
Single-Cell Sequencing: Biological Insight and Potential Clinical Implications in Pediatric Leukemia


Abstract
:Simple Summary
Abstract
1. Introduction
2. Genomic, Epigenomic, and Transcriptomic Background of Pediatric Leukemia

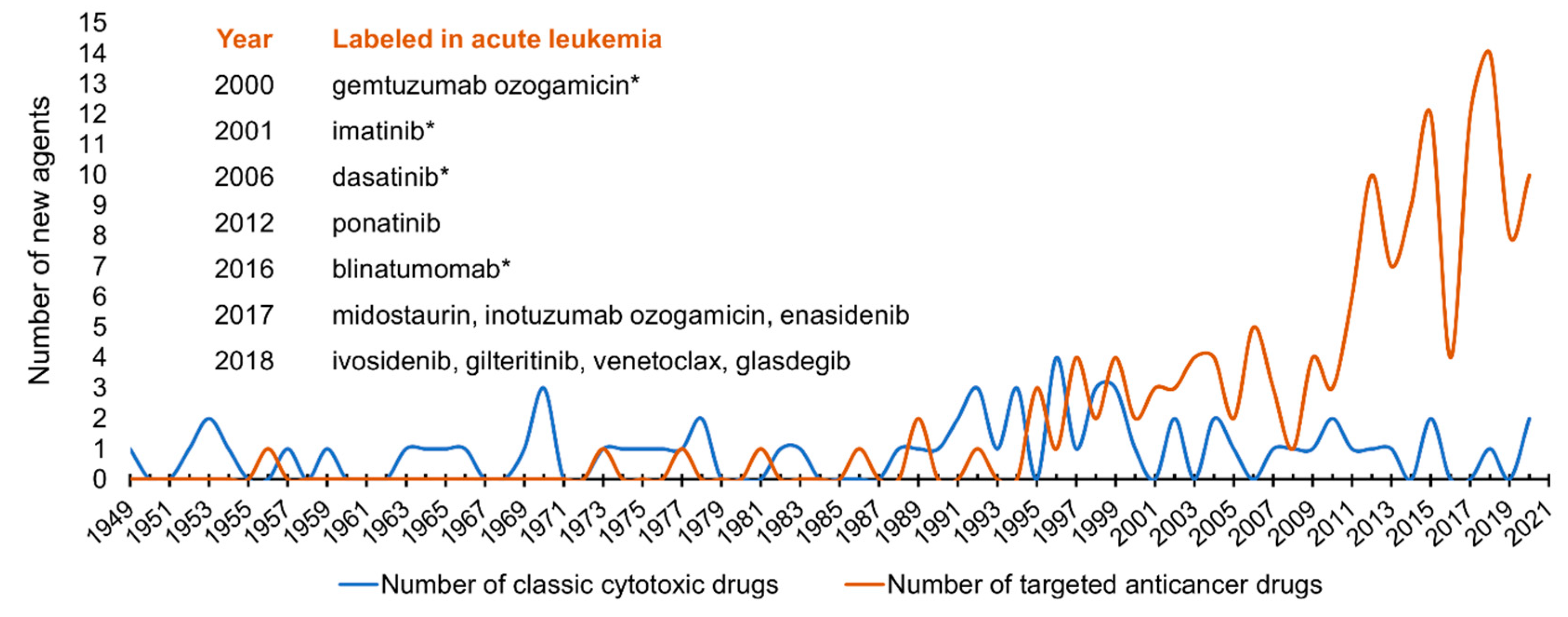{kind=link}
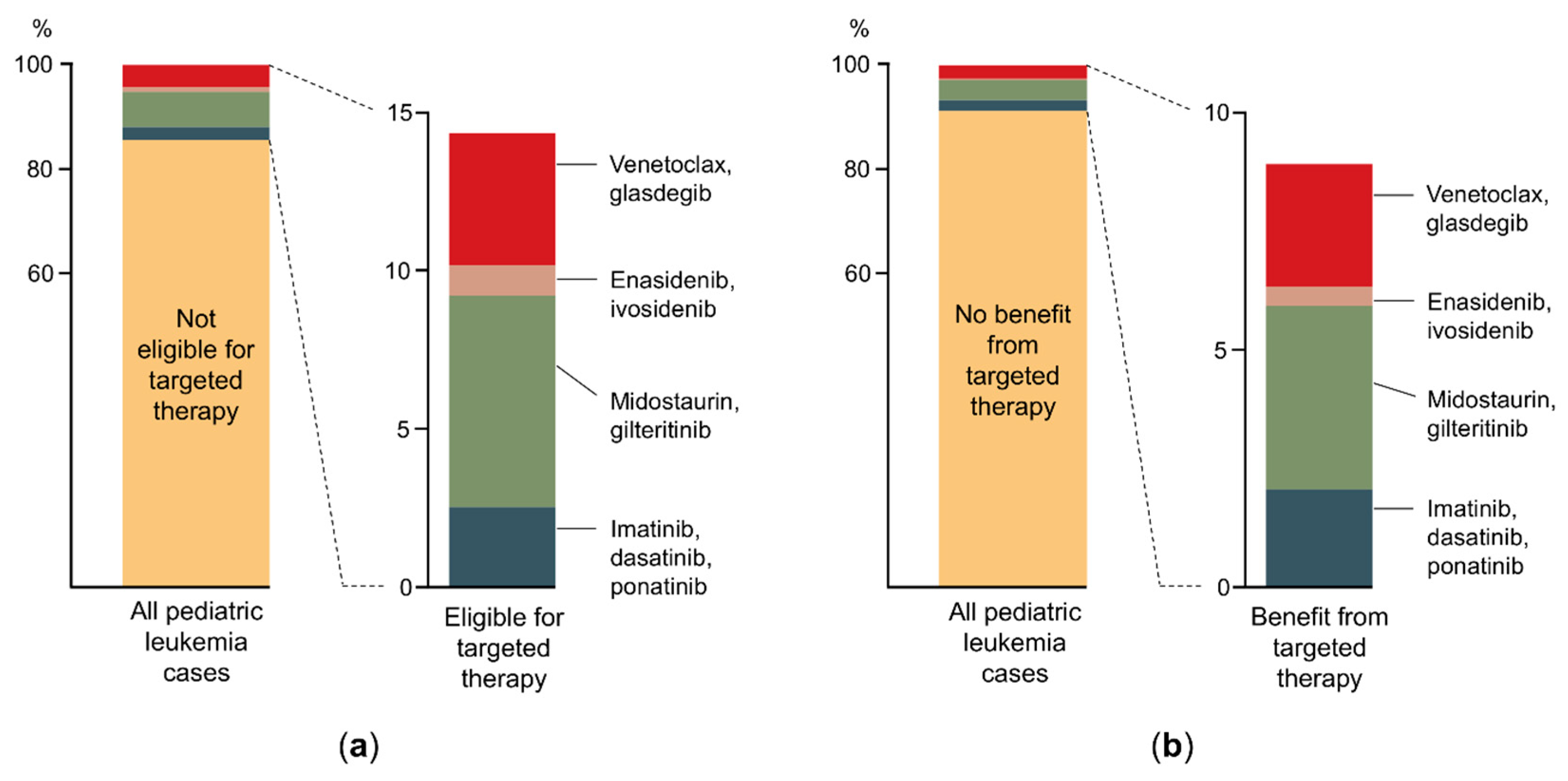{kind=link}
| Genetic Subgroup | Possible Small-Molecule Targeted Agent or Risk Prediction | Refs |
|---|---|---|
| ALL | ||
| Ph+ (BCR-ABL1) | TKI, Aurora kinase (alisertib) | [100] |
| TCF3-PBX1 | PI3Ki (idelalisib), considerable response to chemotherapy | [101] |
| TCF3-HLF | therapy-resistant disease, BCL2i (venetoclax) | [102] |
| KMT2A rearrangement | FLT3i (quizartinib), DOT1L-i (pinometostat), proteasome inhibitor (bortezomib), HDACi ± DNMTi aHSCT when poor response to induction is detected | [103] |
| ETV6-RUNX1, ETV6-RUNX1-like, DUX4 rearrangement (often coincide with ERGdel) | Excellent prognosis, reduced intensity treatment may be considered | [57] |
| MEF2D rearrangement | HDACi (panabinostat), proteasome inhibitor (bortezomib) | [96] |
| ZNF384 rearrangement | FLT3i (sunitinib) | [104] |
| NT5C2 alteration | Anticipate thiopurine resistance, often associated with relapse | [105] |
| CREBBP alteration | Anticipate corticosteroid resistance | [106] |
| Hypodiploid | BCL2i (venetoclax), aHSCT | [107] |
| High hyperdiploid | Low-risk disease | [13] |
| iAMP21 | High-risk disease | [108] |
| NOTCH1+ | PSEN1i (γ-secretase complex inhibition), | [109] |
| IKZF1plus | in the presence of IKZF1 alteration, FAKi potentiates other drugs’ antitumor effect High-risk disease (even with low MRD values) | [110] |
| MYC rearrangement | Resistant disease course | [111] |
| NUP214-ABL1 | TKI | [112] |
| oncogene activation (eg. TAL1, LYL1, LMO1, TLX1, TLX3) by translocation with TCR genes | Varying, but substantial prognostic effect | [113] |
| CDKN2A/CDKN2B deletions | CDK4/CDK6i (palbociclib), | [114] |
| KMT2A-ENL | Despite KMT2A rearrangement, the prognosis is not poor in T-cell, ALL and aHSCT is generally avoidable | [115] |
| Ph-like precursor B-cell ALL | [116,117] | |
| ABL class (ABL1, ABL2, CSF1R, PDGFRA, PDGFRB) | Dasatinib | |
| CRLF2 class (IGH-CRLF2, P2RY8-CRLF2, CRLF2mut) | USP9Xi, HDACi (givinostat) | |
| JAK2/EPOR class (JAK2, EPOR, PAX5) | JAKi (ruxolitinib) | |
| Ras/MAPK class (NRAS, KRAS, PTPN11, NF1) | MEKi (trametinib) | |
| Rare kinase fusions (NTRK3, FGFR1, PTK2B, TYK2, DGKH, LYN) | NTRKi (larotrectinib, NTRK3), ALKi/ROS1i (crizotinib, NTRK3), ponatinib (FGFR1), FAKi (PTK2B), TYK2i (TYK2) | |
| IKZF1 alteration in Ph-like cases | Retinoid use potentiates TKIs’ effect | |
| AML | ||
| KMT2A rearrangement | DOT1Li (pinometostat) | [118] |
| NUP98 rearrangement | CDK6i (palbociclib), BCL2i (navitoclax) High-risk disease (need for aHSCT) | [119,120] |
| CBF-AML (inv(16): CBFB-MYH11, t(8;21): RUNX1-RUNX1T1) | KITi (dasatinib) Low-risk disease | [121] |
| FLT3 activating mutations | FLT3i (midostaurin, crenolanib, gilteritinib, lestaurtinib, quizartinib, sorafenib) | [15] |
| The coincidence anticipates a favorable prognosis, outcomes are better than in FLT3-ITDneg cases | |
| Usually unsuccessful induction, dismal prognosis, need for aHSCT | |
| IDH1/IDH2 mutations (rare in children) | IDHi (enasidenib, ivosidenib) | [122] |
| HIF1A, BRE and CLEC7A expression levels | Drug resistance toward cytarabine, daunorubicin, and/or etoposide | [123] |
3. Pathobiology Uncovered by Single-Cell Analysis
3.1. Pediatric Acute Lymphoblastic Leukemia
3.2. Pediatric Acute Myeloid Leukemia
4. Clinical Aspects of Blast-Level Pathobiological Data
5. Emerging Targeted Therapies and Related Clinical Decision Making
6. High-Resolution Cellular Characterization for Future Clinical Management
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fialkow, P.J. Clonal origin of human tumors. Annu. Rev. Med. 1979, 30, 135–143. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306. [Google Scholar] [CrossRef]
- Alpar, D.; Barber, L.J.; Gerlinger, M. Genetic Intratumor Heterogeneity. Epigenetic Cancer Ther. 2015, 571–593. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 340, 1546–1558. [Google Scholar] [CrossRef]
- Alsagaby, S.A. Omics-based insights into therapy failure of pediatric B-lineage acute lymphoblastic leukemia. Oncol. Rev. 2019, 13, 149–155. [Google Scholar] [CrossRef]
- Arber, D.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.; Le Beau, M.; Bloomfield, C.; Cazzola, M.; Vardiman, J. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Lee Harris, N.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [Green Version]
- Kumar-Sinha, C.; Chinnaiyan, A.M. Precision oncology in the age of integrative genomics. Nat. Biotechnol. 2018, 36, 46. [Google Scholar] [CrossRef]
- Leblanc, V.G.; Marra, M.A. Next-generation sequencing approaches in cancer: Where have they brought us and wherewill they take us? Cancers 2015, 7, 1925–1958. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Pui, C.-H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. [Google Scholar] [CrossRef] [PubMed]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2021, candisc.0316.2021. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Gupta, S.D.; Sachs, Z. Novel single-cell technologies in acute myeloid leukemia research. Transl. Res. 2017, 189, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M. Leukaemia “firsts” in cancer research and treatment. Nat. Rev. Cancer 2016, 16, 163–172. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, Y.; Tan, Y.; Zhao, W.; Tian, Q. Single-Cell RNA Sequencing in Hematological Diseases. Proteomics 2020, 20, 1900228. [Google Scholar] [CrossRef] [PubMed]
- García-sanz, R.; Jiménez, C. Time to move to the single-cell level: Applications of single-cell multi-omics to hematological malignancies and waldenström’s macroglobulinemia—A particularly heterogeneous lymphoma. Cancers (Basel) 2021, 13, 1541. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef] [Green Version]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Dong, X.; Huo, L.; Wei, X.; Wang, F.; Sun, K. The potential roles and advantages of single cell sequencing in the diagnosis and treatment of hematological malignancies. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2018; Volume 1068, pp. 119–133. [Google Scholar]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef]
- Hou, Y.; Song, L.; Zhu, P.; Zhang, B.; Tao, Y.; Xu, X.; Li, F.; Wu, K.; Liang, J.; Shao, D.; et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 2012, 148, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.; Mi, X.; North, K.; Binder, M.; Penson, A.; Lasho, T.; Knorr, K.; Haddadin, M.; Liu, B.; Pangallo, J.; et al. Single-cell genomics reveals the genetic and molecular bases for escape from mutational epistasis in myeloid neoplasms. Blood 2020, 136, 1477–1486. [Google Scholar] [CrossRef]
- Sachs, K.; Sarver, A.L.; Noble-Orcutt, K.E.; LaRue, R.S.; Antony, M.L.; Chang, D.; Lee, Y.; Navis, C.M.; Hillesheim, A.L.; Nykaza, I.R.; et al. Single-cell gene expression analyses reveal distinct self-renewing and proliferating subsets in the leukemia stem cell compartment in acute myeloid leukemia. Cancer Res. 2020, 80, 458–470. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.-S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Stetson, L.; Balasubramanian, D.; Ribeiro, S.; Stefan, T.; Gupta, K.; Xu, X.; Fourati, S.; Roe, A.; Jackson, Z.; Schauner, R.; et al. Single cell RNA sequencing of AML initiating cells reveals RNA-based evolution during disease progression. Leukemia 2021, 35, 2799–2812. [Google Scholar] [CrossRef] [PubMed]
- Van Galen, P.; Hovestadt, V.; Wadsworth, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef] [Green Version]
- Demaree, B.; Delley, C.L.; Vasudevan, H.N.; Peretz, C.A.C.; Ruff, D.; Smith, C.C.; Abate, A.R. Joint profiling of DNA and proteins in single cells to dissect genotype-phenotype associations in leukemia. Nat. Commun. 2021, 12, 1583. [Google Scholar] [CrossRef]
- Louka, E.; Povinelli, B.; Rodriguez-Meira, A.; Buck, G.; Wen, W.X.; Wang, G.; Sousos, N.; Ashley, N.; Hamblin, A.; Booth, C.A.G.; et al. Heterogeneous disease-propagating stem cells in juvenile myelomonocytic leukemia. J. Exp. Med. 2021, 218, e20180853. [Google Scholar] [CrossRef]
- Walter, C.; Pozzorini, C.; Reinhardt, K.; Geffers, R.; Xu, Z.; Reinhardt, D.; von Neuhoff, N.; Hanenberg, H. Single-cell whole exome and targeted sequencing in NPM1/FLT3 positive pediatric acute myeloid leukemia. Pediatr. Blood Cancer 2018, 65, e26848. [Google Scholar] [CrossRef]
- Chen, J.; Kao, Y.R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Ediriwickrema, A.; Aleshin, A.; Reiter, J.G.; Corces, M.R.; Kohnke, T.; Stafford, M.; Liedtke, M.; Medeiros, B.C.; Majeti, R. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 2020, 4, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.E.O.; Magrini, V.; Demeter, R.; Miller, C.A.; Fulton, R.; Fulton, L.L.; Eades, W.C.; Elliott, K.; Heath, S.; Westervelt, P.; et al. Clonal Architecture of Secondary Acute Myeloid Leukemia Defined by Single-Cell Sequencing. PLoS Genet. 2014, 10, e1004462. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef]
- Pellegrino, M.; Sciambi, A.; Treusch, S.; Durruthy-Durruthy, R.; Gokhale, K.; Jacob, J.; Chen, T.X.; Geis, J.A.; Oldham, W.; Matthews, J.; et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018, 28, 1345–1352. [Google Scholar] [CrossRef] [Green Version]
- Petti, A.A.; Williams, S.R.; Miller, C.A.; Fiddes, I.T.; Srivatsan, S.N.; Chen, D.Y.; Fronick, C.C.; Fulton, R.S.; Church, D.M.; Ley, T.J. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat. Commun. 2019, 10, 3660. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc. Natl. Acad. Sci. USA 2014, 111, 17947–17952. [Google Scholar] [CrossRef] [Green Version]
- Ebinger, S.; Özdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Castro Alves, C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, M.T.; Dolgalev, I.; Evensen, N.A.; Ma, C.; Chambers, T.; Roberts, K.G.; Sreeram, S.; Dai, Y.; Tikhonova, A.N.; Lasry, A.; et al. Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2020, 37, 867–882.e12. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jiang, M.; Yu, P.; Li, J.; Ouyang, W.; Feng, C.; Zhao, W.L.; Dai, Y.; Huang, J. Single-Cell Transcriptome Analysis Identifies Ligand–Receptor Pairs Associated With BCP-ALL Prognosis. Front. Oncol. 2021, 11, 322. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, L.; Wu, Y.; Dong, B.; Guo, W.; Wang, M.; Yang, L.; Fan, X.; Tang, Y.; Liu, N.; et al. T-ALL leukemia stem cell ’stemness’ is epigenetically controlled by the master regulator SPI1. Elife 2018, 7, e38314. [Google Scholar] [CrossRef]
- Albertí-Servera, L.; Demeyer, S.; Govaerts, I.; Swings, T.; De Bie, J.; Gielen, O.; Brociner, M.; Michaux, L.; Maertens, J.; Uyttebroeck, A.; et al. Single-cell DNA amplicon sequencing reveals clonal heterogeneity and evolution in T-cell acute lymphoblastic leukemia. Blood 2021, 137, 801–811. [Google Scholar] [CrossRef]
- Anand, P.; Guillaumet-Adkins, A.; Dimitrova, V.; Yun, H.; Drier, Y.; Sotudeh, N.; Rogers, A.; Ouseph, M.M.; Nair, M.; Potdar, S.; et al. Single-cell RNA-seq reveals developmental plasticity with coexisting oncogenic states and immune evasion programs in ETP-ALL. Blood 2021, 137, 2463–2480. [Google Scholar] [CrossRef]
- Caron, M.; St-Onge, P.; Sontag, T.; Wang, Y.C.; Richer, C.; Ragoussis, I.; Sinnett, D.; Bourque, G. Single-cell analysis of childhood leukemia reveals a link between developmental states and ribosomal protein expression as a source of intra-individual heterogeneity. Sci. Rep. 2020, 10, 8079. [Google Scholar] [CrossRef] [PubMed]
- De Bie, J.; Demeyer, S.; Alberti-Servera, L.; Geerdens, E.; Segers, H.; Broux, M.; De Keersmaecker, K.; Michaux, L.; Vandenberghe, P.; Voet, T.; et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 1358–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, J.; Gonzalez-Pena, V.; Yergeau, D.; Ma, X.; Gawad, C. Genome-wide segregation of single nucleotide and structural variants into single cancer cells. BMC Genom. 2017, 18, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Huang, J.-Y. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehtonen, J.; Teppo, S.; Lahnalampi, M.; Kokko, A.; Kaukonen, R.; Oksa, L.; Bouvy-Liivrand, M.; Malyukova, A.; Mäkinen, A.; Laukkanen, S.; et al. Single cell characterization of B-lymphoid differentiation and leukemic cell states during chemotherapy in ETV6-RUNX1-positive pediatric leukemia identifies drug-targetable transcription factor activities. Genome Med. 2020, 12, 99. [Google Scholar] [CrossRef]
- Penter, L.; Gohil, S.H.; Lareau, C.; Ludwig, L.S.; Parry, E.M.; Huang, T.; Li, S.; Zhang, W.; Livitz, D.; Leshchiner, I.; et al. Longitudinal single-cell dynamics of chromatin accessibility and mitochondrial mutations in chronic lymphocytic leukemia mirror disease history. Cancer Discov. 2021, candisc.0276.2021. [Google Scholar] [CrossRef]
- Rendeiro, A.F.; Krausgruber, T.; Fortelny, N.; Zhao, F.; Penz, T.; Farlik, M.; Schuster, L.C.; Nemc, A.; Tasnády, S.; Réti, M.; et al. Chromatin mapping and single-cell immune profiling define the temporal dynamics of ibrutinib response in CLL. Nat. Commun. 2020, 11, 577. [Google Scholar] [CrossRef] [Green Version]
- Roider, T.; Seufert, J.; Uvarovskii, A.; Frauhammer, F.; Bordas, M.; Abedpour, N.; Stolarczyk, M.; Mallm, J.P.; Herbst, S.A.; Bruch, P.M.; et al. Dissecting intratumour heterogeneity of nodal B-cell lymphomas at the transcriptional, genetic and drug-response levels. Nat. Cell Biol. 2020, 22, 896–906. [Google Scholar] [CrossRef]
- Wang, L.; Mo, S.; Li, X.; He, Y.; Yang, J. Single-cell RNA-seq reveals the immune escape and drug resistance mechanisms of mantle cell lymphoma. Cancer Biol. Med. 2020, 17, 726–739. [Google Scholar] [CrossRef]
- Zhang, S.; Jiang, V.C.; Han, G.; Hao, D.; Lian, J.; Liu, Y.; Zhang, R.; McIntosh, J.; Wang, R.; Dang, M.; et al. Longitudinal single-cell profiling reveals molecular heterogeneity and tumor-immune evolution in refractory mantle cell lymphoma. Nat. Commun. 2021, 12, 2877. [Google Scholar] [CrossRef]
- Andor, N.; Simonds, E.F.; Czerwinski, D.K.; Chen, J.; Grimes, S.M.; Wood-Bouwens, C.; Zheng, G.X.Y.; Kubit, M.A.; Greer, S.; Weiss, W.A.; et al. Single-cell RNA-Seq of follicular lymphoma reveals malignant B-cell types and coexpression of T-cell immune checkpoints. Blood 2019, 133, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Haebe, S.; Shree, T.; Sathe, A.; Day, G.; Czerwinski, D.K.; Grimes, S.M.; Lee, H.J.; Binkley, M.S.; Long, S.R.; Martin, B.; et al. Single-cell analysis can define distinct evolution of tumor sites in follicular lymphoma. Blood 2021, 137, 2869–2880. [Google Scholar] [CrossRef]
- Milpied, P.; Cervera-Marzal, I.; Mollichella, M.L.; Tesson, B.; Brisou, G.; Traverse-Glehen, A.; Salles, G.; Spinelli, L.; Nadel, B. Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat. Immunol. 2018, 19, 1013–1024. [Google Scholar] [CrossRef]
- Mitra, A.K.; Mukherjee, U.K.; Harding, T.; Jang, J.S.; Stessman, H.; Li, Y.; Abyzov, A.; Jen, J.; Kumar, S.; Rajkumar, V.; et al. Single-cell analysis of targeted transcriptome predicts drug sensitivity of Longitudinal single-cell profiling reveals molecular heterogeneity and tumor-immune evolution in refractory mantle cell lymphomasingle cells within human myeloma tumors. Leukemia 2016, 30, 1094–1102. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-cell transcriptome analysis reveals disease-defining t-cell subsets in the tumor microenvironment of classic hodgkin lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Buus, T.B.; Willerslev-Olsen, A.; Fredholm, S.; Blümel, E.; Nastasi, C.; Gluud, M.; Hu, T.; Lindahl, L.M.; Iversen, L.; Fogh, H.; et al. Single-cell heterogeneity in Sézary syndrome. Blood Adv. 2018, 2, 2115–2126. [Google Scholar] [CrossRef] [Green Version]
- Rindler, K.; Bauer, W.M.; Jonak, C.; Wielscher, M.; Shaw, L.E.; Rojahn, T.B.; Thaler, F.M.; Porkert, S.; Simonitsch-Klupp, I.; Weninger, W.; et al. Single-Cell RNA Sequencing Reveals Tissue Compartment-Specific Plasticity of Mycosis Fungoides Tumor Cells. Front. Immunol. 2021, 12, 666935. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Geskin, L.J.; Bayan, C.A.; Conway, J.F.; Lafyatis, R.; Fuschiotti, P. Single-cell lymphocyte heterogeneity in advanced cutaneous T-cell lymphoma skin tumors. Clin. Cancer Res. 2019, 25, 4443–4454. [Google Scholar] [CrossRef] [Green Version]
- Jonak, C.; Alkon, N.; Rindler, K.; Rojahn, T.B.; Shaw, L.E.; Porkert, S.; Weninger, W.; Trautinger, F.; Stingl, G.; Tschandl, P.; et al. Single-cell RNAseq profiling in a patient with discordant primary cutaneous B and T cell lymphoma reveals micromilieu-driven immune skewing. Br. J. Dermatol. 2021, 185, 1013–1025. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Dong, R.; Man, J.; Sun, L.; Qian, X.; Zhu, X.; Cao, P.; Yu, Y.; Le, J.; et al. Single-Cell RNA-seq Reveals Characteristics of Malignant Cells and Immune Microenvironment in Subcutaneous Panniculitis-Like T-Cell Lymphoma. Front. Oncol. 2021, 11, 813. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Huang, X.-T.; Li, X.; Qin, P.-Z.; Zhu, Y.; Xu, S.-N.; Chen, J.-P. Technical Advances in Single-Cell RNA Sequencing and Applications in Normal and Malignant Hematopoiesis. Front. Oncol. 2018, 8, 582. [Google Scholar] [CrossRef]
- Ysebaert, L.; Quillet-Mary, A.; Tosolini, M.; Pont, F.; Laurent, C.; Fournié, J.J. Lymphoma Heterogeneity Unraveled by Single-Cell Transcriptomics. Front. Immunol. 2021, 12, 202. [Google Scholar] [CrossRef]
- Gianni, F.; Belver, L.; Ferrando, A. The genetics and mechanisms of T-cell acute lymphoblastic leukemia. Cold Spring Harb. Perspect. Med. 2020, 10, a035246. [Google Scholar] [CrossRef]
- Greaves, M.F.; Maia, A.T.; Wiemels, J.L.; Ford, A.M. Leukemia in twins: Lessons in natural history. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef]
- Greaves, M. Darwin and evolutionary tales in leukemia. The Ham-Wasserman Lecture. Hematol. Am. Soc. Hematol. Educ. Program 2009, 2009, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Dobbins, S.E.; Sherborne, A.L.; Chubb, D.; Galbiati, M.; Cazzaniga, G.; Micalizzi, C.; Tearle, R.; Lloyd, A.L.; Hain, R.; et al. Developmental timing of mutations revealed by whole-genome sequencing of twins with acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 7429–7433. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.; Lutz, C.; Van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef]
- Alpar, D.; Wren, D.; Ermini, L.; Mansur, M.B.; Van Delft, F.W.; Bateman, C.M.; Titley, I.; Kearney, L.; Szczepanski, T.; Gonzalez, D.; et al. Clonal origins of ETV6-RUNX1+ acute lymphoblastic leukemia: Studies in monozygotic twins. Leukemia 2015, 29, 839–846. [Google Scholar] [CrossRef]
- Mullighan, C.; Phillips, L.; Su, X.; Ma, J.; Miller, C.; Shurtleff, S.; Downing, J. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar] [CrossRef] [Green Version]
- Kiss, R.; Gángó, A.; Benard-Slagter, A.; Egyed, B.; Haltrich, I.; Hegyi, L.; de Groot, K.; Király, P.A.; Krizsán, S.; Kajtár, B.; et al. Comprehensive profiling of disease-relevant copy number aberrations for advanced clinical diagnostics of pediatric acute lymphoblastic leukemia. Mod. Pathol. 2020, 33, 812–824. [Google Scholar] [CrossRef]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.J.; et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- Antic, Ž.; Lelieveld, S.H.; Van Der Ham, C.G.; Sonneveld, E.; Hoogerbrugge, P.M.; Kuiper, R.P. Unravelling the sequential interplay of mutational mechanisms during clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Genes (Basel) 2021, 12, 214. [Google Scholar] [CrossRef]
- Yang, F.; Brady, S.W.; Tang, C.; Sun, H.; Du, L.; Barz, M.J.; Ma, X.; Chen, Y.; Fang, H.; Li, X.; et al. Chemotherapy and mismatch repair deficiency cooperate to fuel TP53 mutagenesis and ALL relapse. Nat. Cancer 2021, 2, 819–834. [Google Scholar] [CrossRef]
- Nordlund, J.; Syvänen, A.C. Epigenetics in pediatric acute lymphoblastic leukemia. Semin. Cancer Biol. 2018, 51, 129–138. [Google Scholar] [CrossRef]
- Nordlund, J.; Bäcklin, C.L.; Zachariadis, V.; Cavelier, L.; Dahlberg, J.; Öfverholm, I.; Barbany, G.; Nordgren, A.; Övernäs, E.; Abrahamsson, J.; et al. DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin. Epigenet. 2015, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, A.S.; Lafta, F.M.; Schwalbey, E.C.; Nakjang, S.; Cockell, S.J.; Iliasova, A.; Enshaei, A.; Schwab, C.; Rand, V.; Clifford, S.C.; et al. Epigenetic landscape correlates with genetic subtype but does not predict outcome in childhood acute lymphoblastic leukemia. Epigenetics 2015, 10, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Fleur-Lominy, S.; Evensen, N.A.; Bhatla, T.; Sethia, G.; Narang, S.; Choi, J.H.; Ma, X.; Yang, J.J.; Kelly, S.; Raetz, E.; et al. Evolution of the epigenetic landscape in childhood B Acute lymphoblastic leukemia and its role in drug resistance. Cancer Res. 2020, 80, 5189–5202. [Google Scholar] [CrossRef]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; Von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; Mccastlain, K.; Reshmi, S.C.; Payne-turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef] [Green Version]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Farrar, J.E.; Schuback, H.L.; Ries, R.E.; Wai, D.; Hampton, O.A.; Trevino, L.R.; Alonzo, T.A.; Auvil, J.M.G.; Davidsen, T.M.; Gesuwan, P.; et al. Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Cancer Res. 2016, 76, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, E.; Taylor-Weiner, A.N.; Chang, T.Y.; Gelston, L.C.; Wang, Y.D.; Mazor, T.; Esquivel, E.; Yu, A.; Seepo, S.; Olsen, S.R.; et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 2015, 47, 1326–1333. [Google Scholar] [CrossRef]
- Okabe, S.; Tauchi, T.; Tanaka, Y.; Ohyashiki, K. Therapeutic targeting of Aurora A kinase in Philadelphia chromosome-positive ABL tyrosine kinase inhibitor-resistant cells. Oncotarget 2018, 9, 32496–32506. [Google Scholar] [CrossRef] [Green Version]
- Eldfors, S.; Kuusanmäki, H.; Kontro, M.; Majumder, M.M.; Parsons, A.; Edgren, H.; Pemovska, T.; Kallioniemi, O.; Wennerberg, K. Idelalisib sensitivity and mechanisms of disease progression in relapsed TCF3-PBX1 acute lymphoblastic leukemia. Leukemia 2017, 31, 51–57. [Google Scholar] [CrossRef]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C. Genomics and drug profiling of fatal TCF3-HLF−positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef]
- Janczar, S.; Janczar, K.; Pastorczak, A.; Harb, H.; Paige, A.J.W.; Zalewska-Szewczyk, B.; Danilewicz, M.; Mlynarski, W. The role of histone protein modifications and mutations in histone modifiers in pediatric B-cell progenitor acute lymphoblastic leukemia. Cancers 2017, 9, 2. [Google Scholar] [CrossRef]
- Griffith, M.; Griffith, O.L.; Krysiak, K.; Skidmore, Z.L.; Campbell, K.M.; Lesurf, R.; Hundal, J.; Zhang, J.; Spies, N.C.; Ainscough, B.J.; et al. Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia. Exp. Hematol. 2016, 44, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, T.; Liu, S.; Li, J.; Meyer, J.; Zhao, X.; Yang, W.; Shao, Y.; Heath, R.; Carroll, W.L. Mechanisms of NT5C2-Mediated Thiopurine Resistance in Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2019, 18, 1887–1895. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.; Zhang, J.; Kasper, L.; Lerach, S.; Payne-Turner, D.; Phillips, L.; Heatley, S.; Holmfeldt, L.; Collins-Underwood, J.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Flores, E.; Comeaux, E.Q.; Kim, K.L.; Melnik, E.; Beckman, K.; Davis, K.L.; Wu, K.; Akutagawa, J.; Bridges, O.; Marino, R.; et al. Bcl-2 Is a Therapeutic Target for Hypodiploid B-Lineage Acute Lymphoblastic Leukemia. Cancer Res. 2019, 79, 2339–2351. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.J.; Moorman, A.V.; Schwab, C.; Carroll, A.J.; Raetz, E.A.; Devidas, M.; Strehl, S.; Nebral, K.; Harbott, J. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): Cytogenetic characterization and outcome. Leukemia 2014, 28, 1015–1021. [Google Scholar] [CrossRef] [Green Version]
- Habets, R.A.; De Bock, C.E.; Serneels, L.; Lodewijckx, I.; Verbeke, D.; Nittner, D.; Narlawar, R.; Demeyer, S.; Dooley, J. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci. Transl. Med. 2019, 11, eaau6246. [Google Scholar] [CrossRef] [Green Version]
- Marke, R.; van Leeuwen, F.N.; Scheijen, B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 103, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, K.; Imamura, T.; Ishimaru, S.; Imai, C.; Shimonodan, H.; Fujita, N.; Okada, K.; Taketani, T.; Kanai, R.; Tauchi, H.; et al. Nationwide study of pediatric B-cell precursor acute lymphoblastic leukemia with chromosome 8q24/MYC rearrangement in Japan. Pediatr. Blood Cancer 2020, 67, e28341. [Google Scholar] [CrossRef]
- Deenik, W.; Beverloo, H.B.; van der Poel-van de Luytgaarde, S.C.P.A.M.; Wattel, M.M.; van Esser, J.W.J.; Valk, P.J.M.; Cornelissen, J.J. Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 2009, 23, 627–629. [Google Scholar] [CrossRef] [Green Version]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [Green Version]
- Carrasco Salas, P.; Fernández, L.; Vela, M.; Bueno, D.; González, B.; Valentín, J.; Lapunzina, P.; Pérez-Martínez, A. The role of CDKN2A/B deletions in pediatric acute lymphoblastic leukemia. Pediatr. Hematol. Oncol. 2016, 33, 415–422. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Camitta, B.M.; Mahmoud, H.; Raimondi, S.C.; Carroll, A.J.; Borowitz, M.J.; Shuster, J.J.; Link, M.P.; Pullen, D.J.; Downing, J.R.; et al. Childhood Acute Lymphoblastic Leukemia With the MLL-ENL Fusion and t(11;19)(q23;p13.3) Translocation. J. Clin. Oncol. 2019, 17, 191–196. [Google Scholar] [CrossRef]
- Tran, T.H.; Loh, M.L. Ph-like acute lymphoblastic leukemia. Hematology 2016, 1, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Schmoellerl, J.; Barbosa, I.; Eder, T.; Brandstoetter, T.; Schmidt, L.; Maurer, B.; Troester, S. CDK6 is an essential direct target of NUP98-fusion proteins in acute myeloid leukemia. Blood 2020, 136, 387–400. [Google Scholar] [CrossRef]
- Kivioja, J.L.; Thanasopoulou, A.; Kumar, A.; Kontro, M.; Yadav, B.; Heckman, C.A. Dasatinib and navitoclax act synergistically to target NUP98-NSD1+/FLT3-ITD+ acute myeloid leukemia. Leukemia 2019, 33, 1360–1372. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Weber, D.; Benner, A.; Bullinger, L.; Heuser, M.; Gaidzik, V.I.; Thol, F.; Agrawal, M.; Teleanu, V.; et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia—results of the AMLSG 11-08 trial. Leukemia 2018, 32, 1621–1630. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Cucchi, D.G.J.; Bachas, C.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T.C.J.M.; Kwidama, Z.J.; Schuurhuis, G.J.; Assaraf, Y.G.; de Haas, V.; Kaspers, G.J.L.; Cloos, J. Harnessing gene expression profiles for the identification of ex vivo drug response genes in pediatric acute myeloid leukemia. Cancers 2020, 12, 1247. [Google Scholar] [CrossRef]
- Candelli, T.; Schneider, P.; Castro, P.G.; Jones, L.A.; Bodewes, E.; Rockx-Brouwer, D.; Pieters, R.; Holstege, F.C.P.; Margaritis, T.; Stam, R.W. Acute lymphoblastic leukemia Identification and characterization of relapse-initiating cells in MLL-rearranged infant ALL by single-cell transcriptomics. Leukemia 2021. [Google Scholar] [CrossRef]
- Ma, X.; Edmonson, M.; Yergeau, D.; Muzny, D.M.; Hampton, O.A.; Rusch, M.; Song, G.; Easton, J.; Harvey, R.C.; Wheeler, D.A.; et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat. Commun. 2015, 6, 6604. [Google Scholar] [CrossRef]
- Dobson, S.M.; García-Prat, L.; Vanner, R.J.; Wintersinger, J.; Waanders, E.; Gu, Z.; McLeod, J.; Gan, O.I.; Grandal, I.; Payne-Turner, D.; et al. Relapse-fated latent diagnosis subclones in acute B lineage leukemia are drug tolerant and possess distinct metabolic programs. Cancer Discov. 2020, 10, 568–587. [Google Scholar] [CrossRef] [Green Version]
- Zuna, J.; Cavé, H.; Eckert, C.; Szczepanski, T.; Meyer, C.; Mejstrikova, E.; Fronkova, E.; Muzikova, K.; Clappier, E.; Mendelova, D.; et al. Childhood secondary ALL after ALL treatment. Leukemia 2007, 21, 1431–1435. [Google Scholar] [CrossRef] [Green Version]
- Alpár, D.; Nagy, G.; Hohoff, C.; Kajtár, B.; Bartyik, K.; Hermesz, J.; Jáksó, P.; Andrikovics, H.; Kereskai, L.; Pajor, L. Sex chromosome changes after sex-mismatched allogeneic bone marrow transplantation can mislead the chimerism analysis. Pediatr. Blood Cancer 2010, 55, 1239–1242. [Google Scholar] [CrossRef]
- Rabilloud, T.; Potier, D.; Pankaew, S.; Nozais, M.; Loosveld, M.; Payet-Bornet, D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat. Commun. 2021, 12, 865. [Google Scholar] [CrossRef]
- Zhao, Y.; Aldoss, I.; Qu, C.; Crawford, J.C.; Gu, Z.; Allen, E.K.; Zamora, A.E.; Alexander, T.B.; Wang, J.; Goto, H.; et al. Tumor-intrinsic and -extrinsic determinants of response to blinatumomab in adults with B-ALL. Blood 2021, 137, 471–484. [Google Scholar] [CrossRef]
- Ratti, S.; Lonetti, A.; Follo, M.Y.; Paganelli, F.; Martelli, A.M.; Chiarini, F.; Evangelisti, C. B-all complexity: Is targeted therapy still a valuable approach for pediatric patients? Cancers 2020, 12, 3498. [Google Scholar] [CrossRef]
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021, 2, 326–337. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef]
- Wang, R.; Dang, M.; Harada, K.; Han, G.; Wang, F.; Pool Pizzi, M.; Zhao, M.; Tatlonghari, G.; Zhang, S.; Hao, D.; et al. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat. Med. 2021, 27, 141–151. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; Van De Ven, C.; De Groot-Kruseman, H.A.; De Haas, V.; et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef]
- Antić, Ž.; Yu, J.; Van Reijmersdal, S.V.; Van Dijk, A.; Dekker, L.; Segerink, W.H.; Sonneveld, E.; Fiocco, M.; Pieters, R.; Hoogerbrugge, P.M.; et al. Multiclonal complexity of pediatric acute lymphoblastic leukemia and the prognostic relevance of subclonal mutations. Haematologica 2020. [Google Scholar] [CrossRef]
- Barz, M.J.; Hof, J.; Groeneveld-Krentz, S.; Loh, J.W.; Szymansky, A.; Astrahantseff, K.; von Stackelberg, A.; Khiabanian, H.; Ferrando, A.A.; Eckert, C.; et al. Subclonal NT5C2 mutations are associated with poor outcomes after relapse of pediatric acute lymphoblastic leukemia. Blood 2020, 135, 921–933. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine ClinicalTrials. Available online: https://clinicaltrials.gov (accessed on 9 July 2021).
- Jeha, S.; Coustan-Smith, E.; Pei, D.; Sandlund, J.T.; Rubnitz, J.E.; Howard, S.C.; Inaba, H.; Bhojwani, D.; Metzger, M.L.; Cheng, C.; et al. Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 2014, 120, 1514–1519. [Google Scholar] [CrossRef] [Green Version]
- Porkka, K.; Koskenvesa, P.; Lundán, T.; Rimpiläinen, J.; Mustjoki, S.; Smykla, R.; Wild, R.; Luo, R.; Arnan, M.; Brethon, B.; et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 2008, 112, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Zwaan, C.M.; Söderhäll, S.; Brethon, B.; Luciani, M.; Rizzari, C.; Stam, R.W.; Besse, E.; Dutreix, C.; Fagioli, F.; Ho, P.A.; et al. A phase 1/2, open-label, dose-escalation study of midostaurin in children with relapsed or refractory acute leukaemia. Br. J. Haematol. 2019, 185, 623–627. [Google Scholar] [CrossRef]
- Karol, S.E.; Alexander, T.; Das Gupta, S.; Pounds, S.B.; Canavera, K.; Klco, J.M.; Lacayo, N.J.; Pui, C.-H.; Opferman, J.T.; Rubnitz, J.E. Safety and activity of venetoclax in combination with high-dose cytarabine in children with relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 2019, 37, 10004. [Google Scholar] [CrossRef]
- Lonetti, A.; Pession, A.; Masetti, R. Targeted Therapies for Pediatric AML: Gaps and Perspective. Front. Pediatr. 2019, 7, 463. [Google Scholar] [CrossRef]
- DiNardo, C.; Stein, E.; de Botton, S.; Roboz, G.; Altman, J.; Mims, A.; Swords, R.; Collins, R.; Mannis, G.; DA, P.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Gilteritinib: A novel FLT3 inhibitor for acute myeloid leukemia. Biomark. Res. 2019, 7, 19. [Google Scholar] [CrossRef]
- Dai, H.P.; Yin, J.; Li, Z.; Yang, C.X.; Cao, T.; Chen, P.; Zong, Y.H.; Zhu, M.Q.; Zhu, X.M.; Xiao, S.; et al. Rapid Molecular Response to Dasatinib in a Pediatric Relapsed Acute Lymphoblastic Leukemia With NCOR1-LYN Fusion. Front. Oncol. 2020, 10, 359. [Google Scholar] [CrossRef]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Tasian, S.K.; Sulis, M.L.; Stevenson, K.; Blonquist, T.M.; Apsel Winger, B.; Cooper, T.M.; Pauly, M.; Maloney, K.W.; Burke, M.J.; et al. Matched Targeted Therapy for Pediatric Patients with Relapsed, Refractory, or High-Risk Leukemias: A Report from the LEAP Consortium. Cancer Discov. 2021, 11, 1424–1439. [Google Scholar] [CrossRef]
- Marks, L.J.; Oberg, J.A.; Pendrick, D.; Sireci, A.N.; Glasser, C.; Coval, C.; Zylber, R.J.; Chung, W.K.; Pang, J.; Turk, A.T.; et al. Precision medicine in children and young adults with hematologic malignancies and blood disorders: The Columbia university experience. Front. Pediatr. 2017, 5, 265. [Google Scholar] [CrossRef] [Green Version]
- Khater, F.; Vairy, S.; Langlois, S.; Dumoucel, S.; Sontag, T.; St-Onge, P.; Bittencourt, H.; Dal Soglio, D.; Champagne, J.; Duval, M.; et al. Molecular Profiling of Hard-to-Treat Childhood and Adolescent Cancers. JAMA Netw. Open 2019, 2, e192906. [Google Scholar] [CrossRef]
- Barahona, P.; Fletcher, J.; Fuentes-Bolanos, N.; Gauthier, M.-E.; Haber, M.; Lock, R.B.; Marshall, G.M.; Mayoh, C.; Mould, E.; Nagabushan, S.; et al. Zero Childhood Cancer (ZERO): A comprehensive precision medicine platform for children with high-risk cancer. In Proceedings of the Cancer Research; American Association for Cancer Research (AACR), Los Angeles, CA, USA, 24–29 April 2020; Volume 80, p. A52. [Google Scholar]
- Mody, R.J.; Wu, Y.M.; Lonigro, R.J.; Cao, X.; Roychowdhury, S.; Vats, P.; Frank, K.M.; Prensner, J.R.; Asangani, I.; Palanisamy, N.; et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA—J. Am. Med. Assoc. 2015, 314, 913–925. [Google Scholar] [CrossRef]
- Harris, M.H.; DuBois, S.G.; Bender, J.L.G.; Kim, A.; Crompton, B.D.; Parker, E.; Dumont, I.P.; Hong, A.L.; Guo, D.; Church, A.; et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors. JAMA Oncol. 2016, 2, 608–615. [Google Scholar] [CrossRef] [Green Version]
- McFarland, J.M.; Paolella, B.R.; Warren, A.; Geiger-Schuller, K.; Shibue, T.; Rothberg, M.; Kuksenko, O.; Colgan, W.N.; Jones, A.; Chambers, E.; et al. Multiplexed single-cell transcriptional response profiling to define cancer vulnerabilities and therapeutic mechanism of action. Nat. Commun. 2020, 11, 4296. [Google Scholar] [CrossRef]
- Bartram, J.; Goulden, N.; Wright, G.; Adams, S.; Brooks, T.; Edwards, D.; Inglott, S.; Yousafzai, Y.; Hubank, M.; Halsey, C. High throughput sequencing in acute lymphoblastic leukemia reveals clonal architecture of central nervous system and bone marrow compartments. Haematologica 2018, 103, e110–e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocho, Y.; Liu, J.; Hu, J.; Yang, W.; Dharia, N.V.; Zhang, J.; Shi, H.; Du, G.; John, A.; Lin, T.N.; et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat. Cancer 2021, 2, 284–299. [Google Scholar] [CrossRef]
- Xiao, W.; Miles, L.A.; Bowman, R.L.; Durani, V.; Tian, H.S.; DelGaudio, N.L.; Mishra, T.; Zhu, M.; Zhang, Y.; Stump, S.E.; et al. A JAK2/IDH1-mutant MPN clone unmasked by ivosidenib in an AML patient without antecedent MPN. Blood Adv. 2020, 4, 6034–6038. [Google Scholar] [CrossRef]
- McKenney, A.S.; Lau, A.N.; Hanasoge Somasundara, A.V.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.T.; Patel, M.A.; Papalexi, E.; et al. JAK2/IDH-mutant–driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 789–804. [Google Scholar] [CrossRef]
- NCT05014165. Available online: https://clinicaltrials.gov/ct2/show/NCT05014165 (accessed on 4 November 2021).
- NCT03117751. Available online: https://clinicaltrials.gov/ct2/show/NCT03117751 (accessed on 4 November 2021).
- Zeng, T.; Dai, H. Single-cell RNA sequencing-based computational analysis to describe disease heterogeneity. Front. Genet. 2019, 10, 629. [Google Scholar] [CrossRef] [Green Version]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef]


| First Author | Year | Entity | Number of Patients 1 | Number of Cells Analyzed | Method | Refs |
|---|---|---|---|---|---|---|
| Gawad et al. | 2014 | B-ALL | 6 | 1479 | scDNA-seq | [47] |
| Easton et al. | 2017 | ALL | 1 | 128 | scDNA-seq | [56] |
| Walter et al. | 2017 | AML | 3 | 3 | scDNA-seq (WES) | [38] |
| De Bie et al. | 2018 | T-ALL | 4 | 1507 and 8297 | scDNA-seq and scRNA-seq | [55] |
| Li et al. | 2020 | B-ALL | 1 | 56 | scDNA-seq | [85] |
| Caron et al. | 2020 | T-ALL | 8 | 39,375 | scRNA-seq | [54] |
| Mehtonen et al. | 2020 | B-ALL | 6 | 44,746 | scRNA-seq | [58] |
| Witkowski et al. | 2020 | B-ALL | 9 | 34,407 and 42,621 | scRNA-seq and CITE-seq | [49] |
| Wu et al. 2 | 2021 | B-ALL | 7 | 38,860 | scRNA-seq | [50] |
| Alberti-Servera et al. | 2021 | T-ALL | 8 | 108,188 | scDNA-seq | [52] |
| Demaree et al. | 2021 | AML | 1 | 14,465 | DAb-seq | [36] |
| Candelli et al. | 2021 | B-ALL | 15 | 30,909 | scRNA-seq | [124] |
| Louka et al. | 2021 | JMML | 3 | 645 and 17,547 | scDNA-seq and scRNA-seq | [37] |
| Name of MTB (Country) | Launch Year | New Aspects, Perspectives | Leukemia Included | Refs |
|---|---|---|---|---|
| iCAT (United States) | 2012 |
| No | [154] |
| Peds-MiOncoSeq (United States) | 2012 |
| Yes | [153] |
| PIPseq (United States) | 2014 |
| Yes | [150] |
| TRICEPS (Canada) | 2014 |
| Yes | [151] |
| INFORM (Germany) | 2015 |
| Yes | [148] |
| LEAP (United States) | 2016 |
| No | [149] |
| ZERO (Australia) | 2017 |
| Yes | [152] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpár, D.; Egyed, B.; Bödör, C.; Kovács, G.T. Single-Cell Sequencing: Biological Insight and Potential Clinical Implications in Pediatric Leukemia. Cancers 2021, 13, 5658. https://doi.org/10.3390/cancers13225658
Alpár D, Egyed B, Bödör C, Kovács GT. Single-Cell Sequencing: Biological Insight and Potential Clinical Implications in Pediatric Leukemia. Cancers. 2021; 13(22):5658. https://doi.org/10.3390/cancers13225658
Chicago/Turabian StyleAlpár, Donát, Bálint Egyed, Csaba Bödör, and Gábor T. Kovács. 2021. "Single-Cell Sequencing: Biological Insight and Potential Clinical Implications in Pediatric Leukemia" Cancers 13, no. 22: 5658. https://doi.org/10.3390/cancers13225658