
Rare Germline Variants in Chordoma-Related Genes and Chordoma Susceptibility

 , , , add
Show full author list
, , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
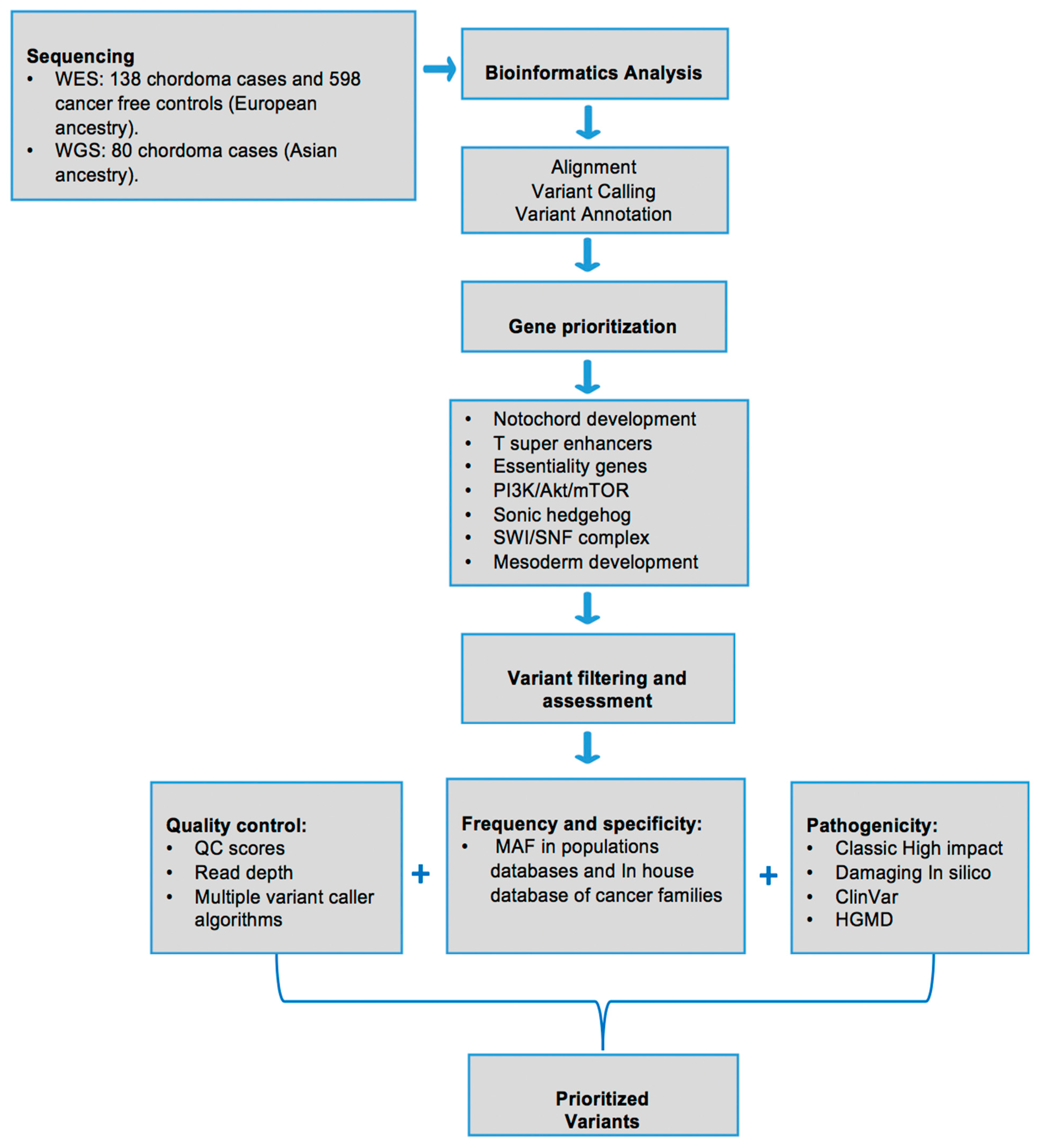
2. Materials and Methods
2.1. Study Populations
2.2. Library Construction, Sequencing and Bioinformatics Analysis
2.3. Rare Variant Burden Test
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wasserman, J.K.; Gravel, D.; Purgina, B. Chordoma of the Head and Neck: A Review. Head Neck Pathol. 2018, 12, 261–268. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Sommer, J. Building a global consensus approach to chordoma: A position paper from the medical and patient community. Lancet Oncol. 2015, 16, e71–e83. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Gronchi, A.; Fossati, P.; Akiyama, T.; Alapetite, C.; Baumann, M.; Blay, J.Y.; Bolle, S.; Boriani, S.; Bruzzi, P.; et al. Best practices for the management of local-regional recurrent chordoma: A position paper by the Chordoma Global Consensus Group. Ann. Oncol. 2017, 28, 1230–1242. [Google Scholar] [CrossRef]
- Jones, P.S.; Aghi, M.K.; Muzikansky, A.; Shih, H.A.; Barker, F.G.; Curry, W.T. Outcomes and patterns of care in adult skull base chordomas from the Surveillance, Epidemiology, and End Results (SEER) database. J. Clin. Neurosci. 2014, 21, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Kispert, A.; Koschorz, B.; Herrmann, B.G. The T protein encoded by Brachyury is a tissue-specific transcription factor. EMBO J. 1995, 14, 4763–4772. [Google Scholar] [CrossRef]
- Yang, X.R.; Ng, D.; Alcorta, D.A.; Liebsch, N.J.; Sheridan, E.; Li, S.; Goldstein, A.M.; Parry, D.M.; Kelley, M. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat. Genet. 2009, 41, 1176–1178. [Google Scholar] [CrossRef]
- Pillay, N.; Plagnol, V.; Tarpey, P.S.; Lobo, S.B.; Presneau, N.; Szuhai, K.; Halai, D.; Berisha, F.; Cannon, S.R.; Mead, S.; et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat. Genet. 2012, 44, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.; Shi, J.; Ballew, B.; Hyland, P.L.; Li, W.-Q.; Rotunno, M.; Alcorta, D.A.; Liebsch, N.J.; Mitchell, J.; Bass, S.; et al. Characterization of T gene sequence variants and germline duplications in familial and sporadic chordoma. Qual. Life Res. 2014, 133, 1289–1297. [Google Scholar] [CrossRef]
- Vujovic, S.; Henderson, S.; Presneau, N.; Odell, E.; Jacques, T.S.; Tirabosco, R.; Boshoff, C.; Flanagan, A.M. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J. Pathol. 2006, 209, 157–165. [Google Scholar] [CrossRef]
- Palena, C.; Polev, D.E.; Tsang, K.Y.; Fernando, R.I.; Litzinger, M.; Krukovskaya, L.L.; Baranova, A.V.; Kozlov, A.P.; Schlom, J. The Human T-Box Mesodermal Transcription Factor Brachyury Is a Candidate Target for T-Cell–Mediated Cancer Immunotherapy. Clin. Cancer Res. 2007, 13, 2471–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarpey, P.S.; Behjati, S.; Young, M.D.; Martincorena, I.; Alexandrov, L.B.; Farndon, S.J.; Guzzo, C.; Hardy, C.; Latimer, C.; Butler, A.P.; et al. The driver landscape of sporadic chordoma. Nat. Commun. 2017, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Presneau, N.; Shalaby, A.; Ye, H.; Pillay, N.; Halai, D.; Idowu, B.; Tirabosco, R.; Whitwell, D.; Jacques, T.S.; Kindblom, L.G.; et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: A genetic and functional-based study. J. Pathol. 2011, 223, 327–335. [Google Scholar] [CrossRef]
- Hsu, W.; Mohyeldin, A.; Shah, S.R.; Ap Rhys, C.M.; Johnson, L.F.; Sedora-Roman, N.I.; Kosztowski, T.A.; Awad, O.A.; McCarthy, E.F.; Loeb, D.M.; et al. Generation of chordoma cell line JHC7 and the identification of Brachyury as a novel molecular target. J. Neurosurg. 2011, 115, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.R.; David, J.; Tippens, N.D.; Mohyeldin, A.; Martinez-Gutierrez, J.C.; Ganaha, S.; Schiapparelli, P.; Hamilton, D.H.; Palena, C.; Levchenko, A.; et al. Brachyury-YAP Regulatory Axis Drives Stemness and Growth in Cancer. Cell Rep. 2017, 21, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohagan, J.K.; Prorok, P.C.; Hayes, R.B.; Kramer, B.S. The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial of the National Cancer Institute: History, organization, and status. Control. Clin. Trials. 2000, 21, 251S–272S. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Qual. Life Res. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol. 2020, 6, 724–734. [Google Scholar] [CrossRef]
- Lee, S.; Emond, M.J.; Bamshad, M.J.; Barnes, K.C.; Rieder, M.J.; Nickerson, D.A.; Christiani, D.C.; Wurfel, M.M.; Lin, X. Optimal Unified Approach for Rare-Variant Association Testing with Application to Small-Sample Case-Control Whole-Exome Sequencing Studies. Am. J. Hum. Genet. 2012, 91, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Ramroop, J.R.; Gerber, M.M.; Toland, A.E. Germline Variants Impact Somatic Events during Tumorigenesis. Trends Genet. 2019, 35, 515–526. [Google Scholar] [CrossRef]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.-Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat. Med. 2019, 25, 292–300. [Google Scholar] [CrossRef]
- Aszódi, A.; Chan, D.; Hunziker, E.; Bateman, J.F.; Fässler, R. Collagen II Is Essential for the Removal of the Notochord and the Formation of Intervertebral Discs. J. Cell Biol. 1998, 143, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-S.; Cohn, M.J.; Harfe, B.D. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: Implications for disk degeneration and chordoma formation. Dev. Dyn. 2008, 237, 3953–3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Scott, M.P.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA. 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [Green Version]
- Hollmann, T.J.; Hornick, J.L. INI1-deficient tumors: Diagnostic features and molecular genetics. Am. J. Surg. Pathol. 2011, 35, e47–e63. [Google Scholar] [CrossRef]
- Yadav, R.; Sharma, M.C.; Malgulwar, P.B.; Pathak, P.; Sigamani, E.; Suri, V.; Sarkar, C.; Kumar, A.; Singh, M.; Sharma, B.S.; et al. Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro-Oncology 2013, 16, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMaster, M.L.; Goldstein, A.M.; Parry, D.M. Clinical features distinguish childhood chordoma associated with tuberous sclerosis complex (TSC) from chordoma in the general paediatric population. J. Med Genet. 2011, 48, 444–449. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| ID | Gender | Age | Morphology | Site | Gene | Chr | Location | SNP IDS | REF | VAR | Variant Type | Protein Change | Variant Impact | Pathogenecity Prediction a | HGMD/ ClinVar | MAF in Control Datasets | Pathway/Process | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| METASVM | METALR | CADD | gnomAD Exomes NFE b | gnomAD Genomes NFE c | ||||||||||||||||
| 1004 | Female | NA | Conventional | 1 | ATP8B2 | 1 | 154305114 | G | T | missense | Arg210Leu | moderate | D | D | 34 | 1.06 × 10−5 | Mesoderm commitment | |||
| 1101 | Male | 37.2 | Conventional | 2 | LYST | 1 | 235922291 | G | A | stop_gained | Arg2288 * | high | Driver | |||||||
| 1164 | Male | 29.7 | Conventional | 1 | TCF7L1 | 2 | 85529694 | rs147750102 | G | A | missense | Gly205Ser | moderate | D | D | 22.4 | 9.67 × 10−5 | 6.49 × 10−5 | Mesoderm commitment | |
| 1040 | Female | 32.7 | Conventional | 1 | EPB41L5 | 2 | 120776677 | rs200315720 | G | A | missense | Arg6His | moderate | D | D | 29.9 | 1.98 × 10−4 | 1.30 × 10−4 | Mesoderm commitment | |
| 1020 | Female | 39.0 | Conventional | 1 | EPB41L5 | 2 | 120833086 | rs766560121 | C | A | missense | Leu148Ile | moderate | D | D | 28.9 | 0 | Mesoderm commitment | ||
| 3001 | NA | NA | NA | NA | LRP2 | 2 | 170038097 | rs137983840 | C | T | missense | Ala3344Thr | moderate | D | D | 26.3 | 6.16 × 10−5 | 6.48 × 10−5 | Sonic Hedgehog | |
| 1035 | Female | 26.1 | Chondroid | 1 | PDK1 | 2 | 173460594 | T | frameshift & stop_gained | Asn424fs | high | PI3K/AKT/mTOR | ||||||||
| 1111 | Male | 51.3 | Chondroid | 1 | NFE2L2 | 2 | 178095743 | C | A | missense | Asp530Tyr | moderate | D | D | 22.5 | Mesoderm commitment | ||||
| 1014 | Female | 44.3 | Conventional | 1 | BMPR2 | 2 | 203420750 | rs146310981 | G | A | missense | Val788Ile | moderate | D | D | 22.7 | 3.52 × 10−5 | Mesoderm commitment | ||
| 1139 | Female | 39.5 | Conventional | 1 | RARB | 3 | 25611341 | A | G | missense | Thr188Ala | moderate | D | D | 22.9 | 8.79 × 10−6 | Mesoderm commitment | |||
| 1053 | Female | 54.6 | Conventional | 1 | HHIP | 4 | 145581068 | ACAC | frameshift | Phe304fs | high | 0 | Sonic Hedgehog | |||||||
| 1127 | Male | 50.7 | Conventional | 3 | SRF | 6 | 43146888 | rs765592889 | T | C | missense | Val496Ala | moderate | D | D | 22.4 | 1.76 × 10−5 | Mesoderm commitment | ||
| 1048/ 5320 | NA | NA | NA | NA | TBXT | 6 | 166571981 | rs368179445 | C | T | missense | Arg377Gln | moderate | D | D | 25.5 | 0 | T super-enhancer | ||
| 1080 | Female | 47.4 | Conventional | 1 | ATP6V1B2 | 8 | 20068082 | C | T | missense & splice region | Arg130Trp | moderate | D | D | 35 | 0 | T super-enhancer | |||
| 1161 | Male | 78.3 | Conventional | 1 | EXT1 | 8 | 118832021 | rs145720047 | G | C | missense | Pro477Arg | moderate | D | D | 22.4 | 1.24 × 10−4 | 6.48 × 10−5 | Mesoderm commitment | |
| 1001 | Female | 47.0 | Conventional | 1 | DEPTOR | 8 | 120977595 | A | frameshift | Glu185fs | high | PI3K/AKT/mTOR | ||||||||
| 1034 | Female | 51.0 | Conventional | 2 | SMARCA2 | 9 | 2039568 | rs774084308 | C | T | missense | Pro153Leu | moderate | D | D | 22.2 | 0 | SWI/SNF complex | ||
| 1080 | Female | 47.4 | Conventional | 1 | JAK2 | 9 | 5078325 | A | G | missense | His671Arg | moderate | D | D | 21.7 | Mesoderm commitment | ||||
| 1154 | Female | 44.1 | Conventional | 2 | PTCH1 | 9 | 98209505 | rs556901417 | G | A | missense | Arg1345Cys | moderate | D | D | 25.2 | 1.09 × 10−4 | 3.89 × 10−4 | Sonic Hedgehog | |
| 1042 | Female | 39.5 | Conventional | 1 | SUFU | 10 | 104309821 | rs34406289 | G | A | missense | Ala138Thr | moderate | D | D | 33 | 2.64 × 10−5 | 0 | Sonic Hedgehog | |
| 1141 | Female | 57.6 | Conventional | 3 | SUFU | 10 | 104375030 | rs79299301 | G | A | missense | Arg343His | moderate | D | D | 20.8 | 1.58 × 10−4 | 6.49 × 10−5 | Sonic Hedgehog | |
| 1006 | Male | 46.2 | Conventional | 1 | PAX6 | 11 | 31823215 | A | T | missense | Val98Glu | moderate | D | D | 27.5 | Mesoderm commitment | ||||
| 1162 | Female | 66.4 | Conventional | 3 | EXT2 | 11 | 44254000 | rs138495222 | C | T | missense | Thr620Met | moderate | D | D | 34 | DM | 9.24 × 10−4 | 8.43 × 10−4 | Mesoderm commitment |
| 1090 | Female | 28.3 | Conventional | 1 | GDF3 | 12 | 7842985 | rs146973734 | C | T | missense | Arg195Gln | moderate | T | T | 0.004 | P/DM | 1.85 × 10−4 | 5.84 × 10−4 | Notochord development |
| 1001 | Female | 47.0 | Conventional | 1 | COL2A1 | 12 | 48380213 | rs201823490 | G | A | missense | Pro478Leu | moderate | D | D | 23.7 | 0 | 0 | Notochord development | |
| 1113 | Female | 27.8 | Conventional | 1 | SOX21 | 13 | 95364265 | G | C | missense | Asn13Lys | moderate | D | D | 21.6 | Mesoderm commitment | ||||
| 1050 | Male | 60.3 | Conventional | 2 | WDHD1 | 14 | 55462357 | rs556223202 | G | A | stop_gained | Arg373 * | high | 8.88 × 10−6 | Mesoderm commitment | |||||
| 1127 | Male | 50.7 | Conventional | 3 | AKT1 | 14 | 105246527 | rs397514644 | G | A | missense | Arg25Cys | moderate | D | T | 29.6 | P/DM | PI3K/AKT/mTOR | ||
| 1091 | Female | 69.9 | Conventional | 2 | TSC2 | 16 | 2130319 | C | T | missense | Ala1184Val | moderate | D | D | 24 | 3.56 × 10−5 | PI3K/AKT/mTOR | |||
| 1028 | Female | 39.7 | Conventional | 1 | TSC2 | 16 | 2136297 | rs373635516 | C | T | missense | Pro1589Leu | moderate | D | D | 21.9 | 0 | PI3K/AKT/mTOR | ||
| 1006 | Male | 46.2 | Conventional | 1 | ACACA | 17 | 35603791 | A | T | missense | Val804Glu | moderate | D | D | 23.7 | Mesoderm commitment | ||||
| 1114 | Male | 44.4 | Conventional | 1 | PRKACA | 19 | 14203391 | rs41296324 | A | T | stop_lost | Ter125Lysext * | high | 0 | 2.60 × 10−4 | Mesoderm commitment | ||||
| 1045 | Female | 31.2 | Conventional | 1 | FOXA2 | 20 | 22563180 | C | T | missense | Gly234Ser | moderate | D | D | 22.3 | Mesoderm commitment | ||||
| 1098 | Female | 13.8 | Conventional | 3 | SMARCB1 | 22 | 24129394 | AA | frameshift | Lys13fs | high | SWI/SNF complex | ||||||||
| ID | Gender | Age | Morphology | Gene | Chr | Location | SNP IDS | REF | VAR | Variant Type | Protein Change | Variant Impact | Pathogenecity Prediction a | HGMD/ ClinVar | MAF in Control Datasets | Pathway/Process | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| METASVM | METALR | CADD | gnomAD Exomes EAS b | gnomAD Genomes EAS c | |||||||||||||||
| 1804 | M | 65 | Conventional | ATP8B2 | 1 | 154317567 | A | C | missense | Lys836Gln | moderate | D | D | 26.2 | 1.74 × 10−4 | 0 | Mesoderm commitment | ||
| 1686 | M | 64 | Conventional | LRP2 | 2 | 170033035 | C | T | missense | Gly3486Glu | moderate | D | D | 33 | 0 | 0 | Sonic Hedgehog | ||
| 1372 | NA | NA | NA | LRP2 | 2 | 170062924 | rs577943281 | T | A | missense | Thr2436Ser | moderate | D | D | 22.7 | 2.90 × 10−4 | 6.17 × 10−4 | Sonic Hedgehog | |
| 1964 | M | 24 | Chondroid | LRP2 | 2 | 170055366 | rs755215116 | A | C | missense | Ile2836Met | moderate | D | D | 24.2 | 3.66 × 10−3 | 6.17 × 10−4 | Sonic Hedgehog | |
| 1442 | NA | NA | NA | PDK1 | 2 | 173460575 | rs745678398 | A | G | structural interaction | High | 0 | 0 | PI3K/AKT/mTOR | |||||
| 1886 | F | 62 | Conventional | TCF7L1 | 2 | 85536476 | C | T | missense | Thr553Ile | moderate | D | D | 23 | 5.82 × 10−5 | 0 | Mesoderm commitment | ||
| 1790 | M | 65 | Conventional | TCF7L1 | 2 | 85531113 | rs373770977 | G | A | missense | Val252Ile | moderate | D | D | 23.4 | 1.25 × 10−4 | 0 | Mesoderm commitment | |
| 1779 | M | 47 | Conventional | TCF7L1 | 2 | 85536536 | rs555810312 | C | T | missense | Pro573Leu | moderate | D | D | 23.2 | 4.19 × 10−4 | 0 | Mesoderm commitment | |
| 2011 | F | 7 | Conventional | TBXT | 6 | 166581010 | rs563349798 | C | G | missense | Val24Leu | moderate | D | D | 29.7 | 0 | 0 | T super-enhancer | |
| 1686 | M | 64 | Conventional | EXT1 | 8 | 118819501 | rs753261171 | G | A | missense | Thr613Met | moderate | D | D | 33 | 0 | 0 | Mesoderm commitment | |
| 776 | M | 54 | Conventional | SUFU | 10 | 104309738 | C | T | structural interaction | High | 5.80 × 10−5 | 0 | Sonic Hedgehog | ||||||
| 1921 | M | 57 | Conventional | EXT2 | 11 | 44193231 | rs767085143 | A | G | missense | Asn448Ser | moderate | D | D | 24.3 | 2.90 × 10−4 | 0 | Mesoderm commitment | |
| 1372 | NA | NA | NA | COL2A1 | 12 | 48369322 | rs995646562 | C | T | missense | Ala1222Thr | moderate | D | D | 24.1 | 0 | 0 | Notochord development | |
| 1909 | M | 26 | Conventional | COL2A1 | 12 | 48372528 | G | T | missense | Pro916His | moderate | D | D | 25 | 0 | 0 | Notochord development | ||
| 1936 | F | 71 | Conventional | TSC2 | 16 | 2134572 | rs45517338 | C | G | missense | Pro1450Arg | moderate | D | D | 25.8 | 2.35 × 10−4 | 0 | PI3K/AKT/mTOR | |
| 1417 | M | 59 | Conventional | TSC2 | 16 | 2134572 | rs45517338 | C | G | missense | Pro1450Arg | moderate | D | D | 25.8 | 2.35 × 10−4 | 0 | PI3K/AKT/mTOR | |
| 1979 | M | 59 | Chondroid | TSC2 | 16 | 2121610 | rs45509392 | G | A | missense | Asp647Asn | moderate | D | D | 32 | DM | 5.80 × 10−4 | 0 | PI3K/AKT/mTOR |
| 1570 | M | 24 | Chondroid | TSC2 | 16 | 2121610 | rs45509392 | G | A | missense | Asp647Asn | moderate | D | D | 32 | DM | 5.80 × 10−4 | 0 | PI3K/AKT/mTOR |
| 462 | F | 62 | Conventional | TSC2 | 16 | 2121610 | rs45509392 | G | A | missense | Asp647Asn | moderate | D | D | 32 | DM | 5.80 × 10−4 | 0 | PI3K/AKT/mTOR |
| 1084 | NA | NA | NA | ACACA | 17 | 35549102 | rs772483773 | C | T | missense | Val1449Met | moderate | D | D | 25.2 | 6.96 × 10−4 | 6.17 × 10−4 | Mesoderm commitment | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yepes, S.; Shah, N.N.; Bai, J.; Koka, H.; Li, C.; Gui, S.; McMaster, M.L.; Xiao, Y.; Jones, K.; Wang, M.; et al. Rare Germline Variants in Chordoma-Related Genes and Chordoma Susceptibility. Cancers 2021, 13, 2704. https://doi.org/10.3390/cancers13112704
Yepes S, Shah NN, Bai J, Koka H, Li C, Gui S, McMaster ML, Xiao Y, Jones K, Wang M, et al. Rare Germline Variants in Chordoma-Related Genes and Chordoma Susceptibility. Cancers. 2021; 13(11):2704. https://doi.org/10.3390/cancers13112704
Chicago/Turabian StyleYepes, Sally, Nirav N. Shah, Jiwei Bai, Hela Koka, Chuzhong Li, Songbai Gui, Mary Lou McMaster, Yanzi Xiao, Kristine Jones, Mingyi Wang, and et al. 2021. "Rare Germline Variants in Chordoma-Related Genes and Chordoma Susceptibility" Cancers 13, no. 11: 2704. https://doi.org/10.3390/cancers13112704