
Germline CDH1 G212E Missense Variant: Combining Clinical, In Vitro and In Vivo Strategies to Unravel Disease Burden

 , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. DNA Extraction and Variant Detection
2.3. In Silico Predictions
2.4. Plasmids
2.5. Cell Culture and Transfection
2.6. Western Blotting
2.7. Immunofluorescence Staining and Expression Profiling
2.8. Matrigel Invasion Assay
2.9. Slow Aggregation Assay
2.10. Cell Network Analysis
2.11. Generation of Drosophila Stocks
2.12. Drosophila Genetics
2.13. Ovary Immunofluorescence and Imaging
2.14. Statistical Analysis
3. Results
3.1. The G212E E-Cadherin Variant Segregates with Diffuse Gastric Cancer within a Large Family Pedigree
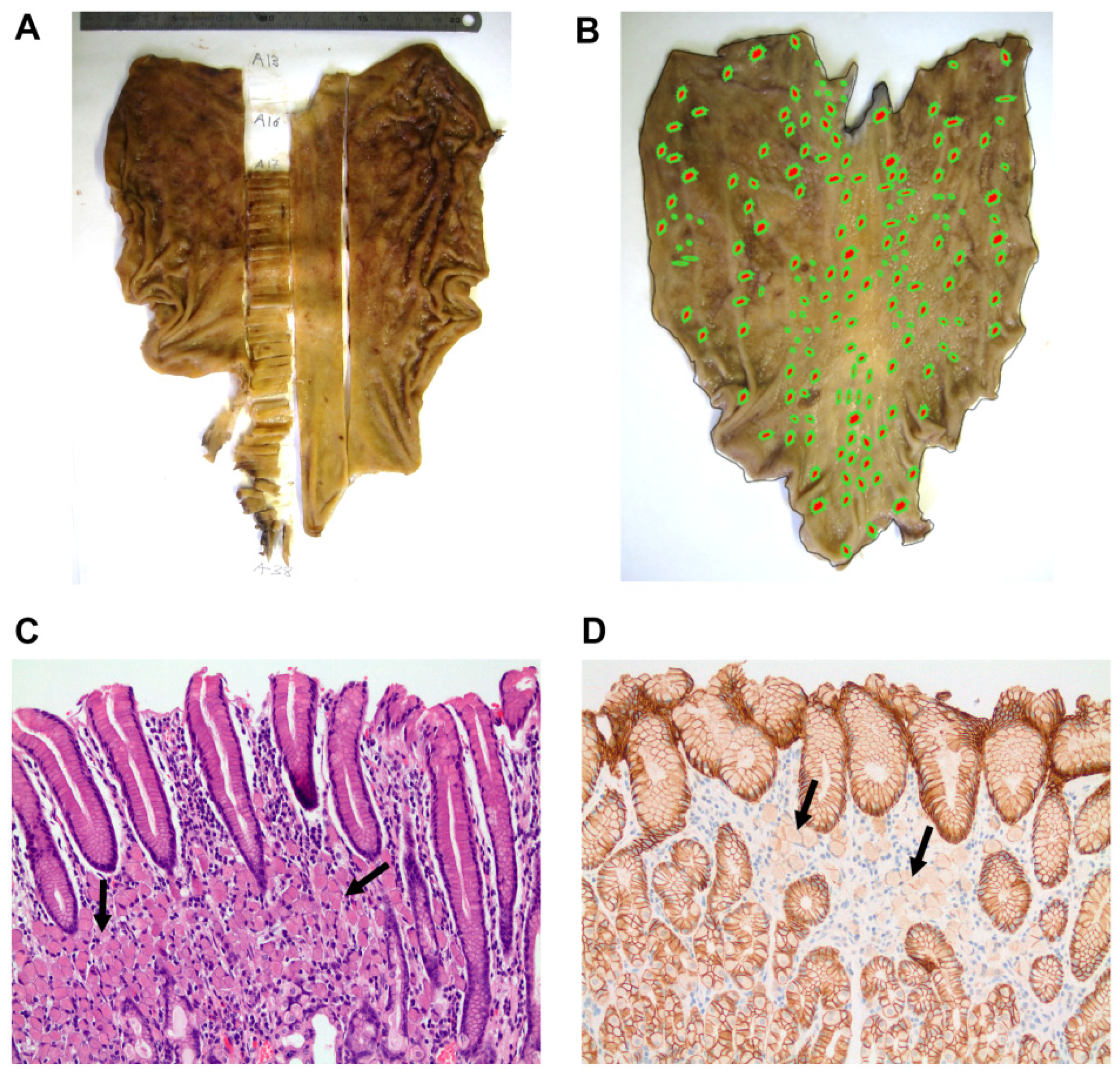
3.2. Histopathogical Findings Are Compatible with HDGC Clinical Presentation
3.3. In Silico Analysis Predicts Destabilization of E-Cadherin Structure by the G212E Variant
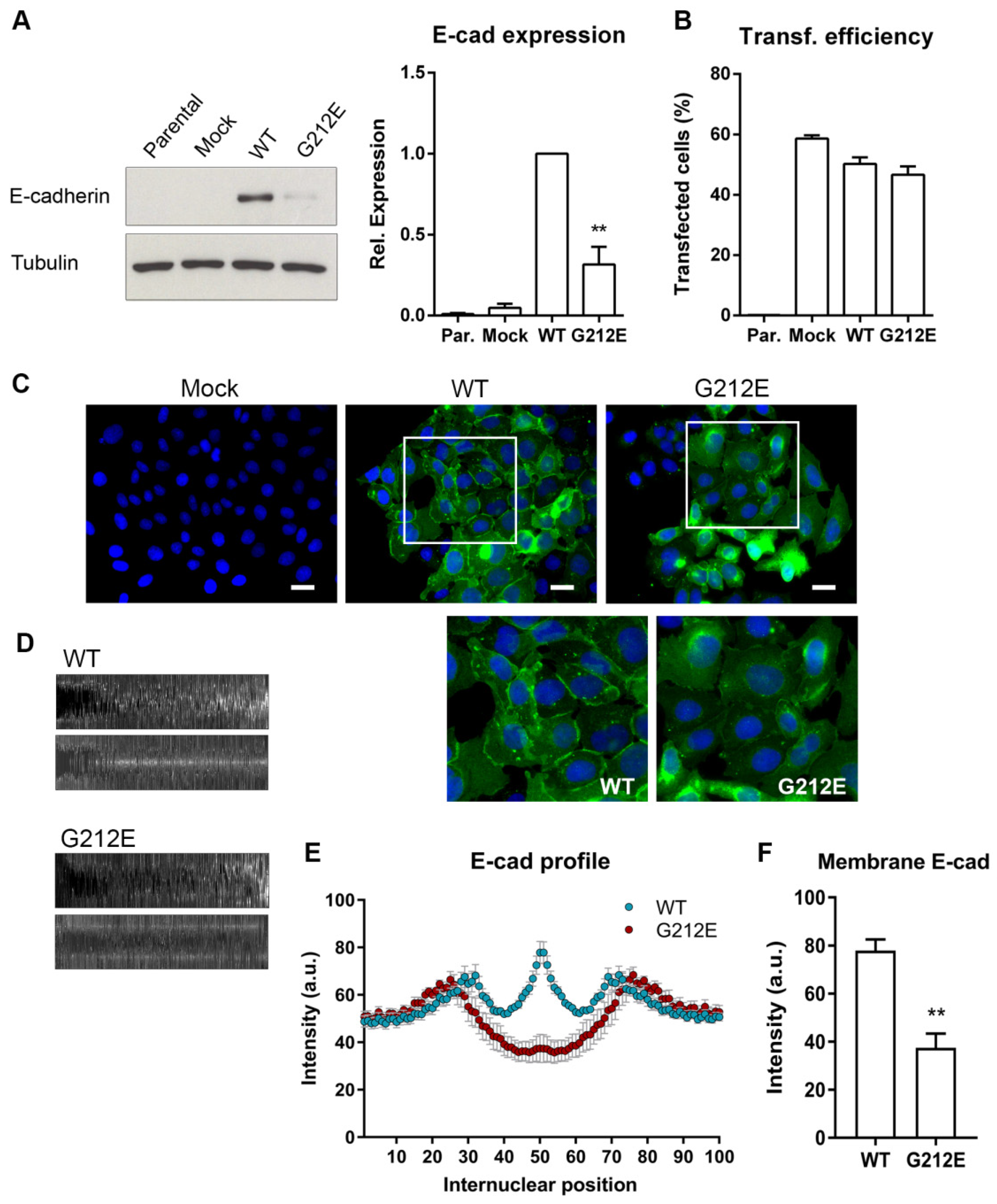
3.4. The CDH1 G212E Variant Yields Abnormal E-Cadherin Levels and Distribution Profiles
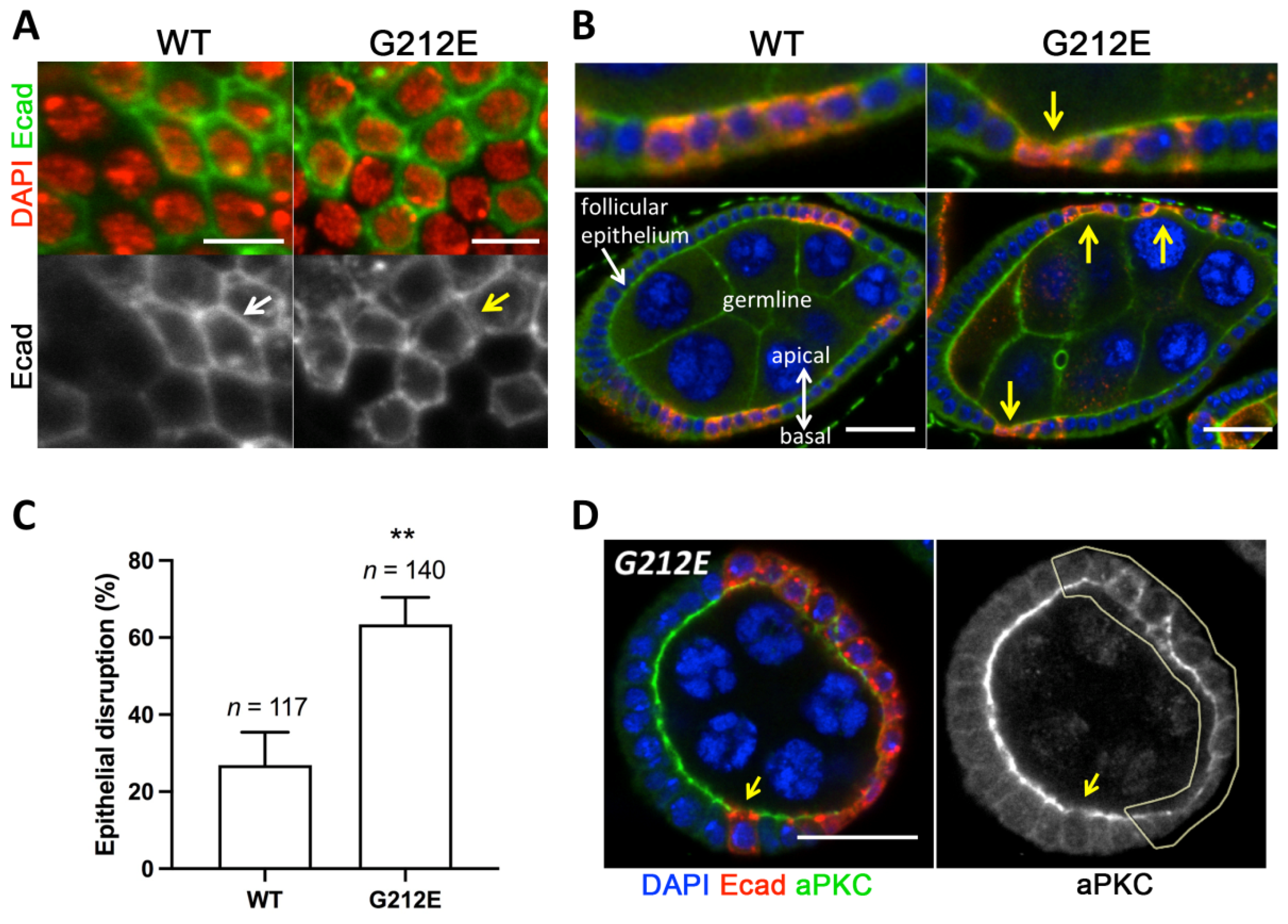
3.5. The G212E Variant Compromises Protein Function and Cell-Cell Adhesion
3.6. Expression of Human G212E Variant Causes Epithelial Disruption in Drosophila
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Favalli, V.; Tini, G.; Bonetti, E.; Vozza, G.; Guida, A.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Machine learning-based reclassification of germline variants of unknown significance: The RENOVO algorithm. Am. J. Hum. Genet. 2021, 108, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Horak, P.; Frohling, S.; Glimm, H. Integrating next-generation sequencing into clinical oncology: Strategies, promises and pitfalls. ESMO Open 2016, 1, e000094. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2012, 13, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Young, D.L.; Fields, S. The role of functional data in interpreting the effects of genetic variation. Mol. Biol. Cell 2015, 26, 3904–3908. [Google Scholar] [CrossRef]
- Corso, G.; Montagna, G.; Figueiredo, J.; La Vecchia, C.; Fumagalli Romario, U.; Fernandes, M.S.; Seixas, S.; Roviello, F.; Trovato, C.; Guerini-Rocco, E.; et al. Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review. Cancers 2020, 12, 1598. [Google Scholar] [CrossRef]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. Familial gastric cancer: Genetic susceptibility, pathology, and implications for management. Lancet. Oncol. 2015, 16, e60–e70. [Google Scholar] [CrossRef]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet. Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef]
- Figueiredo, J.; Melo, S.; Carneiro, P.; Moreira, A.M.; Fernandes, M.S.; Ribeiro, A.S.; Guilford, P.; Paredes, J.; Seruca, R. Clinical spectrum and pleiotropic nature of CDH1 germline mutations. J. Med. Genet. 2019, 56, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, J.P.; Gullo, I.; Wen, X.; Devezas, V.; Baptista, M.; Oliveira, C.; Carneiro, F. Pathological features of total gastrectomy specimens from asymptomatic hereditary diffuse gastric cancer patients and implications for clinical management. Histopathology 2018, 73, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Gullo, I.; Devezas, V.; Baptista, M.; Garrido, L.; Castedo, S.; Morais, R.; Wen, X.; Rios, E.; Pinheiro, J.; Pinto-Ribeiro, I.; et al. Phenotypic heterogeneity of hereditary diffuse gastric cancer: Report of a family with early-onset disease. Gastrointest. Endosc. 2018, 87, 1566–1575. [Google Scholar] [CrossRef]
- Barber, M.E.; Save, V.; Carneiro, F.; Dwerryhouse, S.; Lao-Sirieix, P.; Hardwick, R.H.; Caldas, C.; Fitzgerald, R.C. Histopathological and molecular analysis of gastrectomy specimens from hereditary diffuse gastric cancer patients has implications for endoscopic surveillance of individuals at risk. J. Pathol. 2008, 216, 286–294. [Google Scholar] [CrossRef]
- Suriano, G.; Oliveira, C.; Ferreira, P.; Machado, J.C.; Bordin, M.C.; De Wever, O.; Bruyneel, E.A.; Moguilevsky, N.; Grehan, N.; Porter, T.R.; et al. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum. Mol. Genet. 2003, 12, 575–582. [Google Scholar] [CrossRef]
- Suriano, G.; Seixas, S.; Rocha, J.; Seruca, R. A model to infer the pathogenic significance of CDH1 germline missense variants. J. Mol. Med. 2006, 84, 1023–1031. [Google Scholar] [CrossRef]
- Pereira, P.S.; Teixeira, A.; Pinho, S.; Ferreira, P.; Fernandes, J.; Oliveira, C.; Seruca, R.; Suriano, G.; Casares, F. E-cadherin missense mutations, associated with hereditary diffuse gastric cancer (HDGC) syndrome, display distinct invasive behaviors and genetic interactions with the Wnt and Notch pathways in Drosophila epithelia. Hum. Mol. Genet. 2006, 15, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Simoes-Correia, J.; Figueiredo, J.; Lopes, R.; Stricher, F.; Oliveira, C.; Serrano, L.; Seruca, R. E-cadherin destabilization accounts for the pathogenicity of missense mutations in hereditary diffuse gastric cancer. PLoS ONE 2012, 7, e33783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, J.; Soderberg, O.; Simoes-Correia, J.; Grannas, K.; Suriano, G.; Seruca, R. The importance of E-cadherin binding partners to evaluate the pathogenicity of E-cadherin missense mutations associated to HDGC. Eur. J. Hum. Genet. 2013, 21, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Sanches, J.M.; Figueiredo, J.; Fonseca, M.; Duraes, C.; Melo, S.; Esmenio, S.; Seruca, R. Quantification of mutant E-cadherin using bioimaging analysis of in situ fluorescence microscopy. A new approach to CDH1 missense variants. Eur. J. Hum. Genet. 2015, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestre, T.; Figueiredo, J.; Ribeiro, A.S.; Paredes, J.; Seruca, R.; Sanches, J.M. Quantification of topological features in cell meshes to explore E-cadherin dysfunction. Sci. Rep. 2016, 6, 25101. [Google Scholar] [CrossRef]
- Lee, K.; Krempely, K.; Roberts, M.E.; Anderson, M.J.; Carneiro, F.; Chao, E.; Dixon, K.; Figueiredo, J.; Ghosh, R.; Huntsman, D.; et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 2018, 39, 1553–1568. [Google Scholar] [CrossRef]
- Melo, S.; Figueiredo, J.; Fernandes, M.S.; Goncalves, M.; Morais-de-Sa, E.; Sanches, J.M.; Seruca, R. Predicting the Functional Impact of CDH1 Missense Mutations in Hereditary Diffuse Gastric Cancer. Int. J. Mol. Sci. 2017, 18, 2687. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A graphical interface for the FoldX forcefield. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wilkinson, M.F. Site-directed mutagenesis of large (13-kb) plasmids in a single-PCR procedure. Biotechniques 2000, 29, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, J.; Rodrigues, I.; Ribeiro, J.; Fernandes, M.S.; Melo, S.; Sousa, B.; Paredes, J.; Seruca, R.; Sanches, J.M. Geometric compensation applied to image analysis of cell populations with morphological variability: A new role for a classical concept. Sci. Rep. 2018, 8, 10266. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, A.; Boots, B.; Sugihara, K.; Chiu, S.N.; Kendall, D.G. Definitions and Basic Properties of Voronoi Diagrams, in Spatial Tessellations: Concepts and Applications of Voronoi Diagrams; John Wiley & Sons: Hoboken, NJ, USA, 2000; pp. 43–112. [Google Scholar]
- Bischof, J.; Basler, K. Recombinases and their use in gene activation, gene inactivation, and transgenesis. Methods Mol. Biol. 2008, 420, 175–195. [Google Scholar]
- Corso, G.; Figueiredo, J.; Biffi, R.; Trentin, C.; Bonanni, B.; Feroce, I.; Serrano, D.; Cassano, E.; Annibale, B.; Melo, S.; et al. E-cadherin germline mutation carriers: Clinical management and genetic implications. Cancer Metastasis Rev. 2014, 33, 1081–1094. [Google Scholar] [CrossRef]
- Figueiredo, J.; Seruca, J. Germline missense mutants in hereditary diffuse gastric cancer. Spotlight Fam. Hered. Gastric Cancer 2013, 7, 77–86. [Google Scholar]
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. E-cadherin alterations in hereditary disorders with emphasis on hereditary diffuse gastric cancer. Prog. Mol. Biol. Transl. Sci. 2013, 116, 337–359. [Google Scholar]
- Simoes-Correia, J.; Figueiredo, J.; Oliveira, C.; van Hengel, J.; Seruca, R.; van Roy, F.; Suriano, G. Endoplasmic reticulum quality control: A new mechanism of E-cadherin regulation and its implication in cancer. Hum. Mol. Genet. 2008, 17, 3566–3576. [Google Scholar] [CrossRef] [Green Version]
- Corso, G.; Corso, F.; Bellerba, F.; Carneiro, P.; Seixas, S.; Cioffi, A.; La Vecchia, C.; Magnoni, F.; Bonanni, B.; Veronesi, P.; et al. Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review. Cancers 2021, 13, 1269. [Google Scholar] [CrossRef]
- Mateus, A.R.; Simoes-Correia, J.; Figueiredo, J.; Heindl, S.; Alves, C.C.; Suriano, G.; Luber, B.; Seruca, R. E-cadherin mutations and cell motility: A genotype-phenotype correlation. Exp. Cell Res. 2009, 315, 1393–1402. [Google Scholar] [CrossRef]
- Lo, W.; Zhu, B.; Sabesan, A.; Wu, H.H.; Powers, A.; Sorber, R.A.; Ravichandran, S.; Chen, I.; McDuffie, L.A.; Quadri, H.S.; et al. Associations of CDH1 germline variant location and cancer phenotype in families with hereditary diffuse gastric cancer (HDGC). J. Med. Genet. 2019, 56, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Masciari, S.; Larsson, N.; Senz, J.; Boyd, N.; Kaurah, P.; Kandel, M.J.; Harris, L.N.; Pinheiro, H.C.; Troussard, A.; Miron, P.; et al. Germline E-cadherin mutations in familial lobular breast cancer. J. Med. Genet. 2007, 44, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corso, G.; Figueiredo, J.; La Vecchia, C.; Veronesi, P.; Pravettoni, G.; Macis, D.; Karam, R.; Lo Gullo, R.; Provenzano, E.; Toesca, A.; et al. Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. J. Med. Genet. 2018, 55, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Vogelaar, I.P.; Figueiredo, J.; van Rooij, I.A.; Simoes-Correia, J.; van der Post, R.S.; Melo, S.; Seruca, R.; Carels, C.E.; Ligtenberg, M.J.; Hoogerbrugge, N. Identification of germline mutations in the cancer predisposing gene CDH1 in patients with orofacial clefts. Hum. Mol. Genet. 2013, 22, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Ghoumid, J.; Stichelbout, M.; Jourdain, A.S.; Frenois, F.; Lejeune-Dumoulin, S.; Alex-Cordier, M.P.; Lebrun, M.; Guerreschi, P.; Duquennoy-Martinot, V.; Vinchon, M.; et al. Blepharocheilodontic syndrome is a CDH1 pathway-related disorder due to mutations in CDH1 and CTNND1. Genet. Med. 2017, 19, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.C.; Wheeler, J.M.; Kim, J.C.; Ilyas, M.; Beck, N.E.; Kim, B.S.; Park, K.C.; Bodmer, W.F. The E-cadherin gene (CDH1) variants T340A and L599V in gastric and colorectal cancer patients in Korea. Gut 2000, 47, 262–267. [Google Scholar] [CrossRef]
- Richards, F.M.; McKee, S.A.; Rajpar, M.H.; Cole, T.R.; Evans, D.G.; Jankowski, J.A.; McKeown, C.; Sanders, D.S.; Maher, E.R. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum. Mol. Genet. 1999, 8, 607–610. [Google Scholar] [CrossRef]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef]
- Kleinman, H.K.; McGarvey, M.L.; Liotta, L.A.; Robey, P.G.; Tryggvason, K.; Martin, G.R. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry 1982, 21, 6188–6193. [Google Scholar] [CrossRef]
- Pacquelet, A.; Rorth, P. Regulatory mechanisms required for DE-cadherin function in cell migration and other types of adhesion. J. Cell Biol. 2005, 170, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Rosales-Nieves, A.E.; Gonzalez-Reyes, A. Genetics and mechanisms of ovarian cancer: Parallels between Drosophila and humans. Semin. Cell Dev. Biol. 2014, 28, 104–109. [Google Scholar] [CrossRef]
- Fomicheva, M.; Tross, E.M.; Macara, I.G. Polarity proteins in oncogenesis. Curr. Opin. Cell Biol. 2020, 62, 26–30. [Google Scholar] [CrossRef]
- Humar, B.; Guilford, P. Hereditary diffuse gastric cancer: A manifestation of lost cell polarity. Cancer Sci. 2009, 100, 1151–1157. [Google Scholar] [CrossRef]
- Gloerich, M.; Bianchini, J.M.; Siemers, K.A.; Cohen, D.J.; Nelson, W.J. Cell division orientation is coupled to cell-cell adhesion by the E-cadherin/LGN complex. Nat. Commun. 2017, 8, 13996. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Age at Diagnosis | Surveillance Period | Screening Biopsies up to Gastrectomy (nr) | Paraffin-Embedded Tissue Blocks (nr) | Intramucosal Foci(nr and Size) | Foci Anatomical Location | Visible Tumour (nr) TNM Localization Size | Histological Features |
|---|---|---|---|---|---|---|---|---|
| IV-14 | 59 | 13 y | 23 | 315 | 16 <2 mm | All of the stomach, more frequent in body and fundus | No T1aN0 | Superficial half mucosa |
| IV-37 | 43 | 6 m | 2 | 208 | 9 <5 mm | All of the stomach | No T1aN0 | Superficial half mucosa |
| IV-49 | 46 | no | 1 | 209 | 32 <2 mm | All of the stomach, more frequent in lesser curvature | Yes (1) T2N0 Cardia ulcerated 1.8 cm | Superficial half mucosa |
| IV-51 | 28 | 1 y | 3 | 214 | 169 <2 mm | All of the stomach | No T1aN0 | Superficial half mucosa |
| IV-52 | 27 | 7 m | 2 | 202 | 54 < 8 mm | All of the stomach, more frequent in lesser curvature and posterior wall | No T1aN0 | Superficial half mucosa |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueiredo, J.; Mercadillo, F.; Melo, S.; Barroso, A.; Gonçalves, M.; Díaz-Tasende, J.; Carneiro, P.; Robles, L.; Colina, F.; Ibarrola, C.; et al. Germline CDH1 G212E Missense Variant: Combining Clinical, In Vitro and In Vivo Strategies to Unravel Disease Burden. Cancers 2021, 13, 4359. https://doi.org/10.3390/cancers13174359
Figueiredo J, Mercadillo F, Melo S, Barroso A, Gonçalves M, Díaz-Tasende J, Carneiro P, Robles L, Colina F, Ibarrola C, et al. Germline CDH1 G212E Missense Variant: Combining Clinical, In Vitro and In Vivo Strategies to Unravel Disease Burden. Cancers. 2021; 13(17):4359. https://doi.org/10.3390/cancers13174359
Chicago/Turabian StyleFigueiredo, Joana, Fátima Mercadillo, Soraia Melo, Alicia Barroso, Margarida Gonçalves, José Díaz-Tasende, Patrícia Carneiro, Luis Robles, Francisco Colina, Carolina Ibarrola, and et al. 2021. "Germline CDH1 G212E Missense Variant: Combining Clinical, In Vitro and In Vivo Strategies to Unravel Disease Burden" Cancers 13, no. 17: 4359. https://doi.org/10.3390/cancers13174359