
Cholecystokinin Receptor Antagonist Improves Efficacy of Chemotherapy in Murine Models of Pancreatic Cancer by Altering the Tumor Microenvironment

 ,
,  ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Animal Models
2.2. Study Design
2.3. Tissue Analysis by Histology and Immunohistochemistry (IHC)
2.4. Western Immunoblotting
2.5. RNA Isolation and Analysis by Quantitative Reverse Transcription PCR (q-RT-PCR) and RNA Sequencing (RNAseq)
2.6. Mass Spectrometry
2.7. Blood Biomarker miRNA Panel
2.8. Statistical Analysis
3. Results
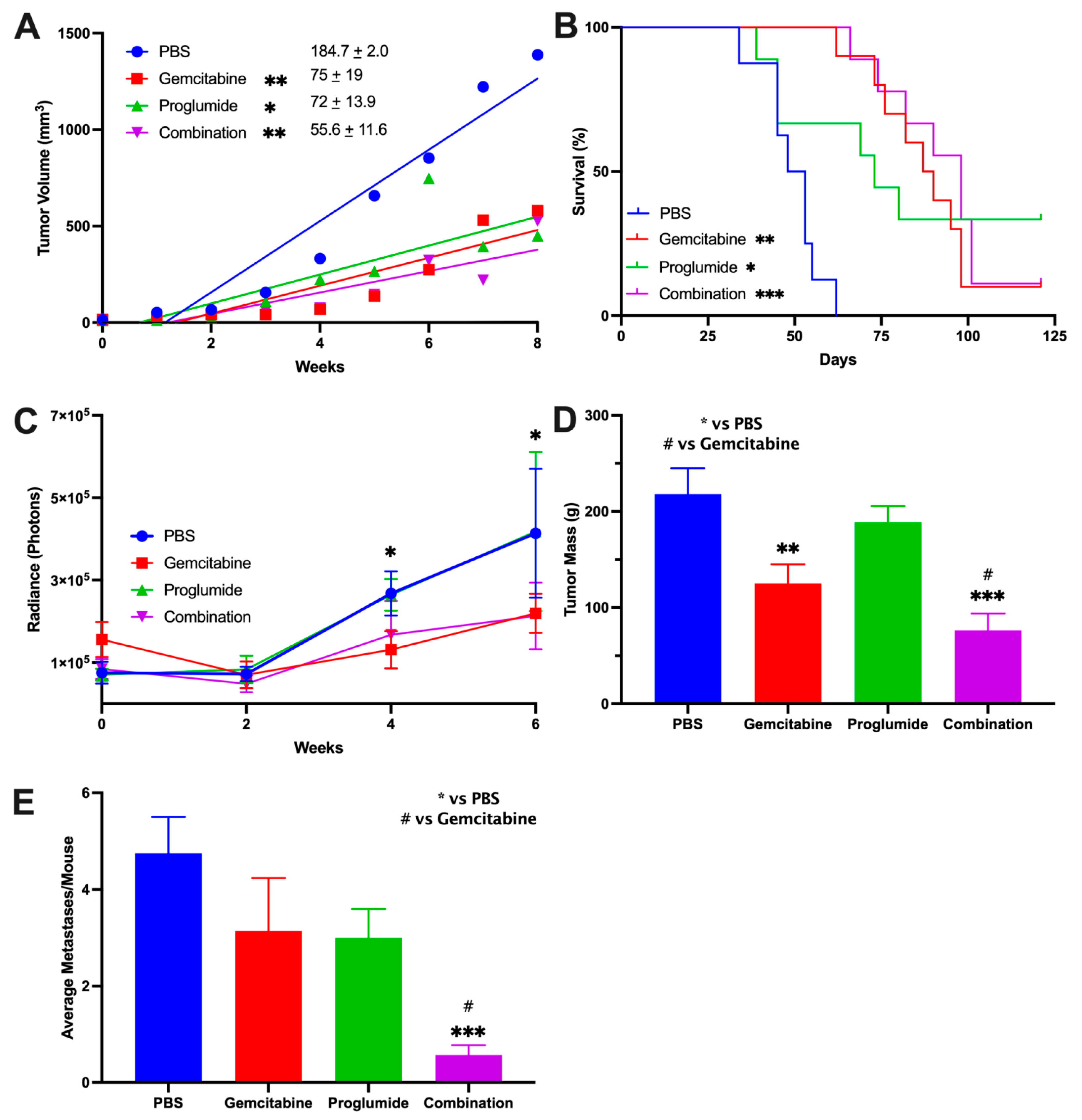
3.1. Combination Therapy with Gemcitabine and a CCK Receptor Antagonist Significantly Slows Pancreatic Tumor Growth, Prolongs Survival, and Decreases Metastases
3.2. CCK Receptor Blockade with Proglumide Decreases Fibrosis in the Pancreatic TME
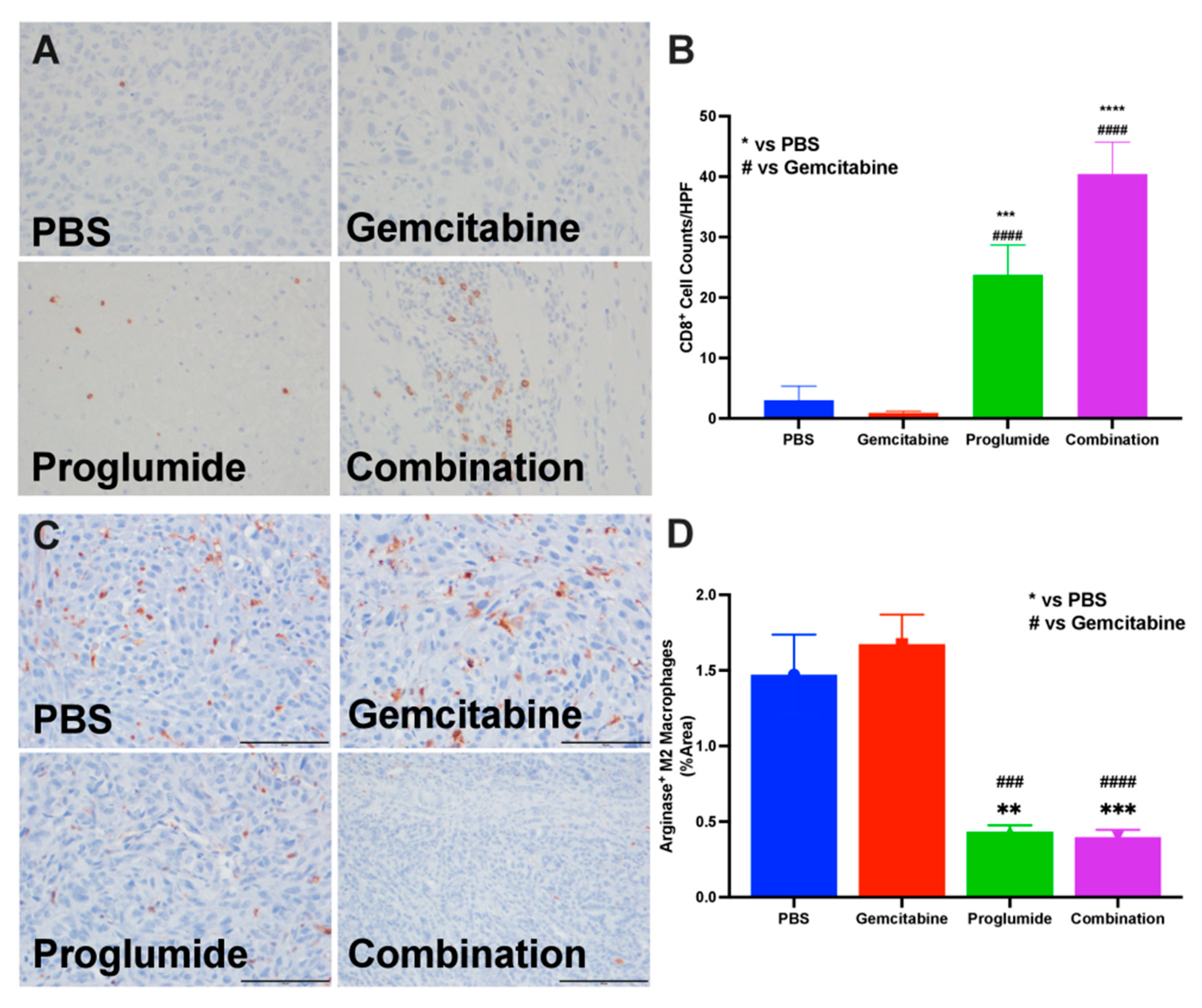
3.3. Proglumide Therapy Alters the Tumor Immune Cell Signature
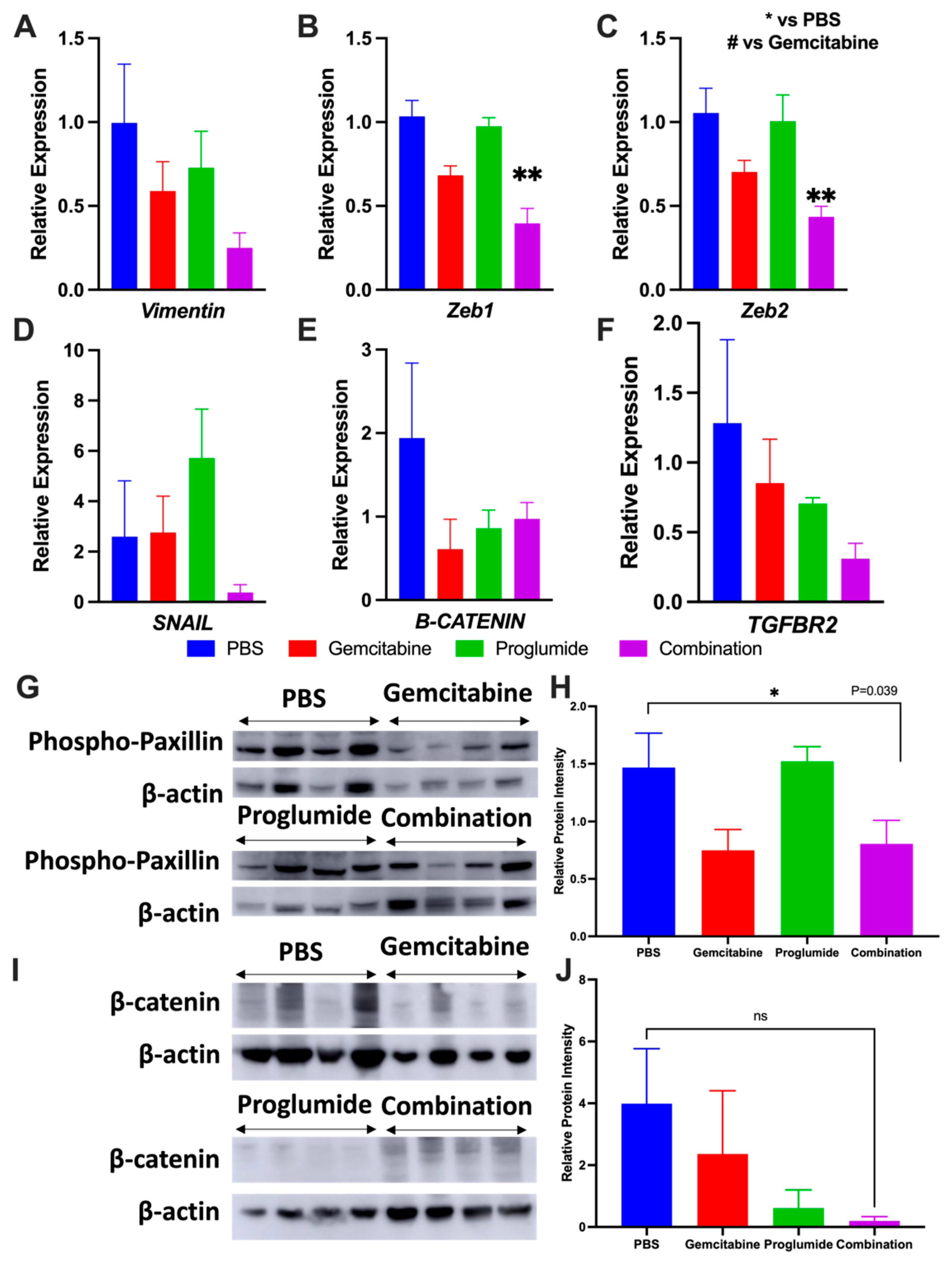
3.4. Combination Therapy with Gemcitabine and Proglumide Decreases Metastases by Preventing Epithelial-to-Mesenchymal Transition (EMT)
3.5. Analysis of Tumors by RNAseq
3.6. Combination Therapy Increases Gemcitabine Uptake into Tumors and Alters microRNA Blood Biomarker Profile
3.7. Measurement of Circulating microRNAs Confirm Decreased Fibrosis and Invasion upon Gemcitabine and Proglumide Combination Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Ansari, D.; Tingstedt, B.; Andersson, B.; Holmquist, F.; Sturesson, C.; Williamson, C.; Sasor, A.; Borg, D.; Bauden, M.; Andersson, R. Pancreatic cancer: Yesterday, today and tomorrow. Future Oncol. 2016, 12, 1929–1946. [Google Scholar] [CrossRef] [Green Version]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Pottler, M.; Lan, B.; Grutzmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Declerck, Y.A. Targeting the tumor microenvironment: From understanding pathways to effective clinical trials. Cancer Res. 2013, 73, 4965–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van, C.E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hirte, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Hecht, J.R.; Papadopoulos, K.P.; Falchook, G.S.; Patel, M.R.; Infante, J.R.; Aljumaily, R.; Wong, D.J.; Autio, K.A.; Wainberg, Z.A.; Bauer, T.M.; et al. Immunologic and tumor responses of pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in pancreatic ductal adenocarcinoma (PDAC). Investig. New Drugs 2021, 39, 182–192. [Google Scholar] [CrossRef]
- Nazemi, M.; Rainero, E. Cross-Talk between the Tumor Microenvironment, Extracellular Matrix, and Cell Metabolism in Cancer. Front. Oncol. 2020, 10, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufresne, M.; Seva, C.; Fourmy, D. Cholecystokinin and gastrin receptors. Physiol. Rev. 2006, 86, 805–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berna, M.J.; Jensen, R.T. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr. Top. Med. Chem. 2007, 7, 1211–1231. [Google Scholar] [CrossRef] [PubMed]
- Hahne, W.F.; Jensen, R.T.; Lemp, G.F.; Gardner, J.D. Proglumide and benzotript: Members of a different class of cholecystokinin receptor antagonists. Proc. Natl. Acad. Sci. USA 1981, 78, 6304–6308. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.P.; Cooper, T.K.; McGovern, C.O.; Gilius, E.L.; Zhong, Q.; Liao, J.; Molinolo, A.A.; Gutknd, J.S.; Matter, G.L. Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas 2014, 43, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.P.; Liu, G.; Soundararajan, V.; Zagon, I.S. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am. J. Physiol. 1994, 266, R277–R283. [Google Scholar] [CrossRef]
- Nadella, S.; Burks, J.; Al-Sabban, A.; Inyang, G.; Wang, J.; Tucker, R.D.; Zamanis, M.E.; Bukowski, W.; Shivapurkar, N.; Smith, J.P. Dietary fat stimulates pancreatic cancer growth and promotes fibrosis of the tumor microenvironment through the cholecystokinin receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G699–G712. [Google Scholar] [CrossRef]
- Smith, J.P.; Solomon, T.E. Cholecystokinin and pancreatic cancer: The chicken or the egg? Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G91–G101. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Owlia, A.; Espeijo, R.; Dai, B. Novel gastrin receptors mediate mitogenic effects of gastrin and processing intermediates of gastrin on Swiss 3T3 fibroblasts. Absence of detectable cholecystokinin (CCK)-A and CCK-B receptors. J. Biol. Chem. 1995, 270, 8429–8438. [Google Scholar] [CrossRef] [Green Version]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Blaker, M.; Koch, J.; Lohse, A.W.; Pace, A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef] [Green Version]
- Phillips, P.A.; Yang, L.; Shulkes, A.; Vonlaufen, A.; Poljak, A.; Bustamante, S.; Warren, A.; Xu, Z.; Guilhaus, M.; Pirola, R.; et al. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 17397–17402. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Titiac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- LaConti, J.J.; Shivapurkar, N.; Preet, A.; Mays, A.D.; Peran, I.; Kim, S.E.; Marshall, J.L.; Riegel, A.T.; Wellstein, A. Tissue and serum microRNAs in the Kras(G12D) transgenic animal model and in patients with pancreatic cancer. PLoS ONE 2011, 6, e20687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivapurkar, N.; Weiner, L.M.; Marshall, J.L.; Madhavan, S.; Mays, A.D.; Juhl, H.; Wellstein, A. Recurrence of early stage colon cancer predicted by expression pattern of circulating microRNAs. PLoS ONE 2014, 9, e84686. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Vietsch, E.E.; Carney, E.; Isaacs, C.; Wellstein, A. Circulating microRNAs in patients with hormone receptor-positive, metastatic breast cancer treated with dovitinib. Clin. Transl. Med. 2017, 6, 37. [Google Scholar] [CrossRef]
- Osborne, N.; Sundseth, R.; Gay, M.D.; Cao, H.; Tucker, R.D.; Nadella, S.; Wang, S.; Liu, X.; Kroemer, A.; Sutton, L.; et al. Vaccine Against Gastrin, Polyclonal Antibody Stimulator, Decreases Pancreatic Cancer Metastases. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G682–G693. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Rodriguez-Aznar, E.; Wiesmuller, L.; Sainz, B., Jr.; Hermann, P.C. EMT and Stemness-Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Huang, S.; Sun, Y.L. Epithelial-Mesenchymal Transition in Pancreatic Cancer: A Review. BioMed Res. Int. 2017, 2017, 2646148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [Green Version]
- Handra-Luca, A.; Hong, S.M.; Walter, K.; Wolfgang, C.; Hruban, R.; Goggins, M. Tumour epithelial vimentin expression and outcome of pancreatic ductal adenocarcinomas. Br. J. Cancer 2011, 104, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef] [Green Version]
- Mylavarapu, S.; Kumar, H.; Kumari, S.; Sravanthi, L.S.; Jain, M.; Basu, A.; Biswas, M.; Mylavarapu, S.V.S.; Das, A.; Roy, M. Activation of Epithelial-Mesenchymal Transition and Altered beta-Catenin Signaling in a Novel Indian Colorectal Carcinoma Cell Line. Front. Oncol. 2019, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Bierie, B.; Moses, H.L. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Bastien, E.; Santiago de Jesus, J.P.; Dierge, R.; Martherus, R.; Linden, C.V.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFbeta2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, G.; Ding, Q.; Li, H.; Zhang, L.; Zhang, L.; He, K.; Wu, L.; Deng, Y.; Yang, D.; Wu, L.; et al. Gastrin stimulates pancreatic cancer cell directional migration by activating the Galpha12/13-RhoA-ROCK signaling pathway. Exp. Mol. Med. 2018, 50, 59. [Google Scholar] [CrossRef] [PubMed]
- Pedone, E.; Marucci, L. Role of beta-Catenin Activation Levels and Fluctuations in Controlling Cell Fate. Genes 2019, 10, 176. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, K.; Yan, Y.; Zhang, X.; Zhang, J.; Zhang, L.; Han, K. Gastrin promotes the metastasis of gastric carcinoma through the beta-catenin/TCF-4 pathway. Oncol. Rep. 2016, 36, 1369–1376. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, H.Q.; Wang, Y.; Yang, Z.S.; Dai, L.J.; Xu, Y.C. Differential expression and therapeutic efficacy of microRNA-346 in diabetic nephropathy mice. Exp. Ther. Med. 2015, 10, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, J.; Wang, S.; Kim, J.; Rao, K.M.; Park, S.Y.; Chung, I.; Ha, C.S.; Kim, S.W.; Yum, Y.H.; Jung, Y. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat. Commun. 2016, 7, 10993. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, S.; Han, M.; Ma, Y.; Feng, S.; Zhao, J.; Lu, H.; Yuan, X.; Cheng, J. miR-185 Inhibits Fibrogenic Activation of Hepatic Stellate Cells and Prevents Liver Fibrosis. Mol. Ther. Nucleic Acids. 2018, 10, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Tao, Q.; Zhou, Y.; Chen, Q.; Li, L.; Hu, S.; Liu, Y.; Zhang, Y.; Shu, J.; Zhang, X.; et al. MicroRNA-708 represses hepatic stellate cells activation and proliferation by targeting ZEB1 through Wnt/beta-catenin pathway. Eur. J. Pharmacol. 2020, 871, 172927. [Google Scholar] [CrossRef]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Allgayer, H. MicroRNA Regulation of Epithelial to Mesenchymal Transition. J. Clin. Med. 2016, 5, 8. [Google Scholar] [CrossRef]
- Yu, K.R.; Lee, S.; Jung, J.W.; Hong, I.S.; Kim, H.S.; Seo, Y.; Shin, T.H.; Kang, K.S. MicroRNA-141-3p plays a role in human mesenchymal stem cell aging by directly targeting ZMPSTE24. J. Cell Sci. 2013, 126, 5422–5431. [Google Scholar] [CrossRef] [Green Version]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser-Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonderheide, R.H.; Bayne, L.J. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol. 2013, 25, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Derissen, E.J.B.; Huitema, A.D.R.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Intracellular pharmacokinetics of gemcitabine, its deaminated metabolite 2′,2′-difluorodeoxyuridine and their nucleotides. Br. J. Clin. Pharmacol. 2018, 84, 1279–1289. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapenyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399. [Google Scholar] [CrossRef]
- Rejiba, S.; Reddy, L.H.; Bigand, C.; Parmentier, C.; Couvreur, P.; Hajri, A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine 2011, 7, 841–849. [Google Scholar] [CrossRef]
- Farrow, B.; Albo, D.; Berger, D.H. The role of the tumor microenvironment in the progression of pancreatic cancer. J. Surg. Res. 2008, 149, 319–328. [Google Scholar] [CrossRef]
- Korc, M. Pancreatic cancer-associated stroma production. Am. J. Surg. 2007, 194, S84–S86. [Google Scholar] [CrossRef] [Green Version]
- Moir, J.A.; Mann, J.; White, S.A. The role of pancreatic stellate cells in pancreatic cancer. Surg. Oncol. 2015, 24, 232–238. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.H.; Zhu, Q.; Li, H.H.; Ra, H.J.; Majumdar, S.; Gulick, D.L.; Jerome, J.A.; Madsen, D.H.; Christofidou-Solomidou, M.; Speicher, D.W.; et al. Fibroblast Activation Protein (FAP) Accelerates Collagen Degradation and Clearance from Lungs in Mice. J. Biol. Chem. 2016, 291, 8070–8089. [Google Scholar] [CrossRef] [Green Version]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, C.; Vaidya, S.; Wadhwan, V.; Hitesh; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, G.; Hu, W.; Yao, Y.; Yu, X.F. Emerging roles and therapeutic value of exosomes in cancer metastasis. Mol. Cancer 2019, 18, 53. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykn, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochun, G.; Koltun, W.; Ma, T.Y. IL1B Increases Intestinal Tight Junction Permeability by Up-regulation of MIR200C-3p, Which Degrades Occludin mRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [CrossRef]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [Green Version]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Ashida, Y.; Tatebayashi, D.; Abe, M.; Himori, K. Cancer Cachexia Induces Preferential Skeletal Muscle Myosin Loss When Combined with Denervation. Front. Physiol. 2020, 11, 445. [Google Scholar] [CrossRef]
- Won, J.H.; Zhang, Y.; Ji, B.; Logsdon, C.D.; Yule, D.I. Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol. Biol. Cell 2011, 22, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Xing, H.B.; Tong, M.T.; Wang, J.; Hu, H.; Zhai, C.Y.; Huang, C.X.; Li, D. Suppression of IL-6 Gene by shRNA Augments Gemcitabine Chemosensitization in Pancreatic Adenocarcinoma Cells. BioMed Res. Int. 2018, 2018, 3195025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Hou, L.; Liang, Y.; Zhang, Q.; Hong, X.; Wang, Y.; Huang, X.; Zhong, T.; Pang, W.; Xu, C.; et al. The p53-inducible CLDN7 regulates colorectal tumorigenesis and has prognostic significance. Neoplasia 2020, 22, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peng, K.; Xie, R.; Zheng, J.; Guo, J.; Wei, R.; Yang, H.; Cai, C.; Wei, Q. Protocadherin gamma-A7 is down-regulated in colorectal cancer and associated with the prognosis in patients with wild-type KRAS. Hum. Pathol. 2019, 83, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, W.; Wang, Y.; Zha, M.; Yao, H.; Jones, P.M.; Sun, Z. Regenerating islet-derived protein 1 inhibits the activation of islet stellate cells isolated from diabetic mice. Oncotarget 2015, 6, 37054–37065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, S.J.; Stolzenberg-Solomon, R.Z.; Kopp, W.; Rager, H.; Virtamo, J.; Albanes, D. Impact of circulating vitamin D binding protein levels on the association between 25-hydroxyvitamin D and pancreatic cancer risk: A nested case-control study. Cancer Res. 2012, 72, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miederer, S.E.; Lindstaedt, H.; Kutz, K.; Wuttke, H. Efficient treatment of gastric ulcer with proglumide (Milid) in outpatients (double blind trial). Acta Hepatogastroenterol. 1979, 26, 314–318. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malchiodi, Z.X.; Cao, H.; Gay, M.D.; Safronenka, A.; Bansal, S.; Tucker, R.D.; Weinberg, B.A.; Cheema, A.; Shivapurkar, N.; Smith, J.P. Cholecystokinin Receptor Antagonist Improves Efficacy of Chemotherapy in Murine Models of Pancreatic Cancer by Altering the Tumor Microenvironment. Cancers 2021, 13, 4949. https://doi.org/10.3390/cancers13194949
Malchiodi ZX, Cao H, Gay MD, Safronenka A, Bansal S, Tucker RD, Weinberg BA, Cheema A, Shivapurkar N, Smith JP. Cholecystokinin Receptor Antagonist Improves Efficacy of Chemotherapy in Murine Models of Pancreatic Cancer by Altering the Tumor Microenvironment. Cancers. 2021; 13(19):4949. https://doi.org/10.3390/cancers13194949
Chicago/Turabian StyleMalchiodi, Zoe X., Hong Cao, Martha D. Gay, Anita Safronenka, Sunil Bansal, Robin D. Tucker, Benjamin A. Weinberg, Amrita Cheema, Narayan Shivapurkar, and Jill P. Smith. 2021. "Cholecystokinin Receptor Antagonist Improves Efficacy of Chemotherapy in Murine Models of Pancreatic Cancer by Altering the Tumor Microenvironment" Cancers 13, no. 19: 4949. https://doi.org/10.3390/cancers13194949