulation of gene expression (GO:0010468) positive regulation of transcription, DNA-templated (GO:0045893) positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 24.1% | 3.6% | [50] |
| PPARGC1A | regulation of gene expression (GO:0010468) positive regulation of transcription, DNA-templated (GO:0045893) positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 41.4% | 10.7% | [51] |
| MAP2K2 | regulation of gene expression (GO:0010468) positive regulation of transcription, DNA-templated (GO:0045893) | 27.6% | 7.1% | [52] |
| BAZ1A | regulation of gene expression (GO:0010468) | 24.1% | 7.1% | [53] |
| DENND4A | regulation of gene expression (GO:0010468) | 24.1% | 7.1% | [54] |
| CHD8 | positive regulation of transcription, DNA-templated (GO:0045893) positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 27.6% | 0.0% | [55] |
| SUPT16H | positive regulation of transcription, DNA-templated (GO:0045893) positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 27.6% | 0.0% | |
| CEBPE | positive regulation of transcription, DNA-templated (GO:0045893) positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 24.1% | 3.6% | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallerani, G.; Rossi, T.; Valgiusti, M.; Angeli, D.; Fici, P.; De Fanti, S.; Bandini, E.; Cocchi, C.; Frassineti, G.L.; Bonafè, M.; et al. CNA Profiling of Single CTCs in Locally Advanced Esophageal Cancer Patients during Therapy Highlights Unexplored Molecular Pathways. Cancers 2021, 13, 6369. https://doi.org/10.3390/cancers13246369
Gallerani G, Rossi T, Valgiusti M, Angeli D, Fici P, De Fanti S, Bandini E, Cocchi C, Frassineti GL, Bonafè M, et al. CNA Profiling of Single CTCs in Locally Advanced Esophageal Cancer Patients during Therapy Highlights Unexplored Molecular Pathways. Cancers. 2021; 13(24):6369. https://doi.org/10.3390/cancers13246369
Chicago/Turabian StyleGallerani, Giulia, Tania Rossi, Martina Valgiusti, Davide Angeli, Pietro Fici, Sara De Fanti, Erika Bandini, Claudia Cocchi, Giovanni Luca Frassineti, Massimiliano Bonafè, and et al. 2021. "CNA Profiling of Single CTCs in Locally Advanced Esophageal Cancer Patients during Therapy Highlights Unexplored Molecular Pathways" Cancers 13, no. 24: 6369. https://doi.org/10.3390/cancers13246369