Comprehensive Analysis of LincRNAs in Classical and Basal-Like Subtypes of Pancreatic Cancer

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
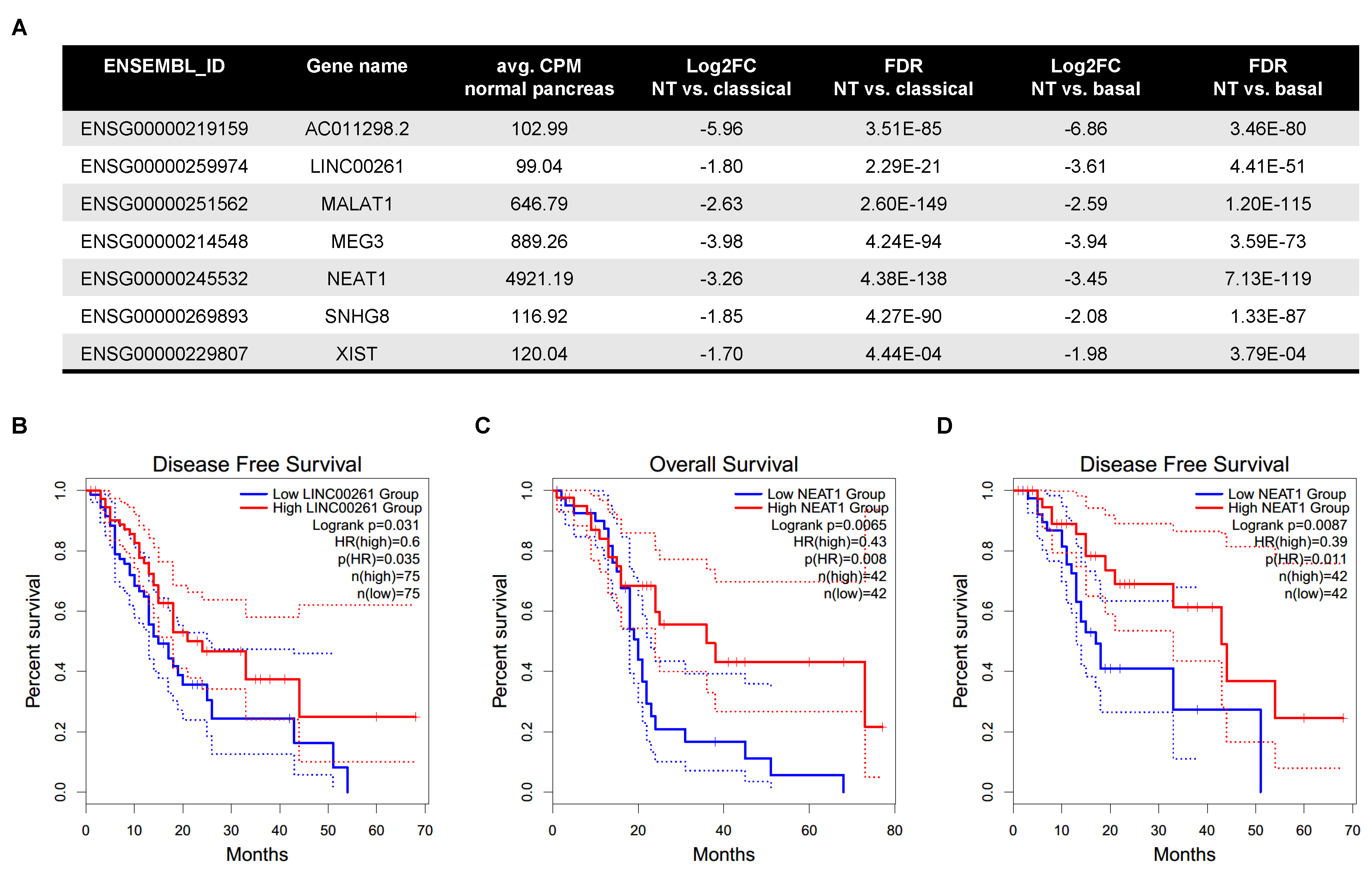
2.1. LincRNA Expression Is Altered in PDAC Compared to Normal Pancreas
2.2. LincRNAs Are Differentially Expressed between PDAC Subtypes
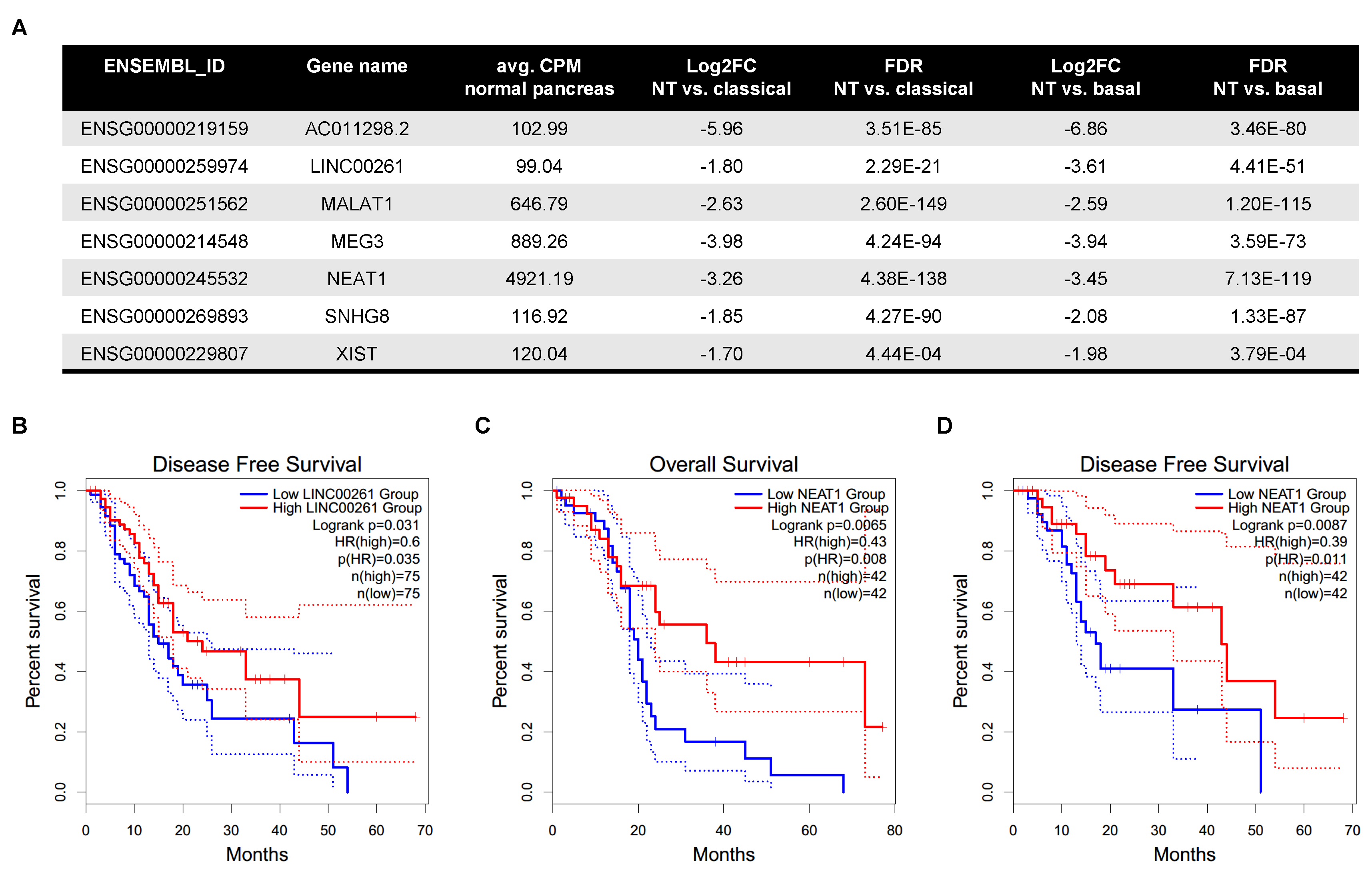
2.3. Putative Transcriptional and Post-Transcriptional Regulators of LincRNA Expression
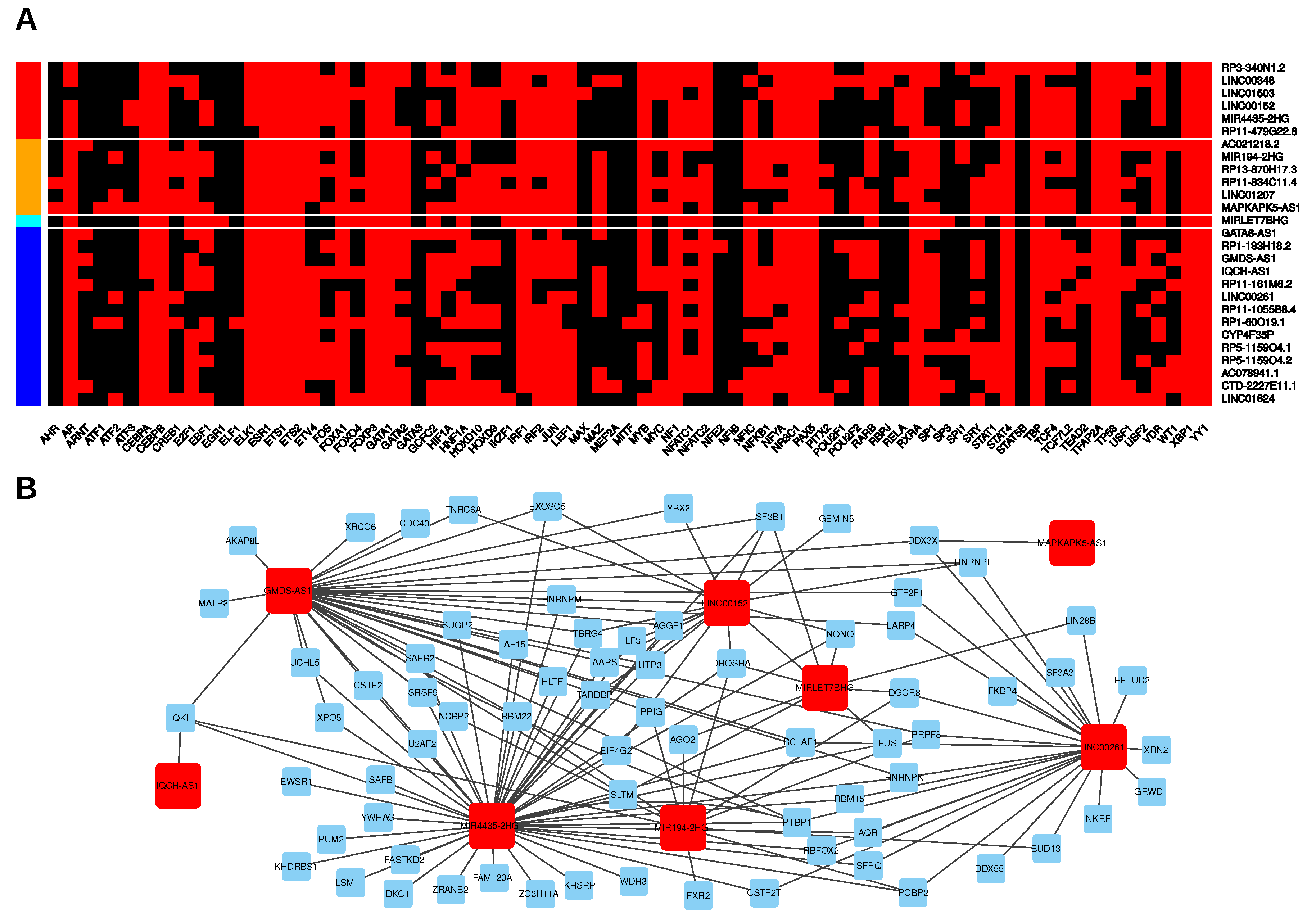
2.4. Putative Signalling Pathways and Direct MRNA Targets Associated with LincRNAs
3. Discussion
4. Materials And Methods
4.1. RNA-Seq Data Processing, Differential Expression and Survival Analysis
4.2. LincRNA Localization Predictions
4.3. Enrichment Analyses
4.4. RBP Binding Predictions
4.5. MiRNA Binding Prediction
4.6. LincRNA-MRNA Target Predictions
4.7. TF Binding Prediction
4.8. Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Chen, C.C.; Chen, L.L.; Li, C.P.; Hsu, Y.T.; Jiang, S.S.; Fan, C.S.; Chua, K.V.; Huang, S.X.; Shyr, Y.M.; Chen, L.T.; et al. Myeloid-derived macrophages and secreted HSP90α induce pancreatic ductal adenocarcinoma development. Oncoimmunology 2018, 7, e1424612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Juiz, N.A.; Iovanna, J.; Dusetti, N. Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front. Oncol. 2019, 9, 246. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Gutschner, T.; Richtig, G.; Haemmerle, M.; Pichler, M. From biomarkers to therapeutic targets-the promises and perils of long non-coding RNAs in cancer. Cancer Metastasis Rev. 2018, 37, 83–105. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Lu, D.; Lin, Y.; Lv, Y.; He, L. A seven-miRNA expression-based prognostic signature and its corresponding potential competing endogenous RNA network in early pancreatic cancer. Exp. Ther. Med. 2019, 18, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lou, W.; Ding, B.; Yang, B.; Lu, H.; Kong, Q.; Fan, W. A novel mRNA-miRNA-lncRNA competing endogenous RNA triple sub-network associated with prognosis of pancreatic cancer. Aging 2019, 11, 2610–2627. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Wang, Q.; Jia, J.; Zhao, H. A competing endogenous RNA network identifies novel mRNA, miRNA and lncRNA markers for the prognosis of diabetic pancreatic cancer. Tumour Biol. 2017, 39, 1010428317707882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Diao, Z.; Yue, X.; Chen, Y.; Zhao, H.; Cheng, L.; Sun, J. Construction and analysis of dysregulated lncRNA-associated ceRNA network identified novel lncRNA biomarkers for early diagnosis of human pancreatic cancer. Oncotarget 2016, 7, 56383–56394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Zhao, Y.; He, R.; Zhou, M.; Pan, S.; Yu, S.; Xie, Y.; Li, X.; Wang, M.; Guo, X.; et al. Three-lncRNA signature is a potential prognostic biomarker for pancreatic adenocarcinoma. Oncotarget 2018, 9, 24248–24259. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Xu, Q.; Zhang, H.; Yin, X.; Zhu, C.; Zhao, K.; Zhu, J. Five key lncRNAs considered as prognostic targets for predicting pancreatic ductal adenocarcinoma. J. Cell. Biochem. 2018, 119, 4559–4569. [Google Scholar] [CrossRef] [Green Version]
- Tahira, A.C.; Kubrusly, M.S.; Faria, M.F.; Dazzani, B.; Fonseca, R.S.; Maracaja-Coutinho, V.; Verjovski-Almeida, S.; Machado, M.C.; Reis, E.M. Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol. Cancer 2011, 10, 141. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Wang, S.; Zhou, Q.; Zhao, J.; Xia, X.; Chen, W.; Zheng, Y.; Xue, M.; Yang, F.; Fu, D.; et al. A Long Non-coding RNA Signature to Improve Prognostic Prediction of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2019, 9, 1160. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, M.; Gutschner, T. Long non-coding RNAs in cancer and development: Where do we go from here? Int. J. Mol. Sci. 2015, 16, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Baas, M.; Diederichs, S. Noncoding RNA gene silencing through genomic integration of RNA destabilizing elements using zinc finger nucleases. Genome Res. 2011, 21, 1944–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhang, S.; Chen, Y.; Wen, X.; Chen, M.; Wang, S.; Zhang, Y. Development of a novel six-long noncoding RNA signature predicting survival of patients with bladder urothelial carcinoma. J. Cell. Biochem. 2019, 120, 19796–19809. [Google Scholar] [CrossRef]
- Song, Y.; Zou, L.; Li, J.; Shen, Z.P.; Cai, Y.L.; Wu, X.D. LncRNA SNHG8 promotes the development and chemo-resistance of pancreatic adenocarcinoma. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8161–8168. [Google Scholar]
- Chen, T.; Lei, S.; Zeng, Z.; Zhang, J.; Xue, Y.; Sun, Y.; Lan, J.; Xu, S.; Mao, D.; Guo, B. Linc00261 inhibits metastasis and the WNT signaling pathway of pancreatic cancer by regulating a miR-552-5p/FOXO3 axis. Oncol. Rep. 2020, 43, 930–942. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Wang, F.; Du, C.; Zhang, Z.; Guo, H.; Xie, X.; Gao, H.; Zhuang, Y.; Kornmann, M.; Gao, H.; et al. Long non-coding RNA MEG3 functions as a tumour suppressor and has prognostic predictive value in human pancreatic cancer. Oncol. Rep. 2018, 39, 1132–1140. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Feng, H.M. MEG3 Suppresses Human Pancreatic Neuroendocrine Tumor Cells Growth and Metastasis by Down-Regulation of Mir-183. Cell. Physiol. Biochem. 2017, 44, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.; Modali, S.D.; Agarwal, S.K. Long Noncoding RNA MEG3 Is an Epigenetic Determinant of Oncogenic Signaling in Functional Pancreatic Neuroendocrine Tumor Cells. Mol. Cell. Biol. 2017, 37, e00278-17. [Google Scholar] [CrossRef] [Green Version]
- Modali, S.D.; Parekh, V.I.; Kebebew, E.; Agarwal, S.K. Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol. Endocrinol. 2015, 29, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Jiang, X.; Jiang, X.; Zhao, H. X-inactive-specific transcript: A long noncoding RNA with complex roles in human cancers. Gene 2018, 679, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Klec, C.; Prinz, F.; Pichler, M. Involvement of the long noncoding RNA NEAT1 in carcinogenesis. Mol. Oncol. 2019, 13, 46–60. [Google Scholar] [CrossRef] [Green Version]
- Gutschner, T.; Haemmerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Yu, K.; Chen, B.; Aran, D.; Charalel, J.; Yau, C.; Wolf, D.M.; van ’t Veer, L.J.; Butte, A.J.; Goldstein, T.; Sirota, M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun. 2019, 10, 3574. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wang, J.O.; Zhou, W.Y.; Chang, X.Y.; Zhang, M.M.; Zhang, Y.; Yang, X.H. Long non-coding RNA LINC01207 silencing suppresses AGR2 expression to facilitate autophagy and apoptosis of pancreatic cancer cells by sponging miR-143-5p. Mol. Cell. Endocrinol. 2019, 493, 110424. [Google Scholar] [CrossRef]
- Dhamija, S.; Becker, A.C.; Sharma, Y.; Myacheva, K.; Seiler, J.; Diederichs, S. LINC00261 and the Adjacent Gene FOXA2 Are Epithelial Markers and Are Suppressed during Lung Cancer Tumorigenesis and Progression. Noncod. RNA 2018, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.F.; Li, W.; Han, Y.D. LINC00261 suppresses cell proliferation, invasion and Notch signaling pathway in hepatocellular carcinoma. Cancer Biomark 2018, 21, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, L.; Zheng, Z.; Chen, S.; Chen, E.; Hu, Y. Long non-coding RNA linc00261 suppresses gastric cancer progression via promoting Slug degradation. J. Cell. Mol. Med. 2017, 21, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Dorn, A.; Glaß, M.; Neu, C.T.; Heydel, B.; Hüttelmaier, S.; Gutschner, T.; Haemmerle, M. LINC00261 Is Differentially Expressed in Pancreatic Cancer Subtypes and Regulates a Pro-Epithelial Cell Identity. Cancers 2020, 12, 1227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liu, Y.; Liu, R.; Zhang, K.; Zhang, Y. The lncRNA DEANR1 facilitates human endoderm differentiation by activating FOXA2 expression. Cell Rep. 2015, 11, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Raulefs, S.; Bruns, P.; Afonso-Grunz, F.; Plötner, A.; Thermann, R.; Jäger, C.; Schlitter, A.M.; Kong, B.; Regel, I.; et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol. Cancer 2015, 14, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yang, J.; Li, Q.; Xu, B.; Lian, Y.; Miao, L. LINC00152: A pivotal oncogenic long non-coding RNA in human cancers. Cell Prolif. 2017, 50, e12349. [Google Scholar] [CrossRef] [Green Version]
- Nötzold, L.; Frank, L.; Gandhi, M.; Polycarpou-Schwarz, M.; Groß, M.; Gunkel, M.; Beil, N.; Erfle, H.; Harder, N.; Rohr, K.; et al. The long non-coding RNA LINC00152 is essential for cell cycle progression through mitosis in HeLa cells. Sci. Rep. 2017, 7, 2265. [Google Scholar] [CrossRef] [Green Version]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target interactions–beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef]
- Messeguer, X.; Escudero, R.; Farré, D.; Nuñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]
- Sarvagalla, S.; Kolapalli, S.P.; Vallabhapurapu, S. The Two Sides of YY1 in Cancer: A Friend and a Foe. Front. Oncol. 2019, 9, 1230. [Google Scholar] [CrossRef] [Green Version]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, C.; Ruemmele, P.; Gehmert, S.; Schenk, H.; Kreutz, M.P.; Mycielska, M.E.; Hackl, C.; Kroemer, A.; Schnitzbauer, A.A.; Stoeltzing, O.; et al. STAT5b as molecular target in pancreatic cancer–inhibition of tumor growth, angiogenesis, and metastases. Neoplasia 2012, 14, 915–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, B.; Yao, J.; Ferri-Borgogno, S.; Zhao, J.; Chen, S.; Wang, Q.; Yan, L.; Zhou, X.; Zhu, C.; Bang, S.; et al. YAP1 oncogene is a context-specific driver for pancreatic ductal adenocarcinoma. JCI Insight 2019, 4, e130811. [Google Scholar] [CrossRef] [Green Version]
- Van Nostrand, E.L.; Pratt, G.A.; Yee, B.A.; Wheeler, E.C.; Blue, S.M.; Mueller, J.; Park, S.S.; Garcia, K.E.; Gelboin-Burkhart, C.; Nguyen, T.B.; et al. Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol. 2020, 21, 90. [Google Scholar] [CrossRef] [Green Version]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Luo, C.; Luo, Y.; Chen, L.; Liu, Y.; Wang, Y.; Han, J.; Zhang, Y.; Wei, N.; Xie, Z.; et al. MRPL33 and its splicing regulator hnRNPK are required for mitochondria function and implicated in tumor progression. Oncogene 2018, 37, 86–94. [Google Scholar] [CrossRef]
- Varia, S.; Cheedu, D.; Markey, M.; Torres-Shafer, K.; Battini, V.P.; Bubulya, A.; Bubulya, P.A. Alignment of Mitotic Chromosomes in Human Cells Involves SR-Like Splicing Factors Btf and TRAP150. Int. J. Mol. Sci. 2017, 18, 1956. [Google Scholar] [CrossRef] [Green Version]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Casey, M.C.; Prakash, A.; Holian, E.; McGuire, A.; Kalinina, O.; Shalaby, A.; Curran, C.; Webber, M.; Callagy, G.; Bourke, E.; et al. Quantifying Argonaute 2 (Ago2) expression to stratify breast cancer. BMC Cancer 2019, 19, 712. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; LaPierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Reon, B.J.; Takao Real Karia, B.; Kiran, M.; Dutta, A. LINC00152 Promotes Invasion through a 3’-Hairpin Structure and Associates with Prognosis in Glioblastoma. Mol. Cancer Res. 2018, 16, 1470–1482. [Google Scholar] [CrossRef] [Green Version]
- Cooke, A.; Schwarzl, T.; Huppertz, I.; Kramer, G.; Mantas, P.; Alleaume, A.M.; Huber, W.; Krijgsveld, J.; Hentze, M.W. The RNA-Binding Protein YBX3 Controls Amino Acid Levels by Regulating SLC mRNA Abundance. Cell Rep. 2019, 27, 3097–3106. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Balda, M.S.; Matter, K. Stress- and Rho-activated ZO-1-associated nucleic acid binding protein binding to p21 mRNA mediates stabilization, translation, and cell survival. Proc. Natl. Acad. Sci. USA 2012, 109, 10897–10902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, L.S.; Bartley, M.A.; Bert, A.; Hunter, J.; Polyak, S.; Diamond, P.; Vadas, M.A.; Goodall, G.J. A multi-protein complex containing cold shock domain (Y-box) and polypyrimidine tract binding proteins forms on the vascular endothelial growth factor mRNA. Potential role in mRNA stabilization. Eur. J. Biochem. 2004, 271, 648–660. [Google Scholar] [CrossRef]
- Dong, X.; Yang, Z.; Yang, H.; Li, D.; Qiu, X. Long Non-coding RNA MIR4435-2HG Promotes Colorectal Cancer Proliferation and Metastasis Through miR-206/YAP1 Axis. Front. Oncol. 2020, 10, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Xin, X.F.; Zhang, J.Y.; Dai, W.; Lv, T.F.; Song, Y. LncRNA GMDS-AS1 inhibits lung adenocarcinoma development by regulating miR-96-5p/CYLD signaling. Cancer Med. 2020, 9, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdȯttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, Y.; Zhang, W.; Zhou, Z.; Wu, J.; Cui, P.; Zhang, Y.; Huang, G. Long non-coding RNA Linc00152 is involved in cell cycle arrest, apoptosis, epithelial to mesenchymal transition, cell migration and invasion in gastric cancer. Cell Cycle 2015, 14, 3112–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Wang, Z.; Wang, S.; Weng, M.; Zhou, D.; Li, C.; Wang, J.; Chen, E.; Quan, Z. Long non-coding RNA LINC00152 promotes gallbladder cancer metastasis and epithelial-mesenchymal transition by regulating HIF-1α via miR-138. Open Biol. 2017, 7, 160247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, T.; Ding, W.; Wang, N.; Huang, H.; Pan, Y.; Wei, A. Long noncoding RNA linc00346 promotes the malignant phenotypes of bladder cancer. Biochem. Biophys. Res. Commun. 2017, 491, 79–84. [Google Scholar] [CrossRef]
- Qu, Y.K.; Qu, X.S.; Chen, G.; Feng, Y.; Teng, X.L.; Liu, W.X.; Cheng, Z.X.; Xu, J.; Guo, L.Q. LINC01503 promotes cell proliferation, invasion and EMT process in cholangio-carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6445–6452. [Google Scholar] [PubMed]
- Peng, W.X.; He, R.Z.; Zhang, Z.; Yang, L.; Mo, Y.Y. LINC00346 promotes pancreatic cancer progression through the CTCF-mediated Myc transcription. Oncogene 2019, 38, 6770–6780. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, C.; Ning, Z.; Hua, Y.; Li, Y.; Chen, L.; Liu, L.; Chen, Z.; Meng, Z. Long non-coding RNA LINC00346 promotes pancreatic cancer growth and gemcitabine resistance by sponging miR-188-3p to derepress BRD4 expression. J. Exp. Clin. Cancer Res. 2019, 38, 60. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [Green Version]
- Terai, G.; Iwakiri, J.; Kameda, T.; Hamada, M.; Asai, K. Comprehensive prediction of lncRNA-RNA interactions in human transcriptome. BMC Genom. 2016, 17 (Suppl. 1), 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Mello, S.S.; Sinow, C.; Raj, N.; Mazur, P.K.; Bieging-Rolett, K.; Broz, D.K.; Imam, J.F.C.; Vogel, H.; Wood, L.D.; Sage, J.; et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Zhang, Y.; Yang, J.; He, S.; Li, M.; Yan, S.; Chen, Y.; Qu, C.; Xu, L. NEAT1 regulates pancreatic cancer cell growth, invasion and migration though mircroRNA-335-5p/c-met axis. Am. J. Cancer Res. 2016, 6, 2361–2374. [Google Scholar]
- Huang, B.; Liu, C.; Wu, Q.; Zhang, J.; Min, Q.; Sheng, T.; Wang, X.; Zou, Y. Long non-coding RNA NEAT1 facilitates pancreatic cancer progression through negative modulation of miR-506-3p. Biochem. Biophys. Res. Commun. 2017, 482, 828–834. [Google Scholar] [CrossRef]
- Luo, Z.; Yi, Z.J.; Ou, Z.L.; Han, T.; Wan, T.; Tang, Y.C.; Wang, Z.C.; Huang, F.Z. RELA/NEAT1/miR-302a-3p/RELA feedback loop modulates pancreatic ductal adenocarcinoma cell proliferation and migration. J. Cell. Physiol. 2019, 234, 3583–3597. [Google Scholar] [CrossRef]
- Feng, Y.; Gao, L.; Cui, G.; Cao, Y. LncRNA NEAT1 facilitates pancreatic cancer growth and metastasis through stabilizing ELF3 mRNA. Am. J. Cancer Res. 2020, 10, 237–248. [Google Scholar]
- Jiao, F.; Hu, H.; Yuan, C.; Wang, L.; Jiang, W.; Jin, Z.; Guo, Z.; Wang, L. Elevated expression level of long noncoding RNA MALAT-1 facilitates cell growth, migration and invasion in pancreatic cancer. Oncol. Rep. 2014, 32, 2485–2492. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Imanirad, P.; Jutooru, I.; Hedrick, E.; Jin, U.H.; Rodrigues Hoffman, A.; Leal de Araujo, J.; Morpurgo, B.; Golovko, A.; Safe, S. Role of metastasis-associated lung adenocarcinoma transcript-1 (MALAT-1) in pancreatic cancer. PLoS ONE 2018, 13, e0192264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ma, L. New Insights into Long Non-Coding RNA MALAT1 in Cancer and Metastasis. Cancers 2019, 11, 216. [Google Scholar] [CrossRef] [Green Version]
- Arun, G.; Spector, D.L. MALAT1 long non-coding RNA and breast cancer. RNA Biol. 2019, 16, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.C.; Dang, Y.W.; Wei, D.M.; Chen, P.; Tang, R.X.; Huang, Q.; Liu, J.H.; Luo, D.Z. Clinical significance and prospective molecular mechanism of MALAT1 in pancreatic cancer exploration: A comprehensive study based on the GeneChip, GEO, Oncomine, and TCGA databases. Onco Targets Ther. 2017, 10, 3991–4005. [Google Scholar] [CrossRef] [Green Version]
- Iacobuzio-Donahue, C.A.; Maitra, A.; Olsen, M.; Lowe, A.W.; van Heek, N.T.; Rosty, C.; Walter, K.; Sato, N.; Parker, A.; Ashfaq, R.; et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Am. J. Pathol. 2003, 162, 1151–1162. [Google Scholar] [CrossRef]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.R.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009, 16, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Yoshida, K.; Yamashita, Y.; Ota, J.; Takada, S.; Kisanuki, H.; Koinuma, K.; Choi, Y.L.; Kaneda, R.; Iwao, T.; et al. Experimental trial for diagnosis of pancreatic ductal carcinoma based on gene expression profiles of pancreatic ductal cells. Cancer Sci. 2005, 96, 387–393. [Google Scholar] [CrossRef]
- Frappart, P.O.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids—A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 8, 2050640620905183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carugo, A.; Genovese, G.; Seth, S.; Nezi, L.; Rose, J.L.; Bossi, D.; Cicalese, A.; Shah, P.K.; Viale, A.; Pettazzoni, P.F.; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.T.; Zhang, X.; Wang, D.W.; Xu, J.; Kou, K.J.; Wang, Z.W.; Yong, G.; Liang, D.S.; Sun, X.Y. Overexpressed lncRNA GATA6-AS1 Inhibits LNM and EMT via FZD4 through the Wnt/β-Catenin Signaling Pathway in GC. Mol. Ther. Nucleic Acids 2020, 19, 827–840. [Google Scholar] [CrossRef]
- Liu, Z.; Dai, J.; Shen, H. Systematic analysis reveals long noncoding RNAs regulating neighboring transcription factors in human cancers. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2785–2792. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [Green Version]
- Gudenas, B.L.; Wang, L. Prediction of LncRNA Subcellular Localization with Deep Learning from Sequence Features. Sci. Rep. 2018, 8, 16385. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Di, C.; Hu, B.; Zhou, M.; Liu, Y.; Song, N.; Li, Y.; Umetsu, J.; Lu, Z.J. CLIPdb: A CLIP-seq database for protein-RNA interactions. BMC Genom. 2015, 16, 51. [Google Scholar] [CrossRef] [Green Version]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glaß, M.; Dorn, A.; Hüttelmaier, S.; Haemmerle, M.; Gutschner, T. Comprehensive Analysis of LincRNAs in Classical and Basal-Like Subtypes of Pancreatic Cancer. Cancers 2020, 12, 2077. https://doi.org/10.3390/cancers12082077
Glaß M, Dorn A, Hüttelmaier S, Haemmerle M, Gutschner T. Comprehensive Analysis of LincRNAs in Classical and Basal-Like Subtypes of Pancreatic Cancer. Cancers. 2020; 12(8):2077. https://doi.org/10.3390/cancers12082077
Chicago/Turabian StyleGlaß, Markus, Agnes Dorn, Stefan Hüttelmaier, Monika Haemmerle, and Tony Gutschner. 2020. "Comprehensive Analysis of LincRNAs in Classical and Basal-Like Subtypes of Pancreatic Cancer" Cancers 12, no. 8: 2077. https://doi.org/10.3390/cancers12082077
APA StyleGlaß, M., Dorn, A., Hüttelmaier, S., Haemmerle, M., & Gutschner, T. (2020). Comprehensive Analysis of LincRNAs in Classical and Basal-Like Subtypes of Pancreatic Cancer. Cancers, 12(8), 2077. https://doi.org/10.3390/cancers12082077