
The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
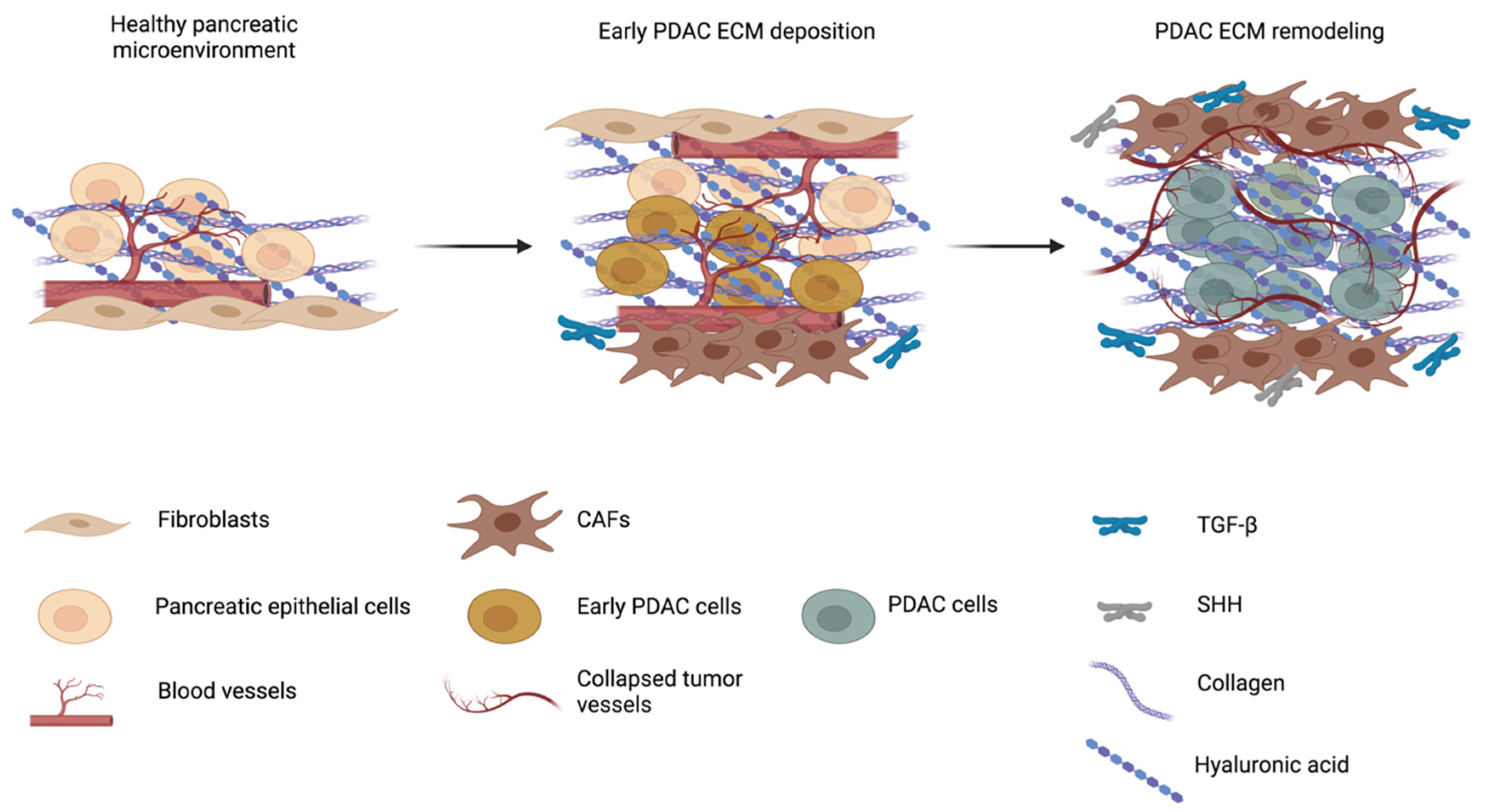
2. Cellular Component of PDAC Microenvironment: Heterogeneity and Plasticity of Cancer-Associated Fibroblasts (CAFs)
3. Physical and Mechanical Modifications of the Matrix in PDAC
3.1. Fibrosis and ECM Remodeling Affect Pancreatic Microenvironment
3.2. ECM Stiffness, Solid Stress and Interstitial Fluid Pressure
3.3. Cellular Response to Stiffness and Solid Stress
4. Pharmacological Tools Targeting the Stromal Barrier: From CAFs to ECM Components and Vessels Normalization
4.1. CAF Targeting as Therapeutic Strategy: A Double-Edged Sword
4.2. Targeting ECM Components
4.3. Reducing the Interstitial Fluid Pressure in the TME
4.4. Tumor Vessels Normalization
5. Identifying New ECM Targets
6. Nanomedicine as Therapeutic Strategy: Improvement of Nanoparticle-Based Systems for by-Passing the ECM
7. Towards Cell Therapy-Based Approaches: Mesenchymal Stem Cells for Drug Delivery
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society, ACS. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- De La Cruz, M.S.D.; Young, A.P.; Ruffin IV, M.T. Diagnosis and management of pancreatic cancer. Am. Fam. Phys. 2014, 89, 626–632. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Kleeff, J.; Reiser, C.; Hinz, U.; Bachmann, J.; Debus, J.; Jaeger, D.; Friess, H.; Büchler, M.W. Surgery for recurrent pancreatic ductal adenocarcinoma. Ann. Surg. 2007, 245, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first- line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Murakami, T.; Hiroshima, Y.; Matsuyama, R.; Homma, Y.; Hoffman, R.M.; Endo, I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 130–137. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef] [Green Version]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Invest. 2007, 117, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; Lobello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nia, H.T.; Munn, L.L.; Jain, R.K. Physical traits of cancer. Science 2020, 370, Issue 6516. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [Green Version]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Tung, J.C.; Barnes, J.M.; Desai, S.R.; Sistrunk, C.; Conklin, M.W.; Schedin, P.; Eliceiri, K.W.; Keely, P.J.; Seewaldt, V.L.; Weaver, V.M. Tumor mechanics and metabolic dysfunction. Free Radic. Biol. Med. 2015, 79, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, A.C. Biomechanical forces shape the tumor microenvironment. Ann. Biomed. Eng. 2011, 39, 1379–1389. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021. [Google Scholar] [CrossRef]
- Neesse, A.; Krug, S.; Gress, T.M.; Tuveson, D.A.; Michl, P. Emerging concepts in pancreatic cancer medicine: Targeting the tumor stroma. Onco. Targets. Ther. 2013, 7, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, J.; Hancock, J.; Kwon, J.; Korc, M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49. [Google Scholar] [CrossRef]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K.; Dandara, C. Architecture of Cancer-Associated Fibroblasts in Tumor Microenvironment: Mapping Their Origins, Heterogeneity, and Role in Cancer Therapy Resistance. Omics J. Integr. Biol. 2020, 24, 314–339. [Google Scholar] [CrossRef]
- Sunami, Y.; Häußler, J.; Klee, J. Cellular heterogeneity of pancreatic stellate cells, mesenchymal stem cells, and cancer-associated fibroblasts in pancreatic cancer. Cancers 2020, 12, 3770. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Gonidec, S.L.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [Green Version]
- Nair, N.; Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.K.K.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Okumura, T.; Ohuchida, K.; Kibe, S.; Iwamoto, C.; Ando, Y.; Takesue, S.; Nakayama, H.; Abe, T.; Endo, S.; Koikawa, K.; et al. Adipose tissue-derived stromal cells are sources of cancer-associated fibroblasts and enhance tumor progression by dense collagen matrix. Int. J. Cancer 2019, 144, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-associated fibroblasts—Heroes or villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Whittle, M.C.; Hingorani, S.R. Fibroblasts in Pancreatic Ductal Adenocarcinoma: Biological Mechanisms and Therapeutic Targets. Gastroenterology 2019, 156, 2085–2096. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tuveson, D.A. Diversity and biology of cancerassociated fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 4, e129212. [Google Scholar] [CrossRef] [Green Version]
- Alkasaliasa, T.; Flaberg, E.; Kashuba, V.; Alexeyenko, A.; Pavlova, T.; Savchenko, A.; Szekely, L.; Klein, G.; Guvena, H. Inhibition of tumor cell proliferation and motility by fibroblasts is both contact and soluble factor dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 17188–17193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoker, M.G.; Shearer, M.; O’Neill, C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J. Cell Sci. 1966, 1, 297–310. [Google Scholar] [CrossRef]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef]
- Gopal, S.; Veracini, L.; Grall, D.; Butori, C.; Schaub, S.; Audebert, S.; Camoin, L.; Baudelet, E.; Adwanska, A.; Beghelli-De La Forest Divonne, S.; et al. Fibronectin-guided migration of carcinoma collectives. Nat. Commun. 2017, 8, 14105. [Google Scholar] [CrossRef]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariani, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-associated fibroblasts lead tumor invasion through integrin-β3-dependent fibronectin asse. J. Cell Biol. 2017, 216, 3509–3520. [Google Scholar] [CrossRef] [Green Version]
- Pausch, T.M.; Aue, E.; Wirsik, N.M.; Freire Valls, A.; Shen, Y.; Radhakrishnan, P.; Hackert, T.; Schneider, M.; Schmidt, T. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the CXCL8 and the CCL2 axes. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Xiao, Y.; Zhang, S.; Wang, J. Advances in crosslinking strategies of biomedical hydrogels. Biomater. Sci. 2019, 7, 843–855. [Google Scholar] [CrossRef]
- Wang, F.T.; Sun, W.E.I.; Zhang, J.T.; Fan, Y.Z. Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer (Review). Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.; Puré, E. Cancer-associated fibroblasts: Key determinants of tumor immunity and immunotherapy. Curr. Opin. Immunol. 2020, 64, 80–87. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadera, B.E.; Li, L.; Toste, P.A.; Wu, N.; Adams, C.; Dawson, D.W.; Donahue, T.R. MicroRNA-21 in Pancreatic Ductal Adenocarcinoma Tumor-Associated Fibroblasts Promotes Metastasis. PLoS ONE 2013, 8, e71978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; He, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wu, J.; Wu, J.; Jing, X.; Du, Q.; Hui, X.; et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut 2021. [Google Scholar] [CrossRef]
- Takesue, S.; Ohuchida, K.; Shinkawa, T.; Otsubo, Y.; Matsumoto, S.; Sagara, A.; Yonenaga, A.; Ando, Y.; Kibe, S.; Nakayama, H.; et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int. J. Oncol. 2020, 56, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Domen, A.; Quatannens, D.; Zanivan, S.; Deben, C.; Van Audenaerde, J.; Smits, E.; Wouters, A.; Lardon, F.; Roeyen, G.; Verhoeven, Y.; et al. Cancer-associated fibroblasts as a common orchestrator of therapy resistance in lung and pancreatic cancer. Cancers 2021, 13, 987. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef]
- Huelsken, J.; Hanahan, D. A Subset of Cancer-Associated Fibroblasts Determines Therapy Resistance. Cell 2018, 172, 643–644. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Lin, Q.; Lu, Y.; Li, G.; Huang, L.; Fu, Z.; Chen, R.; Zhou, Q. Cancer-associated fibroblasts-mediated ATF4 expression promotes malignancy and gemcitabine resistance in pancreatic cancer via the TGF-β1/SMAD2/3 pathway and ABCC1 transactivation. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Artlett, C.M. Inflammasomes in wound healing and fibrosis. J. Pathol. 2013, 229, 157–167. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Barbazán, J.; Matic Vignjevic, D. Cancer associated fibroblasts: Is the force the path to the dark side? Curr. Opin. Cell Biol. 2019, 56, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between tumor and stromal cells in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 5486. [Google Scholar] [CrossRef]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignatelli, C.; Cadamuro, F.; Magli, S.; Rossi, L.; Russo, L.; Nicotra, F. Glycans and hybrid glyco-materials for artificial cell microenvironment fabrication. Carbohydr. Chem. 2021, 44, 250–276. [Google Scholar]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef]
- Kai, F.B.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Dufort, C.C.; Delgiorno, K.E.; Hingorani, S.R. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2016, 150, 1545–1557.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R.; Erler, J.T. Molecular pathways: Connecting fibrosis and solid tumor metastasis. Clin. Cancer Res. 2014, 20, 3637–3643. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Liu, Z.; Zeng, X.; Wu, Q.; Liao, X.; Wang, X.; Han, C.; Yu, T.; Zhu, G.; Qin, W.; et al. Evaluation of the diagnostic ability of laminin gene family for pancreatic ductal adenocarcinoma. Aging 2019, 11, 3679–3703. [Google Scholar] [CrossRef]
- Okada, Y.; Nishiwada, S.; Yamamura, K.; Sho, M.; Baba, H.; Takayama, T.; Goel, A. Identification of laminin γ2 as a prognostic and predictive biomarker for determining response to gemcitabine-based therapy in pancreatic ductal adenocarcinoma. Eur. J. Cancer 2021, 146, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pan, Y.-Z.; Cheung, M.; Cao, M.; Yu, C.; Chen, L.; Zhan, L.; He, Z.-W.; Sun, C.-Y. LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway. Cell Death Dis. 2019, 10, 230. [Google Scholar] [CrossRef] [Green Version]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Guldager Kring Rasmussen, D.; Karsdal, M.A. Laminins. In Biochemistry of Collagens, Laminins and Elastin: Structure, Function and Biomarkers; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 163–196. ISBN 9780128098998. [Google Scholar]
- Huang, C.; Chen, J. Laminin-332 mediates proliferation, apoptosis, invasion, migration and epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma. Mol. Med. Rep. 2021, 23, 11. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, H.; Luo, J.; Wu, X.; Li, X.; Zhao, X.; Zhou, D.; Yu, S. Overexpression of α3, β3 and γ2 chains of laminin-332 is associated with poor prognosis in pancreatic ductal adenocarcinoma. Oncol. Lett. 2018, 16, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Liang, Z.; Shi, X.; Ren, X.; Wang, K.; Liu, T. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 2009, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Bohaumilitzky, L.; Huber, A.K.; Stork, E.M.; Wengert, S.; Woelfl, F.; Boehm, H. A trickster in disguise: Hyaluronan’s ambivalent roles in the matrix. Front. Oncol. 2017, 7, 242. [Google Scholar] [CrossRef] [PubMed]
- Tang, V.W. Collagen, stiffness, and adhesion: The evolutionary basis of vertebrate mechanobiology. Mol. Biol. Cell 2020, 31, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, A.; Malfait, F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin. Genet. 2012, 82, 1–11. [Google Scholar] [CrossRef]
- Tavianatou, A.G.; Caon, I.; Franchi, M.; Piperigkou, Z.; Galesso, D.; Karamanos, N.K. Hyaluronan: Molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J. 2019, 286, 2883–2908. [Google Scholar] [CrossRef]
- Caon, I.; Bartolini, B.; Parnigoni, A.; Caravà, E.; Moretto, P.; Viola, M.; Karousou, E.; Vigetti, D.; Passi, A. Revisiting the hallmarks of cancer: The role of hyaluronan. Semin. Cancer Biol. 2020, 62, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K. Laminin-5 (laminin-332): Unique biological activity and role in tumor growth and invasion. Cancer Sci. 2006, 97, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garciá, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [Green Version]
- Slapak, E.J.; Duitman, J.; Tekin, C.; Bijlsma, M.F.; Spek, C.A. Matrix Metalloproteases in Pancreatic Ductal Adenocarcinoma: Key Drivers of Disease Progression? Biology 2020, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Öhlund, D.; Rickelt, S.; Lidström, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer cell–derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Res. 2020, 80, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Van Arsdall, M.; Tedjarati, S.; McCarty, M.; Wu, W.; Langley, R.; Fidler, I.J. Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J. Natl. Cancer Inst. 2002, 94, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.S.; Landers, R.J.; Underwood, J.C.E.; Harris, A.L.; Lewis, C.E. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J. Pathol. 2000, 192, 150–158. [Google Scholar] [CrossRef]
- Kanda, S.; Landgren, E.; Ljungström, M.; Claesson-Welsh, L. Fibroblast GROWTH factor Receptor 1-Induced Differentiation of Endothelial Cell Line Established from tsA58 Large T Transgenic Mice. Available online: https://pubmed.ncbi.nlm.nih.gov/8838868/ (accessed on 21 July 2021).
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular mediators of angiogenesis. J. Burn Care Res. 2010, 31, 158–175. [Google Scholar] [CrossRef]
- Rubiano, A.; Delitto, D.; Han, S.; Gerber, M.; Galitz, C.; Trevino, J.; Thomas, R.M.; Hughes, S.J.; Simmons, C.S. Viscoelastic properties of human pancreatic tumors and in vitro constructs to mimic mechanical properties. Acta Biomater. 2018, 67, 331–340. [Google Scholar] [CrossRef]
- D’Onofrio, M.; Crosara, S.; De Robertis, R.; Canestrini, S.; Demozzi, E.; Pozzi Mucelli, R. Elastography of the pancreas. Eur. J. Radiol. 2014, 83, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Hirooka, Y.; Kawashima, H.; Ohno, E.; Sugimoto, H.; Hayashi, D.; Morishima, T.; Kawai, M.; Suhara, H.; Takeyama, T.; et al. Quantitative evaluation of pancreatic tumor fibrosis using shear wave elastography. Pancreatology 2016, 16, 1063–1068. [Google Scholar] [CrossRef]
- Nabavizadeh, A.; Payen, T.; Iuga, A.C.; Sagalovskiy, I.R.; Desrouilleres, D.; Saharkhiz, N.; Palermo, C.F.; Sastra, S.A.; Oberstein, P.E.; Rosario, V.; et al. Noninvasive Young’s modulus visualization of fibrosis progression and delineation of pancreatic ductal adenocarcinoma (PDAC) tumors using Harmonic Motion Elastography (HME) in vivo. Theranostics 2020, 10, 4614–4626. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mislati, R.; Ahmed, R.; Vincent, P.; Nwabunwanne, S.F.; Gunn, J.R.; Pogue, B.W.; Doyley, M.M. Elastography can map the local inverse relationship between shear modulus and drug delivery within the pancreatic ductal adenocarcinoma microenvironment. Clin. Cancer Res. 2019, 25, 2136–2143. [Google Scholar] [CrossRef] [Green Version]
- Kreger, S.T.; Voytik-Harbin, S.L. Hyaluronan concentration within a 3D collagen matrix modulates matrix viscoelasticity, but not fibroblast response. Matrix Biol. 2009, 28, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Puls, T.J.; Tan, X.; Whittington, C.F.; Voytik-Harbin, S.L. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE 2017, 12, e0188870. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.R.I.; Wong, H.K.; Nai, S.; Chua, C.K.; Tan, N.S.; Tan, L.P. A 3D biomimetic model of tissue stiffness interface for cancer drug testing. Mol. Pharm. 2014, 11, 2016–2021. [Google Scholar] [CrossRef]
- Miroshnikova, Y.A.; Jorgens, D.M.; Spirio, L.; Auer, M.; Sarang-Sieminski, A.L.; Weaver, V.M. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Phys. Biol. 2011, 8, 026013. [Google Scholar] [CrossRef] [Green Version]
- Mason, B.N.; Starchenko, A.; Williams, R.M.; Bonassar, L.J.; Reinhart-King, C.A. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomater. 2013, 9, 4635–4644. [Google Scholar] [CrossRef] [Green Version]
- Nia, H.T.; Munn, L.L.; Jain, R.K. Mapping physical tumor microenvironment and drug delivery. Clin. Cancer Res. 2019, 25, 2024–2026. [Google Scholar] [CrossRef] [Green Version]
- Kalli, M.; Stylianopoulos, T. Defining the role of solid stress and matrix stiffness in cancer cell proliferation and metastasis. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Nia, H.T.; Liu, H.; Seano, G.; Datta, M.; Jones, D.; Rahbari, N.; Incio, J.; Chauhan, V.P.; Jung, K.; Martin, J.D.; et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat. Biomed. Eng. 2017, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nieskoski, M.D.; Marra, K.; Gunn, J.R.; Kanick, S.C.; Doyley, M.M.; Hasan, T.; Pereira, S.P.; Stuart Trembly, B.; Pogue, B.W. Separation of Solid Stress from Interstitial Fluid Pressure in Pancreas Cancer Correlates With Collagen Area Fraction. J. Biomech. Eng. 2017, 139, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Boucher, Y.; Jain, R.K. Delivery of molecular and nanoscale medicine to tumors: Transport barriers and strategies. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 281–298. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Scott Brockenbrough, J.; et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys. J. 2016, 110, 2106–2119. [Google Scholar] [CrossRef] [Green Version]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Ling, J.Y.; Chen, W.C.; Lin, H.H.; Tang, M.J. Mechanotransduction of matrix stiffness in regulation of focal adhesion size and number: Reciprocal regulation of caveolin-1 and β1 integrin. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Rath, N.; Olson, M.F. Regulation of pancreatic cancer aggressiveness by stromal stiffening. Nat. Med. 2016, 22, 462–463. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Njah, K.; Pobbati, A.V.; Lim, Y.B.; Raju, A.; Lakshmanan, M.; Tergaonkar, V.; Lim, C.T.; Hong, W. Agrin as a Mechanotransduction Signal Regulating YAP through the Hippo Pathway. Cell Rep. 2017, 18, 2464–2479. [Google Scholar] [CrossRef] [Green Version]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkovskaya, A.; Buffone, A.; Žídek, M.; Weaver, V.M. Proteoglycans as Mediators of Cancer Tissue Mechanics. Front. Cell Dev. Biol. 2020, 8, 1–21. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Río Hernández, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnevale, I.; Capula, M.; Giovannetti, E.; Schmidt, T.; Coppola, S. A mechanical memory of pancreatic cancer cells. bioRxiv 2019, 730960. [Google Scholar] [CrossRef]
- Coppola, S.; Carnevale, I.; Danen, E.H.J.; Peters, G.J.; Schmidt, T.; Assaraf, Y.G.; Giovannetti, E. A mechanopharmacology approach to overcome chemoresistance in pancreatic cancer. Drug Resist. Updat. 2017, 31, 43–51. [Google Scholar] [CrossRef]
- Kim, M.K.; Jang, J.W.; Bae, S.C. DNA binding partners of YAP/TAZ. BMB Rep. 2018, 51, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, L.; Ramirez, N.; Kim, J. Context-dependent roles of YAP/TAZ in stem cell fates and cancer. Cell. Mol. Life Sci. 2021, 78, 4201–4219. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; Secone Seconetti, A.; De Santis, G.; Pellacani, G.; Hirsch, T.; Rothoeft, T.; Teig, N.; Pellegrini, G.; Bauer, J.W.; De Luca, M. Laminin 332-Dependent YAP Dysregulation Depletes Epidermal Stem Cells in Junctional Epidermolysis Bullosa. Cell Rep. 2019, 27, 2036–2049.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarper, M.; Cortes, E.; Lieberthal, T.J.; Del Río Hernández, A. ATRA modulates mechanical activation of TGF-β by pancreatic stellate cells. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lachowski, D.; Cortes, E.; Pink, D.; Chronopoulos, A.; Karim, S.A.; Morton, J.P.; Del Río Hernández, A.E. Substrate Rigidity Controls Activation and Durotaxis in Pancreatic Stellate Cells. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Nyberg, K.D.; Scott, M.B.; Welsh, A.M.; Nguyen, A.H.; Wu, N.; Hohlbauch, S.V.; Geisse, N.A.; Gibb, E.A.; Robertson, A.G.; et al. Stiffness of pancreatic cancer cells is associated with increased invasive potential. Integr. Biol. 2016, 8, 1232–1245. [Google Scholar] [CrossRef]
- Kalli, M.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Solid Stress Facilitates Fibroblasts Activation to Promote Pancreatic Cancer Cell Migration. Ann. Biomed. Eng. 2018, 46, 657–669. [Google Scholar] [CrossRef]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Melissari, M.T.; Chalkidi, N.; Sarris, M.E.; Koliaraki, V. Fibroblast Reprogramming in Gastrointestinal Cancer. Front. Cell Dev. Biol. 2020, 8, 630. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, T.; Shen, S.; She, X.; Tuo, Y.; Hu, Y.; Pang, Z.; Jiang, X. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials 2016, 103, 12–21. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- Han, X.; Zhang, W.H.; Wang, W.Q.; Yu, X.J.; Liu, L. Cancer-associated fibroblasts in therapeutic resistance of pancreatic cancer: Present situation, predicaments, and perspectives. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188444. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, J.; Korc, M. Pancreatic Cancer Stroma: Friend or Foe? Cancer Cell 2014, 25, 711–712. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duperret, E.K.; Trautz, A.; Ammons, D.; Perales-Puchalt, A.; Wise, M.C.; Yan, J.; Reed, C.; Weiner, D.B. Alteration of the tumor stroma using a consensus DNA vaccine targeting Fibroblast Activation Protein (FAP) synergizes with antitumor vaccine therapy in Mice. Clin. Cancer Res. 2018, 24, 1190–1201. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, M.; Krüger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Investig. 2006, 116, 1955–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakarla, S.; Chow, K.K.H.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Wang, L.C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Mayorga, G.; Gómez-López, G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Muñoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef]
- Froeling, F.E.M.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wntβ-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [Green Version]
- Dolor, A.; Szoka, F.C. Digesting a Path Forward: The Utility of Collagenase Tumor Treatment for Improved Drug Delivery. Mol. Pharm. 2018, 15, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Koren, L.; Adir, O.; Poley, M.; Alyan, M.; Yaari, Z.; Noor, N.; Krinsky, N.; Simon, A.; Gibori, H.; et al. Collagenase Nanoparticles Enhance the Penetration of Drugs into Pancreatic Tumors. ACS Nano 2019, 13, 11008–11021. [Google Scholar] [CrossRef] [PubMed]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Zion, O.; Genin, O.; Kawada, N.; Yoshizato, K.; Roffe, S.; Nagler, A.; Iovanna, J.L.; Halevy, O.; Pines, M. Inhibition of transforming growth factor β signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas 2009, 38, 427–435. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (Fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Lampi, M.C.; Reinhart-King, C.A. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci. Transl. Med. 2018, 10, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Lin, C.Y.; Liu, Y.; Staveley-OCarroll, K.F.; Li, G.; Cheng, K. Silencing PCBP2 normalizes desmoplastic stroma and improves the antitumor activity of chemotherapy in pancreatic cancer. Theranostics 2021, 11, 2182–2200. [Google Scholar] [CrossRef]
- Chen, S.T.; Kuo, T.C.; Liao, Y.Y.; Lin, M.C.; Tien, Y.W.; Huang, M.C. Silencing of MUC20 suppresses the malignant character of pancreatic ductal adenocarcinoma cells through inhibition of the HGF/MET pathway. Oncogene 2018, 37, 6041–6053. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Rose, J.L.; Wang, W.; Seth, S.; Jiang, H.; Taguchi, A.; Liu, J.; Yan, L.; Kapoor, A.; Hou, P.; et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 2019, 568, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xu, Y.; Geranpayehvaghei, M.; Anderson, G.J.; Li, Y.; Nie, G. Emerging nanomedicines for anti-stromal therapy against desmoplastic tumors. Biomaterials 2020, 232, 119745. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vita, J.; Sánchez-López, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-β-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Jia, F.; Li, Y.; Deng, Y.; Huang, Y.; Liu, W.; Jin, Q.; Ji, J. Nitric oxide-induced stromal depletion for improved nanoparticle penetration in pancreatic cancer treatment. Biomaterials 2020, 246. [Google Scholar] [CrossRef]
- Beyer, C.; Zenzmaier, C.; Palumbo-Zerr, K.; Mancuso, R.; Distler, A.; Dees, C.; Zerr, P.; Huang, J.; Maier, C.; Pachowsky, M.L.; et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFβ signalling. Ann. Rheum. Dis. 2015, 74, 1408–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, K.C.; Bernier, S.G.; Jacobson, S.; Liu, G.; Zhang, P.Y.; Sarno, R.; Catanzano, V.; Currie, M.G.; Masferrer, J.L. SGC stimulator praliciguat suppresses stellate cell fibrotic transformation and inhibits fibrosis and inflammation in models of NASH. Proc. Natl. Acad. Sci. USA 2019, 166, 11057–11062. [Google Scholar] [CrossRef] [Green Version]
- Zenzmaier, C.; Kern, J.; Heitz, M.; Plas, E.; Zwerschke, W.; Mattesich, M.; Sandner, P.; Berger, P. Activators and stimulators of soluble guanylate cyclase counteract myofibroblast differentiation of prostatic and dermal stromal cells. Exp. Cell Res. 2015, 338, 162–169. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Storm, G.; Prakash, J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 2018, 129, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Figlin, R.A.; Kondagunta, G.V.; Yazji, S.; Motzer, R.J.; Bukowski, R.M. Phase II study of volociximab (M200), an α5β1 anti-integrin antibody in refractory metastatic clear cell renal cell cancer (RCC). J. Clin. Oncol. 2006, 24, 4535. [Google Scholar] [CrossRef]
- Evans, T.; Ramanathan, R.K.; Yazji, S.; Glynne-Jones, R.; Anthoney, A.; Berlin, J.; Valle, J.W. Final results from cohort 1 of a phase II study of volociximab, an anti-α5β1 integrin antibody, in combination with gemcitabine (GEM) in patients (pts) with metastatic pancreatic cancer (MPC). J. Clin. Oncol. 2007, 25, 4549. [Google Scholar] [CrossRef]
- Yoon, S.O.; Shin, S.; Karreth, F.A.; Blenis, J.; Karreth, F.A.; Jedrychowski, M.P.; Gygi, S.P.; Plas, D.R.; Dedhar, S.; Roux, P.P.; et al. Focal Adhesion- and IGF1R-Dependent Survival and Migratory Pathways Mediate Tumor Resistance to mTORC1/2 Inhibition. Mol. Cell 2017, 67, 512–527.e4. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Hamm, J.; Dancey, J.; Eisenberg, P.D.; Dagenais, M.; Fields, A.; Hagan, K.; Greenberg, B.; Colwell, B.; Zee, B.; et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2003, 21, 3296–3302. [Google Scholar] [CrossRef]
- Evans, J.D.; Stark, A.; Johnson, C.D.; Daniel, F.; Carmichael, J.; Buckels, J.; Imrie, C.W.; Brown, P.; Neoptolemos, J.P. A phase II trial of marimastat in advanced pancreatic cancer. Br. J. Cancer 2001, 85, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Mikels-Vigdal, A.J.; Stefanutti, E.; Schwarz, M.A.; Monahan, S.; Smith, V.; Schwarz, R.E. Therapeutic efficacy of anti-MMP9 antibody in combination with nab-paclitaxel-based chemotherapy in pre-clinical models of pancreatic cancer. J. Cell. Mol. Med. 2019, 23, 3878–3887. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.S.; Haldar, M.K.; Nahire, R.R.; Katti, P.; Ambre, A.H.; Muhonen, W.W.; Shabb, J.B.; Padi, S.K.R.; Singh, R.K.; Borowicz, P.P.; et al. MMP-9 responsive PEG cleavable nanovesicles for efficient delivery of chemotherapeutics to pancreatic cancer. Mol. Pharm. 2014, 11, 2390–2399. [Google Scholar] [CrossRef] [Green Version]
- Welter, M.; Rieger, H. Interstitial Fluid Flow and Drug Delivery in Vascularized Tumors: A Computational Model. PLoS ONE 2013, 8, 70395. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B.; Shepard, H.M.; O’Connor, P.M.; Kadhim, S.; Jiang, P.; Osgood, R.J.; Bookbinder, L.H.; Li, X.; Sugarman, B.J.; Connor, R.J.; et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol. Cancer Ther. 2010, 9, 3052–3064. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; MacArulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef] [PubMed]
- Maione, F.; Molla, F.; Meda, C.; Latini, R.; Zentilin, L.; Giacca, M.; Seano, G.; Serini, G.; Bussolino, F.; Giraudo, E. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J. Clin. Investig. 2009, 119, 3356–3372. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Destouches, D.; El Khoury, D.; Hamma-Kourbali, Y.; Krust, B.; Albanese, P.; Katsoris, P.; Guichard, G.; Briand, J.P.; Courty, J.; Hovanessian, A.G. Suppression of tumor growth and angiogenesis by a specific antagonist of the cell-surface expressed nucleolin. PLoS ONE 2008, 3, e2518. [Google Scholar] [CrossRef] [Green Version]
- Hovanessian, A.G.; Soundaramourty, C.; El Khoury, D.; Nondier, I.; Svab, J.; Krust, B. Surface expressed nucleolin is constantly induced in tumor cells to mediate calcium-dependent ligand internalization. PLoS ONE 2010, 5, e15787. [Google Scholar] [CrossRef] [Green Version]
- Christian, S.; Pilch, J.; Akerman, M.E.; Porkka, K.; Laakkonen, P.; Ruoslahti, E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J. Cell Biol. 2003, 163, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilles, M.E.; Maione, F.; Cossutta, M.; Carpentier, G.; Caruana, L.; Di Maria, S.; Houppe, C.; Destouches, D.; Shchors, K.; Prochasson, C.; et al. Nucleolin targeting impairs the progression of pancreatic cancer and promotes the normalization of tumor vasculature. Cancer Res. 2016, 76, 7181–7193. [Google Scholar] [CrossRef] [Green Version]
- Teranishi, N.; Naito, Z.; Ishiwata, T.; Tanaka, N.; Furukawa, K.; Seya, T.; Shinji, S.; Tajiri, T. Identification of Neovasculature Using Nestin in Colorectal Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/17273760/ (accessed on 21 July 2021).
- Mokrý, J.; Čížková, D.; Filip, S.; Ehrmann, J.; Österreicher, J.; Kolář, Z.; English, D. Nestin expression by newly formed human blood vessels. Stem Cells Dev. 2004, 13, 658–664. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Naito, Z. Nestin in gastrointestinal and other cancers: Effects on cells and tumor angiogenesis. World J. Gastroenterol. 2011, 17, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Yamahatsu, K.; Matsuda, Y.; Ishiwata, T.; Uchida, E.; Naito, Z. Nestin as a novel therapeutic target for pancreatic cancer via tumor angiogenesis. Int. J. Oncol. 2012, 40, 1345–1357. [Google Scholar] [CrossRef] [Green Version]
- Tanigawa, N.; Amaya, H.; Matsumura, M.; Shimomatsuya, T. Association of tumour vasculature with tumour progression and overall survival of patients with non-early gastric carcinomas. Br. J. Cancer 1997, 75, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Tucker, S.L.; Kitadai, Y.; Koura, A.N.; Bucana, C.D.; Cleary, K.R.; Ellis, L.M. Vessel counts and expression of vascular endothelial growth factor as prognostic factors in node-negative colon cancer. Arch. Surg. 1997, 132, 541–546. [Google Scholar] [CrossRef]
- Matsuda, Y.; Hagio, M.; Ishiwata, T. Nestin: A novel angiogenesis marker and possible target for tumor angiogenesis. World J. Gastroenterol. 2013, 19, 42–48. [Google Scholar] [CrossRef]
- Filipe, E.C.; Chitty, J.L.; Cox, T.R. Charting the unexplored extracellular matrix in cancer. Int. J. Exp. Pathol. 2018, 99, 58–76. [Google Scholar] [CrossRef] [Green Version]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Mani, D.R.; Carr, S.A.; Hynes, R.O. Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression. Sci. Rep. 2017, 7, 40495. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Engel, J. Domain structure and organisation in extracellular matrix proteins. Matrix Biol. 2002, 21, 115–128. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Caprioli, R.M.; Farmer, T.B.; Gile, J. Molecular Imaging of Biological Samples: Localization of Peptides and Proteins Using MALDI-TOF MS. Anal. Chem. 1997, 69, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- DeKeyser, S.S.; Kutz-Naber, K.K.; Schmidt, J.J.; Barrett-Wilt, G.A.; Li, L. Imaging mass spectrometry of neuropeptides in decapod crustacean neuronal tissues. J. Proteome Res. 2007, 6, 1782–1791. [Google Scholar] [CrossRef] [Green Version]
- Gessel, M.; Spraggins, J.M.; Voziyan, P.; Hudson, B.G.; Caprioli, R.M. Decellularization of intact tissue enables MALDI imaging mass spectrometry analysis of the extracellular matrix. J. Mass Spectrom. 2015, 50, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- Fischer, I.; Westphal, M.; Rossbach, B.; Bethke, N.; Hariharan, K.; Ullah, I.; Reinke, P.; Kurtz, A.; Stachelscheid, H. Comparative characterization of decellularized renal scaffolds for tissue engineering. Biomed. Mater. 2017, 12. [Google Scholar] [CrossRef]
- Mayorca-Guiliani, A.E.; Madsen, C.D.; Cox, T.R.; Horton, E.R.; Venning, F.A.; Erler, J.T. ISDoT: In situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix. Nat. Med. 2017, 23, 890–898. [Google Scholar] [CrossRef]
- Mateescu, B.; Kowal, E.J.K.; van Balkom, B.W.M.; Bartel, S.; Bhattacharyya, S.N.; Buzás, E.I.; Buck, A.H.; de Candia, P.; Chow, F.W.N.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA—An ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, M.I.; Amorim, M.G.; Gadelha, C.; Milic, I.; Welsh, J.A.; Freitas, V.M.; Nawaz, M.; Akbar, N.; Couch, Y.; Makin, L.; et al. Technical challenges of working with extracellular vesicles. Nanoscale 2018, 10, 881–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, M.; Camussi, G.; Valadi, H.; Nazarenko, I.; Ekström, K.; Wang, X.; Principe, S.; Shah, N.; Ashraf, N.M.; Fatima, F.; et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 2014, 11, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Lozito, T.P.; Tuan, R.S. Endothelial cell microparticles act as centers of matrix metalloproteinsase-2 (MMP-2) activation and vascular matrix remodeling. J. Cell. Physiol. 2012, 227, 534–549. [Google Scholar] [CrossRef] [Green Version]
- Koeck, E.S.; Iordanskaia, T.; Sevilla, S.; Ferrante, S.C.; Hubal, M.J.; Freishtat, R.J.; Nadler, E.P. Adipocyte exosomes induce transforming growth factor beta pathway dysregulation in hepatocytes: A novel paradigm for obesity-related liver disease. J. Surg. Res. 2014, 192, 268–275. [Google Scholar] [CrossRef]
- Shimoda, M.; Khokha, R. Proteolytic factors in exosomes. Proteomics 2013, 13, 1624–1636. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Bandari, S.K.; Vlodavsky, I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biol. 2019, 75–76, 160–169. [Google Scholar] [CrossRef]
- Ginestra, A.; Monea, S.; Seghezzi, G.; Dolo, V.; Nagase, H.; Mignatti, P.; Vittorelli, M.L. Urokinase plasminogen activator and gelatinases are associated with membrane vesicles shed by human HT1080 fibrosarcoma cells. J. Biol. Chem. 1997, 272, 17216–17222. [Google Scholar] [CrossRef] [Green Version]
- Dolo, V.; Ginestra, A.; Cassará, D.; Ghersi, G.; Nagase, H.; Vittorelli, M.L. Shed membrane vesicles and selective localization of gelatinases and MMP-9/TIMP-1 complexes. Proc. Ann. N. Y. Acad. Sci. 1999, 878, 497–499. [Google Scholar] [CrossRef] [PubMed]
- McAtee, C.O.; Booth, C.; Elowsky, C.; Zhao, L.; Payne, J.; Fangman, T.; Caplan, S.; Henry, M.D.; Simpson, M.A. Prostate tumor cell exosomes containing hyaluronidase Hyal1 stimulate prostate stromal cell motility by engagement of FAK-mediated integrin signaling. Matrix Biol. 2019, 78–79, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.W.; Newgreen, D.F. Carcinoma invasion and metastasis: A role for epithelial-mesenchymal transition? Cancer Res. 2005, 65, 5991–5995. [Google Scholar] [CrossRef] [Green Version]
- Banyard, J.; Bielenberg, D.R. The role of EMT and MET in cancer dissemination. Connect. Tissue Res. 2015, 56, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Diepenbruck, M.; Christofori, G. Epithelial-mesenchymal transition (EMT) and metastasis: Yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.; Li, Q.; Li, S.; He, J.; Cao, W.; Lan, J.; Sun, B.; Zou, H.; Wang, C.; Liu, R.; et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, L.J. The emerging role of exosomes in epithelial-mesenchymal-transition in cancer. Front. Oncol. 2014, 4, 361. [Google Scholar] [CrossRef] [Green Version]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef]
- Galindo-Hernandez, O.; Serna-Marquez, N.; Castillo-Sanchez, R.; Salazar, E.P. Extracellular vesicles from MDA-MB-231 breast cancer cells stimulated with linoleic acid promote an EMT-like process in MCF10A cells. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 299–310. [Google Scholar] [CrossRef]
- Zheng, J.; Hernandez, J.M.; Doussot, A.; Bojmar, L.; Zambirinis, C.P.; Costa-Silva, B.; van Beek, E.J.A.H.; Mark, M.T.; Molina, H.; Askan, G.; et al. Extracellular matrix proteins and carcinoembryonic antigen-related cell adhesion molecules characterize pancreatic duct fluid exosomes in patients with pancreatic cancer. Hpb 2018, 20, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V.P.; Jiang, W.; Popovíc, Z.; Jain, R.K.; Bawendi, M.G.; Fukumura, D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 2426–2431. [Google Scholar] [CrossRef] [Green Version]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef]
- Lieleg, O.; Baumgärtel, R.M.; Bausch, A.R. Selective filtering of particles by the extracellular matrix: An electrostatic bandpass. Biophys. J. 2009, 97, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Stylianopoulos, T.; Soteriou, K.; Fukumura, D.; Jain, R.K. Cationic nanoparticles have superior transvascular flux into solid tumors: Insights from a mathematical model. Ann. Biomed. Eng. 2013, 41, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Fadeel, B.; Feliu, N.; Vogt, C.; Abdelmonem, A.M.; Parak, W.J. Bridge over troubled waters: Understanding the synthetic and biological identities of engineered nanomaterials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 111–129. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Lucero-Acuña, A.; Guzmán, R. Nanoparticle encapsulation and controlled release of a hydrophobic kinase inhibitor: Three stage mathematical modeling and parametric analysis. Int. J. Pharm. 2015, 494, 249–257. [Google Scholar] [CrossRef]
- Fan, F.; Jin, L.; Yang, L. pH-Sensitive Nanoparticles Composed Solely of Membrane-Disruptive Macromolecules for Treating Pancreatic Cancer. ACS Appl. Mater. Interfaces 2021, 13, 12824–12835. [Google Scholar] [CrossRef] [PubMed]
- Colby, A.H.; Oberlies, N.H.; Pearce, C.J.; Herrera, V.L.M.; Colson, Y.L.; Grinstaff, M.W. Nanoparticle drug-delivery systems for peritoneal cancers: A case study of the design, characterization and development of the expansile nanoparticle. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Qian, W.; Uckun, F.M.; Wang, L.; Wang, Y.A.; Chen, H.; Kooby, D.; Yu, Q.; Lipowska, M.; Staley, C.A.; et al. IGF1 Receptor Targeted Theranostic Nanoparticles for Targeted and Image-Guided Therapy of Pancreatic Cancer. ACS Nano 2015, 9, 7976–7991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peled, N.; Wynes, M.W.; Ikeda, N.; Ohira, T.; Yoshida, K.; Qian, J.; Ilouze, M.; Brenner, R.; Kato, Y.; Mascaux, C.; et al. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell. Oncol. 2013, 36, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Mizuuchi, H.; Sato, K.; Takemoto, T.; Iwasaki, T.; Mitsudomi, T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int. J. Cancer 2014, 135, 1002–1006. [Google Scholar] [CrossRef]
- Zhou, H.; Qian, W.; Uckun, F.M.; Zhou, Z.; Wang, L.; Wang, A.; Mao, H.; Yang, L. IGF-1 receptor targeted nanoparticles for image-guided therapy of stroma-rich and drug resistant human cancer. In Proceedings of the Micro- and Nanotechnology Sensors, Systems, and Applications VIII; SPIE: Baltimore, MD, USA, 2016; Volume 9836, p. 983610. [Google Scholar]
- Ji, T.; Lang, J.; Wang, J.; Cai, R.; Zhang, Y.; Qi, F.; Zhang, L.; Zhao, X.; Wu, W.; Hao, J.; et al. Designing Liposomes to Suppress Extracellular Matrix Expression to Enhance Drug Penetration and Pancreatic Tumor Therapy. ACS Nano 2017, 11, 8668–8678. [Google Scholar] [CrossRef]
- Flores, A.I. Nanotechnology and Mesenchymal Stem Cells for Regenerative Medicine. Glob. J. Nanomed. 2017, 1. [Google Scholar] [CrossRef]
- Pittenger, M.F. Mesenchymal stem cells from adult bone marrow. Methods Mol. Biol. 2008, 449, 27–44. [Google Scholar] [CrossRef]
- Miana, V.V.; Prieto González, E.A. Adipose tissue stem cells in regenerative medicine. Ecancermedicalscience 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Arutyunyan, I.; Elchaninov, A.; Makarov, A.; Fatkhudinov, T. Umbilical Cord as Prospective Source for Mesenchymal Stem Cell-Based Therapy. Stem Cells Int. 2016, 2016, 901286. [Google Scholar] [CrossRef] [Green Version]
- De Coppi, P.; Bartsch, G.; Siddiqui, M.M.; Xu, T.; Santos, C.C.; Perin, L.; Mostoslavsky, G.; Serre, A.C.; Snyder, E.Y.; Yoo, J.J.; et al. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 2007, 25, 100–106. [Google Scholar] [CrossRef] [PubMed]
- MacIas, M.I.; Grande, J.; Moreno, A.; Domnguez, I.; Bornstein, R.; Flores, A.I. Isolation and characterization of true mesenchymal stem cells derived from human term decidua capable of multilineage differentiation into all 3 embryonic layers. Am. J. Obstet. Gynecol. 2010, 203, 495.e9–495.e23. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Underhill, L.H.; Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Moniri, M.R.; Sun, X.Y.; Rayat, J.; Dai, D.; Ao, Z.; He, Z.; Verchere, C.B.; Dai, L.J.; Warnock, G.L. TRAIL-engineered pancreas-derived mesenchymal stem cells: Characterization and cytotoxic effects on pancreatic cancer cells. Cancer Gene Ther. 2012, 19, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Zischek, C.; Niess, H.; Ischenko, I.; Conrad, C.; Huss, R.; Jauch, K.W.; Nelson, P.J.; Bruns, C. Targeting tumor stroma using engineered mesenchymal stem cells reduces the growth of pancreatic carcinoma. Ann. Surg. 2009, 250, 747–752. [Google Scholar] [CrossRef]
- Menon, L.G.; Picinich, S.; Koneru, R.; Gao, H.; Lin, S.Y.; Koneru, M.; Mayer-Kuckuk, P.; Glod, J.; Banerjee, D. Differential Gene Expression Associated with Migration of Mesenchymal Stem Cells to Conditioned Medium from Tumor Cells or Bone Marrow Cells. Stem Cells 2007, 25, 520–528. [Google Scholar] [CrossRef]
- Dwyer, R.M.; Potter-Beirne, S.M.; Harrington, K.A.; Lowery, A.J.; Hennessy, E.; Murphy, J.M.; Barry, F.P.; O’Brien, T.; Kerin, M.J. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin. Cancer Res. 2007, 13, 5020–5027. [Google Scholar] [CrossRef] [Green Version]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; Van De Water, J.A.J.M.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4822–4827. [Google Scholar] [CrossRef] [Green Version]
- Son, B.-R.; Marquez-Curtis, L.A.; Kucia, M.; Wysoczynski, M.; Turner, A.R.; Ratajczak, J.; Ratajczak, M.Z.; Janowska-Wieczorek, A. Migration of Bone Marrow and Cord Blood Mesenchymal Stem Cells In Vitro Is Regulated by Stromal-Derived Factor-1-CXCR4 and Hepatocyte Growth Factor-c-met Axes and Involves Matrix Metalloproteinases. Stem Cells 2006, 24, 1254–1264. [Google Scholar] [CrossRef]
- Schmidt, N.O.; Przylecki, W.; Yang, W.; Ziu, M.; Teng, Y.; Kim, S.U.; Black, P.M.; Aboody, K.S.; Carroll, R.S. Brain tumor tropism of transplanted human neural stem cells is induced by vascular endothelial growth factor. Neoplasia 2005, 7, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.; Caldwell, L.; Dietrich, M.; Samudio, I.; Spaeth, E.L.; Watson, K.; Shi, Y.; Abbruzzese, J.; Konopleva, M.; Andreeff, M.; et al. Mesenchymal stromal cells alone or expressing interferon-β suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment. Cytotherapy 2010, 12, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Pessina, A.; Bonomi, A.; Coccè, V.; Invernici, G.; Navone, S.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Viganò, L.; Locatelli, A.; et al. Mesenchymal stromal cells primed with paclitaxel provide a new approach for cancer therapy. PLoS ONE 2011, 6, e28321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonomi, A.; Coccè, V.; Cavicchini, L.; Sisto, F.; Dossena, M.; Balzarini, P.; Portolani, N.; Ciusani, E.; Parati, E.; Alessandri, G.; et al. Adipose Tissue-Derived Stromal Cells Primed in Vitro with Paclitaxel Acquire Anti-Tumor Activity. Int. J. Immunopathol. Pharmacol. 2013, 26, 33–41. [Google Scholar] [CrossRef]
- Mizuno, H.; Tobita, M.; Uysal, A.C. Concise review: Adipose-derived stem cells as a novel tool for future regenerative medicine. Stem Cells 2012, 30, 804–810. [Google Scholar] [CrossRef]
- Tobita, M.; Orbay, H.; Mizuno, H. Adipose-derived stem cells: Current findings and future perspectives. Discov. Med. 2011, 11, 160–170. [Google Scholar]
- Mariani, E.; Facchini, A. Clinical Applications and Biosafety of Human Adult Mesenchymal Stem Cells. Curr. Pharm. Des. 2012, 18, 1821–1845. [Google Scholar] [CrossRef]
- Bonomi, A.; Sordi, V.; Dugnani, E.; Ceserani, V.; Dossena, M.; Coccè, V.; Cavicchini, L.; Ciusani, E.; Bondiolotti, G.; Piovani, G.; et al. Gemcitabine-releasing mesenchymal stromal cells inhibit in vitro proliferation of human pancreatic carcinoma cells. Cytotherapy 2015, 17, 1687–1695. [Google Scholar] [CrossRef] [Green Version]
- Kabashima-Niibe, A.; Higuchi, H.; Takaishi, H.; Masugi, Y.; Matsuzaki, Y.; Mabuchi, Y.; Funakoshi, S.; Adachi, M.; Hamamoto, Y.; Kawachi, S.; et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. 2013, 104, 157–164. [Google Scholar] [CrossRef]
- Clavreul, A.; Pourbaghi-Masouleh, M.; Roger, E.; Lautram, N.; Montero-Menei, C.N.; Menei, P. Human mesenchymal stromal cells as cellular drug-delivery vectors for glioblastoma therapy: A good deal? J. Exp. Clin. Cancer Res. 2017, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Dennis, J.E.; Muzic, R.F.; Lundberg, M.; Caplan, A.I. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs 2001, 169, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev. 2009, 18, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Larson, B.L.; Smith, J.R.; Pochampally, R.; Cui, J.; Prockop, D.J. Expansion of Human Adult Stem Cells from Bone Marrow Stroma: Conditions that Maximize the Yields of Early Progenitors and Evaluate Their Quality. Stem Cells 2002, 20, 530–541. [Google Scholar] [CrossRef]
- Von Bahr, L.; Batsis, I.; Moll, G.; Hägg, M.; Szakos, A.; Sundberg, B.; Uzunel, M.; Ringden, O.; Le Blanc, K. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells 2012, 30, 1575–1578. [Google Scholar] [CrossRef]
- Fouillard, L.; Chapel, A.; Bories, D.; Bouchet, S.; Costa, J.M.; Rouard, H.; Hervé, P.; Gourmelon, P.; Thierry, D.; Lopez, M.; et al. Infusion of allogeneic-related HLA mismatched mesenchymal stem cells for the treatment of incomplete engraftment following autologous haematopoietic stem cell transplantation. Leukemia 2007, 21, 568–570. [Google Scholar] [CrossRef]
- Parekkadan, B.; Milwid, J.M. Mesenchymal stem cells as therapeutics. Annu. Rev. Biomed. Eng. 2010, 12, 87–117. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Hou, S.; Ding, H.; Liu, Z.; Song, M.; Qin, X.; Wang, X.; Yu, M.; Sun, Z.; Liu, J.; et al. In Vivo Tracking of Systemically Administered Allogeneic Bone Marrow Mesenchymal Stem Cells in Normal Rats through Bioluminescence Imaging. Stem Cells Int. 2016, 2016, 3970942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forest, V.F.; Tirouvanziam, A.M.; Perigaud, C.; Fernandes, S.; Fusellier, M.S.; Desfontis, J.C.; Toquet, C.S.; Heymann, M.F.; Crochet, D.P.; Lemarchand, P.F. Cell distribution after intracoronary bone marrow stem cell delivery in damaged and undamaged myocardium: Implications for clinical trials. Stem Cell Res. Ther. 2010, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.K.; Riegler, J.; Wu, J.C. Stem cell imaging: From bench to bedside. Cell Stem Cell 2014, 14, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, E.A.; Henary, M.; El Fakhri, G.; Choi, H.S. Tissue-Specific Near-Infrared Fluorescence Imaging. Acc. Chem. Res. 2016, 49, 1731–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-Y.; Choi, J.-Y.; Jeong, J.-H.; Jang, E.-S.; Kim, A.-S.; Kim, S.-G.; Kweon, H.Y.; Jo, Y.-Y.; Yeo, J.-H. Low molecular weight silk fibroin increases alkaline phosphatase and type I collagen expression in MG63 cells. BMB Rep. 2010, 43, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Godoy, J.A.P.; Paiva, R.M.A.; Souza, A.M.; Kondo, A.T.; Kutner, J.M.; Okamoto, O.K. Clinical Translation of Mesenchymal Stromal Cell Therapy for Graft Versus Host Disease. Front. Cell Dev. Biol. 2019, 7, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.C.; Lu, Y.B.; Yang, Y.N.; Kang, X.W.; Wang, Y.G.; Ma, B.; Xing, S. Progress in clinical trials of cell transplantation for the treatment of spinal cord injury: How many questions remain unanswered? Neural Regen. Res. 2021, 16, 405–413. [Google Scholar] [PubMed]
- Guo, Y.; Yu, Y.; Hu, S.; Chen, Y.; Shen, Z. The therapeutic potential of mesenchymal stem cells for cardiovascular diseases. Cell Death Dis. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.W.; Wang, B. Cartilage repair by mesenchymal stem cells: Clinical trial update and perspectives. J. Orthop. Transl. 2017, 9, 76–88. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug and Study | Target | Design | Status | Phase | PDAC Tumor Stage |
|---|---|---|---|---|---|
| PEGPH20 | HA | ||||
| NCT01959139 | PEGPH20 + FOLFIRINOX | Active | I/II | Metastatic | |
| NCT03193190 | PEGPH20 + atezolizumab | Active | I/II | Metastatic | |
| NCT01839487 | PEGPH20 + GCB/nab | Completed | II | Metastatic | |
| NCT04058964 | PEGPH20 + pembrolizumab | Withdrawn | II | Metastatic | |
| NCT02921022 | PEGPH20 + GCB/nab + rivaroxaban | Active | NA | Advanced | |
| Paricalcitol | CAFs | ||||
| NCT03520790 | Paricalcitol + GCB/nab | Active | I/II | Metastatic | |
| NCT03300921 | Paricalcitol + pembrolizumab | Active | I | Resectable | |
| NCT03883919 | Liposomal Paricalcitol + 5-FU/leucovorin | Active | I | Advanced progressed on GCB-based therapy | |
| NCT03519308 | Paricalcitol + nivolumab + GCB/nab | Recruiting | I | Resectable | |
| NCT04617067 | Paricalcitol + GCB/nab | Recruiting | II | Advanced | |
| NCT04524702 | Paricalcitol + Hydroxychloroquine + nab | Recruiting | II | Advanced metastatic | |
| NCT02930902 | Paricalcitol + pembrolizumab + GCB/nab | Active | I | Resectable | |
| NCT03138720 | Paricalcitol + nab + GCB + Cisplastin | Active | II | Untreated, resectable, borderline and locally advanced | |
| NCT03415854 | Paricalcitol + Cisplatin + GCB/nab (single arm) | Active | II | Metastatic | |
| Xydroxychloro-quine | CAFs | ||||
| NCT01494155 | Hydroxychloroquine + Capecitabine + Radiation (single arm) | Active | II | Resectable | |
| NCT04132505 | Hydroxychloroquine + binimetinib | Recruiting | I | KRAS mutated metastatic | |
| NCT03825289 | Hydroxychloroquine + trametinib | Recruiting | I | Stage II,III,IV unresectable and metastatic | |
| NCT04386057 | Hydroxychloroquine + LY3214996 | Recruiting | II | PDAC | |
| NCT01506973 | Hydroxychloroquine + GCB/nab | Active | I/II | Advanced and metastatic | |
| ATRA | CAFs | ||||
| NCT04241276 | ATRA + GCB/nab | NA | II | PDAC | |
| Losartan | Collagen | ||||
| NCT03563248 | Losartan+nivolumab + SBRT | recruiting | II | Localized | |
| NCT01821729 | Losartan + radiation | active | II | PDAC | |
| NCT04106856 | Losartan + Rx after chemiotherapy | recruiting | I | Borderline resectable or locally advanced | |
| Volociximab | α5β1 integrin | ||||
| NCT00401570 | Volociximab + GCB | Completed | II | Metastatic | |
| IPI-926 | SHH | ||||
| NCT01130142 | IPI-926 + GCB | Completed | I/II | Metastatic | |
| NCT01383538 | IPI-926 + FOLFIRINOX | Completed | I | Advanced |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options. Cancers 2021, 13, 4442. https://doi.org/10.3390/cancers13174442
Ferrara B, Pignatelli C, Cossutta M, Citro A, Courty J, Piemonti L. The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options. Cancers. 2021; 13(17):4442. https://doi.org/10.3390/cancers13174442
Chicago/Turabian StyleFerrara, Benedetta, Cataldo Pignatelli, Mélissande Cossutta, Antonio Citro, José Courty, and Lorenzo Piemonti. 2021. "The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options" Cancers 13, no. 17: 4442. https://doi.org/10.3390/cancers13174442