HMGA Proteins in Hematological Malignancies
by

Angela Minervini
†, 
Nicoletta Coccaro
†,
Luisa Anelli
,
Antonella Zagaria
,
Giorgina Specchia
and
Francesco Albano
*
Department of Emergency and Organ Transplantation (D.E.T.O.), Hematology Section, University of Bari, 70124 Bari, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to the work.
Cancers 2020, 12(6), 1456; https://doi.org/10.3390/cancers12061456
Submission received: 22 April 2020
/
Revised: 25 May 2020
/
Accepted: 1 June 2020
/
Published: 3 June 2020
(This article belongs to the Section Molecular Cancer Biology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The high mobility group AT-Hook (HMGA) proteins are a family of nonhistone chromatin remodeling proteins known as “architectural transcriptional factors”. By binding the minor groove of AT-rich DNA sequences, they interact with the transcription apparatus, altering the chromatin modeling and regulating gene expression by either enhancing or suppressing the binding of the more usual transcriptional activators and repressors, although they do not themselves have any transcriptional activity. Their involvement in both benign and malignant neoplasias is well-known and supported by a large volume of studies. In this review, we focus on the role of the HMGA proteins in hematological malignancies, exploring the mechanisms through which they enhance neoplastic transformation and how this knowledge could be exploited to devise tailored therapeutic strategies.
1. Introduction
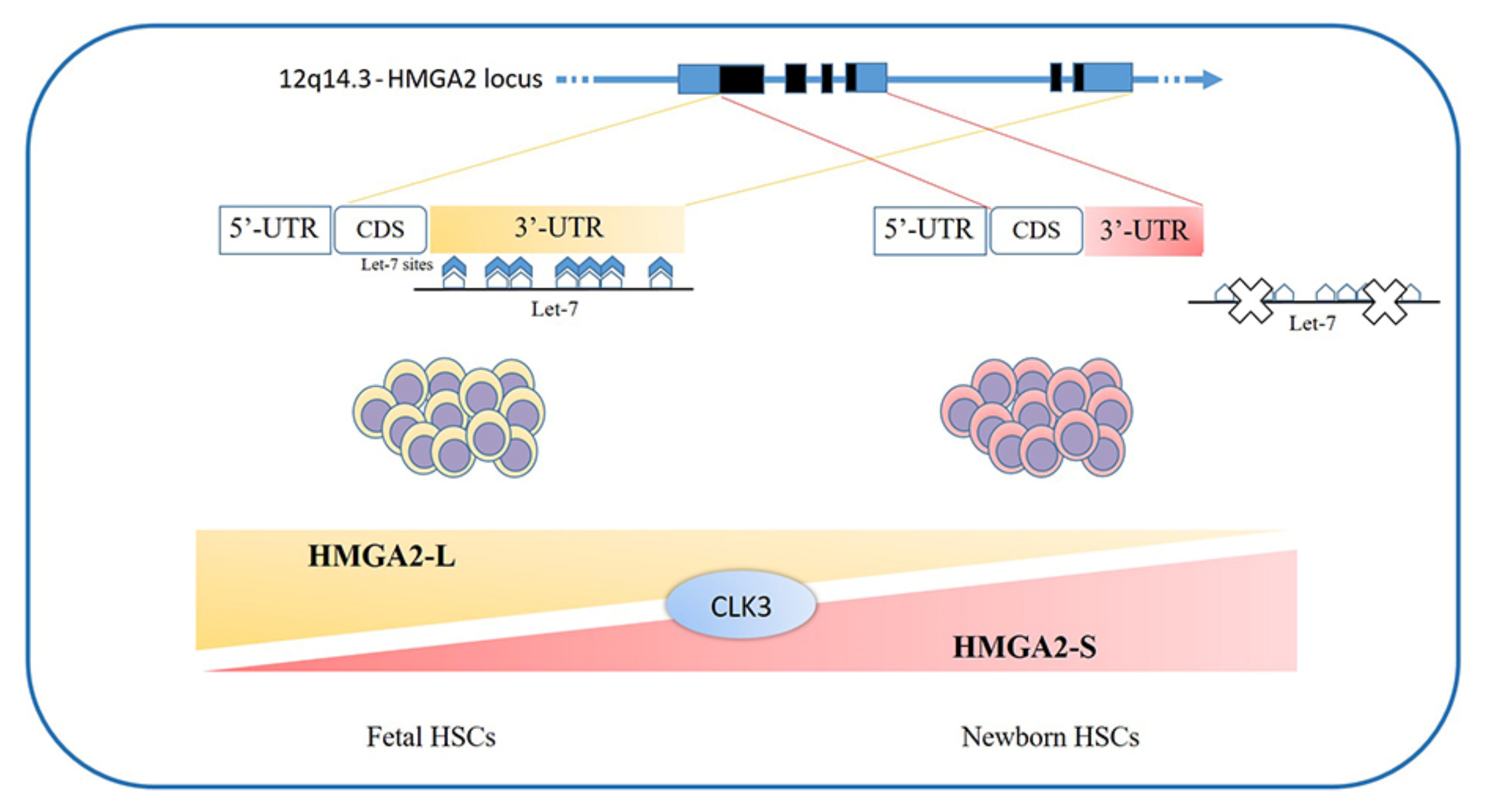
The high mobility group AT-Hook (HMGA) proteins are a family of nonhistone chromatin remodeling proteins regulating gene expression, known as “architectural transcriptional factors” [1,2,3,4]. The gene family comprises the HMGA1 and HMGA2 genes; the former is located on chromosome 6 (6p21), is about 10 kb large, and consists of eight exons [5]; the latter is located on chromosome 12 (12q14-15), is ≥160 kb in size, and consists of five exons [6]. Alternative splicing of the HMGA1 gene produces three messenger RNA (mRNA) and three corresponding proteins, HMGA1a (formerly HMG-I), HMGA1b (formerly HMG-Y), and HMGA1c (formerly HMG-I/R) [7]. The HMGA1b isoform differs from the 1a isoform in that it lacks 11 amino acids; instead, the HMGA1c isoform is produced by alternative splicing of the HMGA1 gene using noncanonical acceptor and donor sites. As regards HMGA2, recently, two isoforms (HMGA2-L and HMGA2-S) have been described for which expression seems to be confined to the hematopoietic compartment and to depend on the stage of development of the hematopoietic stem cell (HSC) (see below) [8].
Sequence alignment showed that HMGA2 has a homology of about 55% with HMGA1 (A and B isoforms), including AT-hooks that are located in separate exons [6]. The AT-hook motif is positively charged and consists of a stretch of nine amino acids, containing the Arg-Gly-Arg-Pro repetition. This motif allows the protein to recognize and bind the AT-rich sequences in the minor groove of B-form DNA, rendering it able to recognize a structure rather than a specific sequence [6,9], so that some data report the ability of the HMGA1 protein to bind DNA even in regions without a rich AT content [10,11,12]. Once bound to DNA, the AT-hook undergoes a conformational change [13], and simultaneous binding of two or more AT-hooks to different binding sites leads to an increase in the strength of HMGA interactions with DNA [14].
By binding the minor groove of AT-rich DNA sequences, HMGA proteins interact with the transcription apparatus, altering the chromatin modeling and regulating gene expression by either enhancing or suppressing the binding of the more usual transcriptional activators and repressors, although they do not themselves have any transcriptional activity [2,6,15,16,17,18]. Their function is critical for the formation of higher-order nucleoprotein complexes (called enhanceosomes) in the promoter region, allowing the efficient transcriptional activation of a gene [19] thanks to their multiple surfaces capable of protein–protein interactions [6,13]. Through such interactions, HMGA proteins may directly or indirectly control transcription, inducing conformational changes in chromatin or in the transcription factors themselves. In such ways, HMGA proteins are able to influence a wide variety of normal biological processes, such as cell growth, proliferation, differentiation, and death [2,6]. They show an active role in the division cycle, and data indicate that both histone H1 and HMGA1 are necessary for chromatin condensation [20,21].
HMGA1 and 2 display different target genes; for example, the BRCA1 gene is negatively regulated by HMGA1 but not by HMGA2 [22]. Instead, HMGA2 has several other targets, including cycle regulators, one of which is the CDKN2A gene, encoding the two proteins p16INK4A and p14ARF. With the RNA-binding proteins LIN28A/B and the microRNA (miRNA) let-7b, HMGA2 and CDKN2A form an axis that controls stem cell aging in both normal and pathological conditions [23,24,25].
2. HMGA Proteins in Oncology
HMGA expression seems to be ubiquitous and high during embryogenesis and null or hardly detectable in adult differentiated tissues [1,2,3,4,26,27,28,29,30]. Several studies demonstrated the involvement of these proteins in embryonic development [31], adipocytes cell growth, and differentiation. Mice lacking the HMGA2 gene showed a pygmy phenotype with a drastic reduction of fat tissue [32]; instead, HMGA2 overexpression results in giant, obese mice [33]. On the contrary, HMGA1 inhibits adipocytes cell growth and triggers differentiation [34].
Overexpression of both genes is observed in most human malignant neoplasias, linking their alteration to carcinogenesis [2,6,35,36,37,38,39], while blocking their expression prevents thyroid cell transformation and triggers the death of malignant cells [40,41,42]. Various in vitro and in vivo studies have demonstrated the oncogenic potential of HMGA proteins; their overexpression transforms mouse and rat fibroblasts [43,44], and in transgenic mice, the development of natural killer T-cell lymphomas and pituitary adenomas has been observed [45,46,47,48]. There are high expression levels of HMGA transcripts in embryonic stem (ES) cells [49,50], HSCs [50,51,52,53], leukemic stem cells [30], and poorly differentiated or refractory tumors [1,2,3,4,30,44,46,51,52,54,55,56,57,58,59,60,61,62]. In cultured human lymphoid cells, the ectopic expression of HMGA1a triggers leukemic transformation [44,62] and, in transgenic mice, the development of an aggressive T-cell lymphoid neoplasia [46,47,58,63,64]. In particular, HMGA1 is overexpressed in several high-grade or refractory neoplasias, including hematologic malignancies and solid tumors [3,46,52,58,60,62,63,65]. Indeed, HMGA1 proteins are the most abundant nonhistone, chromatin-binding proteins in tumor cells [3].
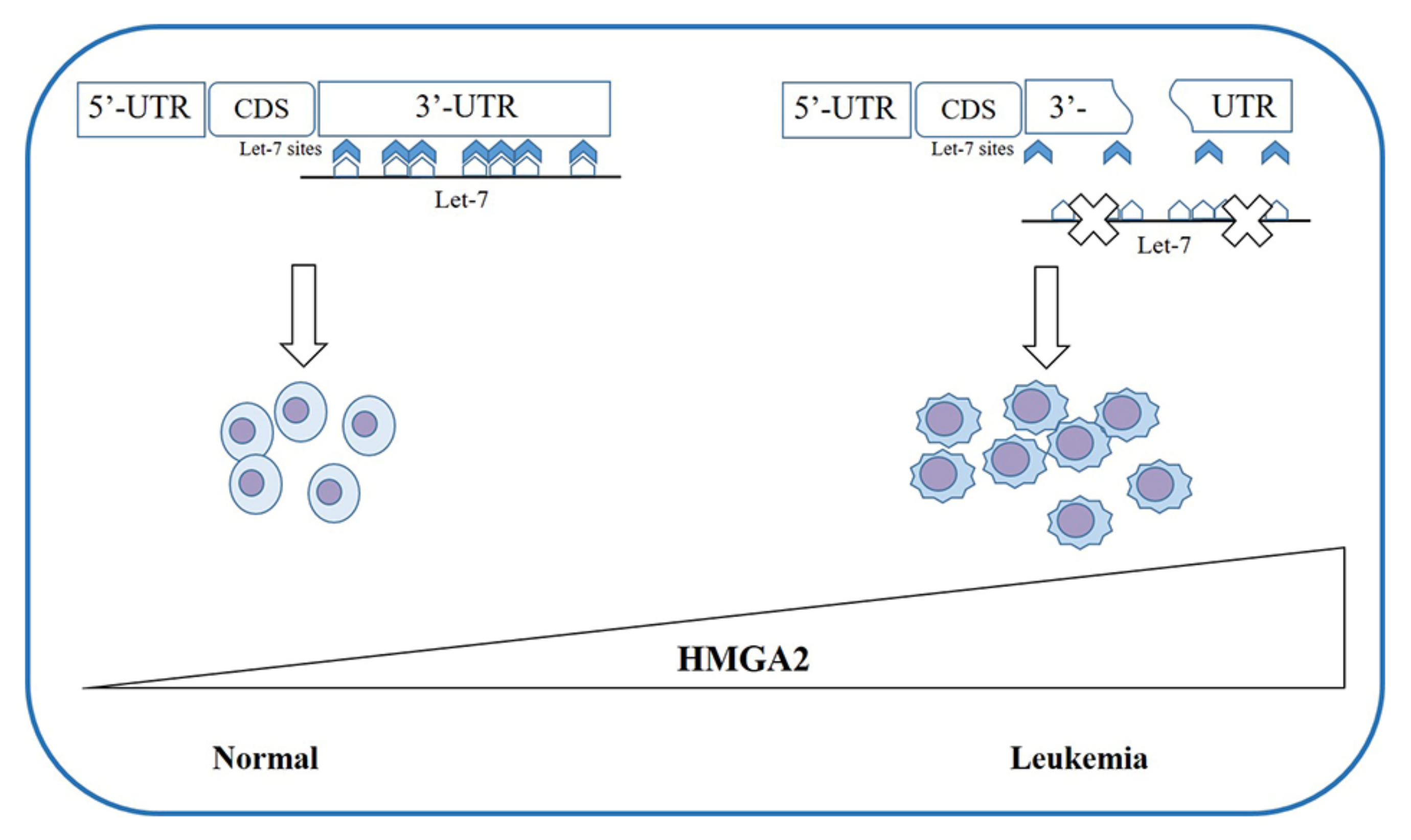
Chromosomal duplications or translocations cause HMGA1 overexpression in rare cases; more frequently, the translocations involve HMGA2 (chromosomal region 12q14-15) in benign tumors, such as lipomas [66] and uterine fibroids [67,68,69]. With Fluorescent in Situ Hybridization (FISH) analysis, it has been observed that, in most cases, the breakpoint in the HMGA2 locus involves exons 1-2 or 1-3, leading to the loss of the C-terminal tail [70], separating the DNA-binding domains from the acid tails, and merging them with ectopic sequences. This seems to be sufficient for tumor development. Further studies revealed the presence of multiple binding sites for miRNA let-7 in the 3′-Untranslated Region (3′-UTR) of HMGA2 [71]. This evidence corroborates the observation that, in tumors with rearrangements destroying 3′-UTR of HMGA2, there were increased levels of the intact protein [68,72,73]; this suggests the presence of functional sites for the binding of miRNAs (such as let-7, mir-98, and miR-33) [74,75,76,77,78,79,80,81,82,83,84,85,86] that are important in regulating the gene expression.
3. HMGA Proteins in Hematopoiesis
In the hematologic field, HMGA proteins seem to play a pivotal role in both physiological and pathogenic conditions.
The loss of HMGA2 in HSCs does not allow a correct fetal hematopoietic process, causing slower self-renewal and proliferation rates, while it seems not to be indispensable in adult hematopoiesis [93]. Indeed, in 2013, in mouse models, Copley et al. demonstrated that the fine equilibrium between levels of the RNA-binding protein LIN28B and let-7 strikingly influence HSC developmentally timed changes, observing that HSC self-renewal potentials reduced when LIN28B decreased and let-7 increased [93]. As HMGA2 expression is high during embryogenesis, being particularly upregulated in fetal HSCs and gradually decreasing during the transition to adult hematopoiesis, experiments in transplanted irradiated hosts to define the role of HMGA2 and LIN28 in HSC functions showed that both LIN28- and HMGA2-induced overexpression increased the self-renewal activity of adult HSCs. However, HMGA2 overexpression in adult HSCs did not seem sufficient to mimic all the effects of elevated LIN28B that triggers a fetal lymphoid differentiation program. Accordingly, HMGA2 seems to be a downstream modulator of the HSC self-renewal ability; its function is regulated by LIN28B, that acts as a master regulator of developmentally timed changes of HSC [93]. However, this regulatory mechanism seems to be even finer tuned [91]. In a recent work, Cesana et al. employed high-throughput genomic approaches to profile miRNAs, long intergenic non coding RNAs (lincRNAs), and mRNAs in HSCs during development in order to characterize transcriptional and posttranscriptional changes [8]. These analyses highlighted distinct alternative splicing patterns of HSCs in various key hematopoietic regulators, one of which is HMGA2, for which an alternative isoform, escaping miRNA-mediated control, was identified. It seems that the splicing kinase CLK3 conserves HMGA2 function in response to an increase of let-7 miRNA levels by regulating HMGA2 splicing. These data delineate a CLK3/HMGA2 functional axis regulating HSC functions during development [8]. In humans, HMGA2 presents alternative splicing isoforms, HMGA2-L, which is sensitive to degradation by let-7, being the dominant isoform in fetal HSCs, whereas newborn HSCs express the HMGA2-S isoform, that is resistant to degradation by let-7 [8] (Figure 1). In support of the crucial role of HMGA2 in regulating HSCs properties, it has been found in gene therapy experiments that a deliberate overexpression of HMGA2, in parallel with that of the transgene of interest, might be needed to obtain the therapeutic effect [94,95], particularly in cases where a large fraction of corrected HSCs must be obtained to gain the benefit. In the case reported in 2010 by Cavazzana et al. of an adult patient with severe βE/β0-thalassaemia treated with gene therapy, the presence of a dominant, myeloid-biased cell clone was demonstrated, in which the integrated vector transferring the β-globin gene caused the transcriptional activation of HMGA2 in erythroid cells and further increased the expression of a truncated HMGA2 mRNA insensitive to degradation by let-7 miRNAs [94].
There is evidence in mice that Hmga2 is also a regulator of gamma-globin mRNA, and that it moderately increased HbF levels in human adult erythroblasts in vitro [96]. Targeted inhibition of let-7 is sufficient for specific developmental changes to occur in gamma-globin transcription and HbF levels through increased HMGA2 levels [97].
As regards the LIN28B-let7-HMGA2 axis, it is also involved in the megakaryocyte and platelet lineage. LIN28B expression in fetal myelo-erythroid progenitors suggests that it could have a role in the prenatal platelet-forming lineages; indeed, megakaryocytes derived from human ES cells express higher levels of LIN28B as compared to adult controls [98]. This implies that LIN28B may also have an active function in megakaryocyte development and thus platelet function [99].
In vitro experiments on ES cells lacking one or both HMGA1 alleles showed that these cells showed a lesser differentiation in T-cell precursors than Wild Type (WT) ES cells and preferentially in B cells, maybe because of an increased interleukin 6 and decreased interleukin 2 expression [100]. Furthermore, there was evidence of an aberrant hemopoietic differentiation, with a reduction of the monocyte/macrophage population and an increase in megakaryocyte precursors, erythropoiesis, and globin gene expression. The restoration of HMGA1 expression reestablished the WT phenotype. These studies highlighted that HMGA1 proteins directly control the transcription of GATA-1, a key transcription factor which regulates red blood cell differentiation, that is overexpressed in HMGA1−/− ES cells. This could offer an explanation, at least in part, of the alteration of the lineages of megakaryocytes/erythrocytes [100]. These observations add weight to those of Pierantoni et al., who observed that the induction of HMGA1 protein synthesis appears to play a role in the differentiation of megakaryocytes while its inhibition may be necessary in the differentiation of erythrocytes or macrophages [101].
Regarding lymphoid compartment, animal experiments showed HMGA2 involvement in mouse thymopoiesis, as it is highly expressed in fetal and neonatal early T cell progenitors (ETP), the most intrathymic precursors, while it results almost undetectable 5 weeks after birth [102]. In the same way, the number of thymic cells increases during the first two weeks after birth, stabilizes, and then declines by seven weeks of age. In fact, HMGA2-deficient mice showed deficient ETPs after birth, and also the total thymocyte number was fewer as compared to WT. Bearing in mind that, also in human fetal thymic progenitors, HMGA2 expression is high and decreases during growth, it is possible that a similar mechanism takes place [102].
As regards HMGA1, studies using HMGA1-green fluorescent protein fusion explored HMGA1 expression in undifferentiated and differentiated populations of hematopoietic cells, demonstrating that HMGA1 is highly expressed in CD4/CD8-double negative (DN) cells and transiently downregulated in CD4/CD8-double positive (DP) cells during early T cell development in the thymus [103]. Indeed, HMGA1 binds directly to cis-regulatory elements in the CD4/CD8 loci in DN cells but not in DP cells. Moreover, in DN leukemic cells, CD4/CD8 expression is induced by the inhibition of HMGA1, and T leukemic cells lacking HMGA1 showed a markedly decreased proliferation [103].
HMGA1 indirectly controls µ enhancer activity during B lymphocyte development, acting on the combinatorial activity of transcription factors [104,105,106,107]. Various studies revealed that HMGA1 interacts with both PU.1 and ETS proto-oncogene 1 transcription factor 1 (Ets-1) in solution [108,109]; HMGA1 interaction with PU.1 likely induces a change in PU.1 structure, increasing PU.1/µ enhancer binding. This augmented binding renders HMGA1 able to boost PU.1/Ets-1 functional synergy on the µ enhancer [108,110].
In view of the role of HMGA proteins in driving both benign and malignant tumors [2] and the evidence regarding their main functions in various hematopoietic physiological mechanisms, various research studies have explored their involvement in hematological malignancies.
4. HMGAs Involvement in Myeloid Malignancies
4.1. HMGAs Involvement in Myeloproliferative Neoplasms
Myeloproliferative neoplasms (MPN) are a group of clonal diseases of the hematopoietic system which feature an excessive production of myeloid cells. This group includes polycythemia vera (PV) essential thrombocythemia (ET), primary myelofibrosis (PMF), and chronic myeloid leukemia (CML) [111]. These four diseases share clinical features [112], but from a biological point of view, it is possible to subdivide them into Philadelphia chromosome (Ph) positive (Ph+) (CML) and Ph negative (Ph-) (PV, ET, and PMF) diseases. CML is characterized by the Ph [113], derived from the balanced translocation of chromosome 9 and chromosome 22 [114]; driver mutations of JAK2, MPL, and CALR together account for approximately 90% of Ph-MPNs [115]. The most frequent mutation is the point mutation of the Janus kinase 2 (JAK2) gene (JAK2V617F) that can be observed in most patients with PV (95%), ET (50%), and PMF (60%) [116,117,118,119]. The JAK2V617F involves the constitutive activation of the JAK-STAT signal pathway which confers a blood cells growth advantage. It has been hypothesized that the cooperation of different genetic accidents is necessary to allow the initiation and progression of the disease; a mutation alone is not sufficient for the development of the disease in humans [120]. In some cases of MPN, an overexpression of HMGA2 provides an important contribution to the mechanisms of cell proliferation and differentiation [70,121,122,123,124,125]. The first group to relate the upregulation of HMGA2 and MPN was Guglielmelli et al.; studying the molecular profile in PMF of CD34+ cells, they observed the presence of the JAK2V617F mutation that influenced the abnormal expression of HMGA2 [125].
As stated above, the HMGA2 gene possesses, at the level of the 3′-UTR, seven complementary sequences to let-7 which negatively regulate the protein expression [126]. Deletion or truncation of HMGA2 3′-UTR determines the loss of the let-7-binding sites, which leads to overexpression of the full-length or truncated protein with a conserved DNA binding capacity and a high likelihood of promoting tumors [127,128]. In their 2011 study, Ikeda et al. [129] created a HMGA transgenic mouse with 3′-UTR deletion and therefore overexpression of the HMGA2 mRNA (ΔHMGA2 mouse); a similar genetic alteration had already been observed in MPN [70,73,121,122,123,124,125]. Their data showed a clinical scenario similar to that of MPN, with an increase of all blood cells, splenomegaly, hypercellularity of the bone marrow (BM), an increased formation, and growth of erythroid colonies, independently of erythropoietin. From a biological point of view, the ΔHMGA2 mice showed an increased expression of JAK2 mRNA and phosphorylation of STAT3 and AKT, independently of the cytokines. Therefore, overexpression of HMGA2 through the constitutive JAK-STAT pathways and AKT activation may provide a proliferative advantage to the hematopoietic stem and progenitor cells [129].
Experiments with ΔHMGA2 mice have shown that the expression of HMGA2 promotes clonal growth in MPNs by improving the self-renewal and functionality of HSC [129] as well as other types of stem cells [24,130,131]. Further data confirming this hypothesis were provided by the Ueda et al. group, that created a ΔHMGA2/JAK2V617F transgenic mouse which overexpresses HMGA2 following the deletion of the 3′-UTR. Compared to JAK2V617F mice, these mice showed a more serious leukocytosis, splenomegaly, and anemia with reduced survival; on the other hand, the severity of the myelofibrosis was comparable. In BM serial transplantation experiments, ΔHMGA2/JAK2V617F cells conferred a greater repopulation capacity that emulated a severe MPN, as compared to JAK2V617F cells; this establishes that HMGA2 is able to promote the progression of MPN at the HSC level [132].
The work by Chen et al. in 2017 emphasized a survival advantage conferred by the JAK2 mutation, given that apoptosis is inhibited in these cells. Patients with HMGA2 overexpression are more likely to develop ET and to experience thrombotic episodes [115]. Numerous other studies have highlighted the increased platelet count in this group of patients and the possibility that they may belong to the ET subtype; already in 2012, Oguro et al. had observed that, in HSC, the overexpression of HMGA2 triggered a myeloproliferative condition in mice, with increased megakaryopoiesis [133], and in 2016, Yang et al. observed the same in mice with JAK2 mutation [134]. While BM cells that upregulated HMGA2 have a proliferative advantage over control competitive repopulation assay in mice and BM transplant models [129], it is possible that overexpressed HMGA2 triggers a hematopoietic proliferation mechanism with a preference for platelets and inhibits the apoptosis mechanism [133,134]. All these works have raised the hypothesis that alteration of the HMGA2 protein could confer a proliferative advantage to altered progenitors, triggering the pathogenesis of MPN or other clonal hematological diseases [127], but how HMGA2 intervenes directly in the pathogenesis of MPN remains to be clarified.
The overexpression of HMGA2 associated with the 3′-UTR deletion was found not only in MPN patients but also in those with myelodysplastic syndrome (MDS) and MDS/MPN [70,121,122,123,124,125] as well as in patients with paroxysmal nocturnal hemoglobinuria (PNH) [73] (Figure 2). PNH is characterized by the absence on the cell surface of the glycosyl phosphatidylinositol protein on the stem cell. All cell lines deriving from this stem cell show the same alteration and an increased expression of HMGA2 due to the truncation of the 3′-UTR [73,127].
Although several studies have confirmed that the alteration involving chromosome 12 leads to the overexpression of HMGA2 in patients with MPN [122,135], data from the work by Chen et al. demonstrated that the regulation of let-7a miRNA has a very important impact on the expression of HMGA2 in patients with MPN. These data are in line with a paper reporting that a reduced expression of let-7 miRNA is the main cause of the alteration of HMGA2 mRNA expression in MPN [36]. Meyer et al. observed an increase in the level of HMGA2 in the accelerated phase and in the blast crisis of CML compared to the chronic phase; furthermore, the expression of the protein is inversely correlated with let-7 [136,137]. Several works have correlated the altered miRNA expression with the genetic complexity of MPNs, including let-7a, miR-150, let-7g, let-7f, miR_4319, and miR-149 [138,139,140,141].
By gene expression profiling of the granulocytes of patients with PV, Bruchova et al. observed that the frequency of the JAK2 mutation is inversely proportional to the expression of let-7a. However, the overexpression of HMGA2 has been observed in patients with PMF but not in patients with PV, among which a correlation between the expression levels of HMGA2 and let-7a was not observed [138]; most likely, this figure was influenced by the small group of patients enrolled in the study [115]. Although an inverse relationship between the expression of HMGA2 and let-7a has been reported, Harada-Shirado et al. failed to observe any possible relationship between the JAK2V617F mutation and the expression of HMGA2 [36]. Furthermore, the upregulation of HMGA2 has also been reported in some MPN patients without the JAK2 mutation [142]. Chen et al. also highlighted that patients who have increased HMGA2 expression are more likely to have one of the MPNs driver mutations [115]. However, there are still too few data available in these papers to be able to draw conclusions. The interaction between HMGA2 and JAK2V617F as well as whether, in patients with JAK2-mutated MPN, HMGA2 overexpression has a specific role in the disease pathogenesis have yet to be clarified [115].
The improvement on the HSC properties due to the expression of HMGA2 has also been observed in some human gene therapy studies using lenti-viral or retro-viral for the transduction of human transgenes [94,95]. In some cases, the virus containing the gene of interest integrated into the genome at the level of the HMGA2 locus, consequently causing the loss of binding sites for the let-7 miRNA and the clonal expansion of the hematopoietic cells. This intervention leads to a long-term effect, which in some patients with severe thalassemia results in independence from continuous blood transfusions [94].
The increased expression or/and truncation of HMGA2 has been observed in patients with both MDS and PNH, in which the common feature is marrow failure [70,73,143]; the failure is in part due to the immunological injury of HSC orchestrated by the self-reactive cytotoxic T lymphocytes (CTL) that secrete the Tumor Necrosis Factor-α (TNF-α) and Interferon-γ (INF-γ) [144,145,146]. It has been hypothesized that an altered hematopoietic clone may survive the attack of IFN-γ produced by CTLs in these pathologies [147], suggesting that a genetic event following the HSC lesion is necessary until the altered cell acquires a clonal growth advantage and can expand (two hit-hypothesis) [148]. As already observed in MDS and PNH, it has recently been shown that TNF-α can induce clonal selection of JAK2V617F+ cells in MPN patients, probably because the action of TNF-α collides with the survival advantage of these cells [149]. It is clear that the pathogenesis of MPN is linked to the orchestrated action of genetic anomalies and immunological or humoral mechanisms. As evidence of this, the use of JAK2 inhibitors shows benefits in patients with PMF by reducing the size of the spleen and in part by reducing the concentration of cytokines [150]. At present, there is no clear information about the correlation between HMGA2 and cytokine production, although the microarray analysis performed in the 2011 study showed the activation of the immune response-related pathway in HSC of ΔHMGA2 mice [129].
Therefore, HMGA2 could be an excellent candidate as a therapeutic target because it is involved at various levels in the pathogenesis of MPN, including the regulation of gene expressions and the hematopoietic clonal proliferation [127].
4.2. HMGAs Involvement in Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is one of the most common blood cancers to affect adults, accounting for approximately 3% of all cancer cases and approximately 25% of all leukemias [151].
AML is a heterogeneous neoplasm, in which clonal proliferation of HSCs is observed, characterized by a blocked or severely compromised differentiation process and a progressive accumulation of blasts at various stages of maturation at the level of the BM, peripheral blood (PB), and other tissues. All this translates into the destruction of the hematopoietic system [151,152]. The prognosis for AML is severe, and the 5-year survival rate is estimated to be around 20%. The etiology of the disease is very complex, and there are many risk factors. Several studies have highlighted recurrent genetic and chromosomal alterations that may underlie the mechanisms of the disease [153,154,155]. Different molecular biology techniques are used to identify the most common genetic alterations, such as NPM1, FLT3, IDH, CEBPA, RUNX1, ASXL1, and TP53. The identification of the presence/absence of these genetic alterations and the cytogenetic data allow not only to perform the risk stratification and to better define the prognosis but also to facilitate the best therapeutic choice [156,157,158,159,160,161].
A new biomarker has recently been identified that has made an important contribution to the prognostic stratification of patients with AML. An increased expression of HMGA2 is found in approximately 22% of patients and in 60% of patients with a mutation related to an unfavorable prognosis. High expression levels of HMGA2 in patients with AML are related to a lower frequency of complete remission (about 59% vs. 83%), relapse-free survival (about 11% vs. 44%), and a poor 3-year overall survival (OS) (13.2% vs. 43%) [162].
Nyquist et al. [163] showed that t(12;13)(q14;q31) is associated with an upregulation of HMGA2 in AMLs, but the role played by the altered HMGA2 expression in the AML mechanism remains unknown. In a 2016 study, Tan et al. aimed to define this relationship by studying the expression of the gene and its biological function in the blasts of patients with de novo AML and leukemic cell lines. Comparing mRNAs levels of HMGA2, they observed significantly higher amounts in the patient samples and cell lines than in normal cells; they also noted that the majority of patients who did not achieve remission had increased HMGA2 mRNA and protein expression [164].
There are several signaling pathways that are known to be protagonists in the progression of leukemias, such as the PI3K/Akt/mTOR, Wnt/β-catenin, JAK/STAT, and NF-κB pathways [165,166,167,168]. The PI3K/Akt/mTOR signal pathway appears to play an important role in the proliferation effect mediated by HMGA2 [164]; this signal pathway is constitutively active in various types of cancer and is crucial in regulating the growth of malignant cells [169]. It is known that the PI3K/Akt/mTOR pathway plays a fundamental role in a vast number of different physiological processes such as cell cycle progression, transcription, translation, metabolism, differentiation, and apoptosis, all of which are reflected on growth, proliferation, survival, and tumorigenesis [170]. The altered expression of HMGA2 in AMLs is coherent with the already known oncogenic role of mTOR and Akt in leukemias. In fact, data show that the reduced expression of HMGA2 inhibits the PI3K/Akt signaling pathway and therefore inhibits cell proliferation through the downregulation of mTOR expression. It is therefore deduced that the PI3K/Akt/mTOR pathway is closely associated with the expression of HMGA2 [164]. Tan et al. demonstrated, in vivo and in vitro, the oncogenic role of HMGA2 in AML since its downregulation hinders proliferation in AML cell lines [164].
Several studies have revealed the involvement of HMGA2 in different signaling pathways [9,164,171], but Yang et al. [172] were the first to observe the effect of HMGA2 expression on the WNT/β-catenin pathway in AML, while this effect had already been observed in other types of tumors [173,174]. Yang at al. used plasmids that overexpressed or silenced HMGA2 in different cell lines, showing that silencing of HMGA2 inhibits Wnt and the levels of active β-catenin, reducing cell viability, invasion, and migration and improving cell apoptosis. The overexpression of HMGA2 elicited inverse results. Furthermore, in this scenario, the X-linked inhibitor of apoptosis and Bcl-2 mRNA and protein levels are mitigated while Bax and cleaved-caspase-3 are overexpressed [172].
Daunorubicin (DNR) is one of the chemotherapy drugs used in the first line. DNR is an anthracycline-based chemotherapy with potent antitumor characteristics [175], but some patients exhibit drug resistance [176]. Yang at al. showed that inhibition of AML cells by DNR improved with HMGA2 silencing and that overexpression causes an adverse effect [172].
The expression of HMGA2 was observed in CD34 + stem cells from healthy donors and in PB from AML patients, while it was not observed in normal blood samples. The high expression of HMGA2 is related to the non-differentiated state of the leukemic cells [136,177]. Furthermore, there are data showing that the altered expression of HMGA2 has a main role in malignant evolution by directly inducing the transformation of myeloid cells [64]. Several papers have fueled the suspicion that the deregulation of HMGA2 expression is sufficient to interfere with the differentiation mechanism of cells in AML [178,179]. Indeed, in experiments involving HMGA2 silencing, the induction of the monocytic-granulocytic differentiation mechanism in leukemic cells was observed. Although this correlation has been observed [171], the mechanism through which it occurs has not yet been clarified.
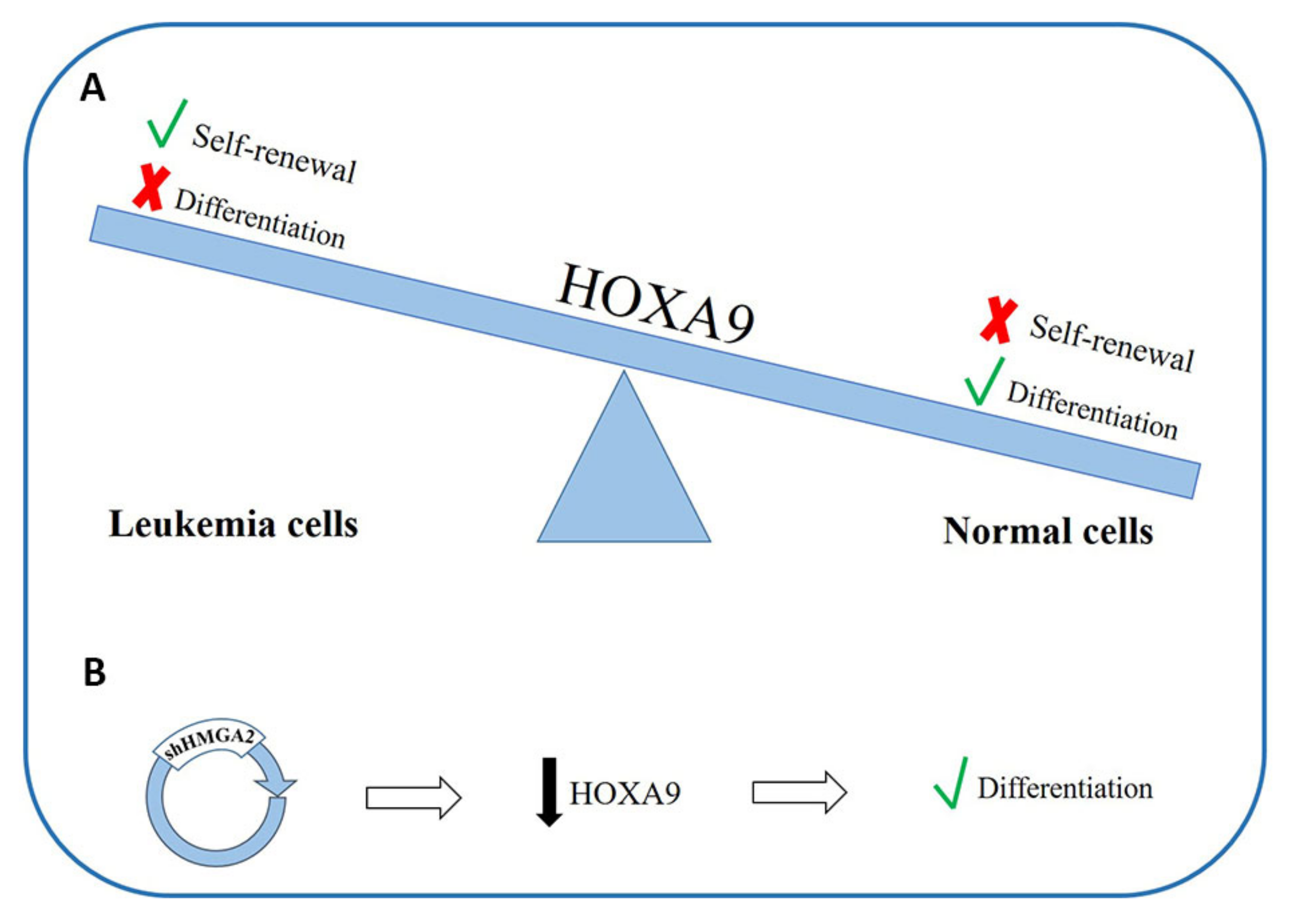
There are data showing that Homeobox A9 (HOXA9) downregulation is fundamental for normal myeloid differentiation [180] (Figure 3A); moreover, its overexpression is able to preserve the self-renewal capacity of leukemic stem cells and to block the differentiation processes leading to leukemogenesis. Different authors have explored the possible role of HOXA9 in the pathogenesis of AML [178,179]. Furthermore, in a 2013 work, it was observed that HMGA2 can regulate HOXA9 in the growth stage of breast cancer and the formation of metastases [181]. In about 70% of patients with AML, HOXA9 is overexpressed, probably due to the important differentiation block that occurs in leukemic cells [182,183]. The Tan group, by means of experiments on cell lines, observed an inhibition of HOXA9 expression in HMGA2 knockdown cells, while the downregulation of HOXA9 supports differentiation in cells with silenced HMGA2 [171] (Figure 3B). These data suggest that HOXA9 and HMGA2 both play an important role in the differentiation process in myeloid cells and that HOXA9 could be a new target for HMGA2 regulation [171]. Given the known ability of HMGA2 to modulate the expression of genes through direct interactions with DNA or by interacting with different transcription factors, it is possible that HMGA2 can influence the expression of HOXA9 by interacting directly with the promoter of the latter or indirectly through other mediators by inducing a cascade of signals.
An interesting work has further highlighted the oncogenic role of HMGA2 in AML but, at the same time, shown that it to be an important modulator of the disease. The study was based on the profiling of miRNAs expression and was conducted in a de novo pediatric AML cohort. It revealed that four miRNAs were downregulated in AML with inv(16), t(8;21), or mixed lineage leukemia (MLL) rearrangements. By inducing the forced expression of one of these miRNA (miR-9), a reduction in leukemic growth and the initiation of monocytic differentiation in AML t(8;21) cells was observed, both in vivo and in vitro. The suppressive role of miR-9 is due to the collaboration with let-7 in order to inhibit the oncogenic LIN28B/HMGA2 axis [184]. LIN28B belongs to a group of factors that can induce somatic reprogramming of cells into pluripotent stem cells [185] and is overexpressed in different types of cancer [186]. This protein selectively represses the expression of the let-7 miRNA [187,188,189,190]. Let-7 miRNAs are tumor suppressors that perform their function by repressing oncogenes such as MYC, RAS [78], and HMGA2; many tumors have a downregulation of these miRNAs [191]. The concerted action of let-7 and miR-9 allows a greater knockdown effect of HMGA2, unlocking the differentiation process and blocking uncontrolled growth. These data may open up the possibility of using miR-9 regulation as a therapeutic option in AML t(8;21) [184].
Gerritsen et al. subdivided AML patients on the basis of two recurrent genetic alterations, the RUNX1 mutation and t(8;21), and observed that, as compared to t(8;21), the RUNX1 mutation is able to initiate a specific transcriptional program that provides an important contribution to leukemic transformation. Among the genes showing a differential expression are MEIS1 [192], TCF4 [193], and HMGA2 [194], already described as upregulated in leukemogenesis and that can potentially contribute to the clinical differences between patients who have the RUNX1 mutation and those with t(8;21) [195].
The studies that followed have in recent years allowed us to consider HMGA2 as a promising molecular marker for the diagnosis of AML [172]; moreover, intervening in the balance of different signal pathways, it can be an important modulator of the main cellular mechanisms and, therefore, a potential therapeutic target in the treatment of AML [164,171,172]. A recent study conducted in the Canadian population showed that using a prognostic test based on the expression of HMGA2 allows a better stratification of AML patients in different risk groups. This can reduce unnecessary treatments and can allow early scheduling of patients in first remission for allogeneic transplant, improving AML patients’ leukemia-free survival and OS. All this not only leads to economic savings but also above all facilitates better patient management and disease outcomes [196].
5. HMGAs Involvement in Lymphoid Malignancies
Acute lymphoblastic leukemia (ALL) is the main cause of cancer death in children and adults and the most frequent lymphoid malignancy in children, with B-cell ALL (B-ALL) accounting for 25% of all childhood cancers [197]. T-cell ALL (T-ALL), instead, accounts for 15% of all ALL and is a very aggressive leukemia featuring the rapid accumulation of T lymphocytes precursors, with a higher risk of relapse as compared to B-ALL [198]. Unluckily, existing stratification risk criteria are narrow and are based primarily on clinical characteristics and underlying genetic lesions [199]. Unlike B-lineage ALL, no risk stratification exists for T-ALL, but a few possible prognostic factors do. Many recurrent genetic alterations have been discovered as oncogene activators on the basis of mutation or overexpression [200]; one example is the activation of NOTCH1 mutations, identified in almost 50% of human T-ALLs [201]. Considering the number of relapses, clarifying the molecular basis of refractory disease is an urgent need in order to promptly recognize patients destined to relapse and to schedule them for more intensive chemotherapeutic treatments.
Several studies examined the HMGAs’ role in lymphoid disease pathogenesis.
Both in the mouse transgenic model and in human T- and B-ALLs, HMGA1a is expressed at levels two- to 10-fold above the normal T- and B- cells [46]. Misexpression of the human HMGA1b isoform in mouse led to the development of a T-cell leukemia with a natural killer (NK) phenotype [47]. Indeed, mice with a complete absence of HMGA1 showed decreased numbers of T-cells and B-cell and myeloid expansion and in some cases developed B-cell leukemic transformation and B-cell lymphomas [202]. In these mice, spleen tissues revealed an increased expression of the Recombination Activating Gene 1/2 (RAG1/2) endonuclease that might be responsible for the high rate of abnormal IGH rearrangements observed in these neoplasias [202].
As regards the interaction of HMGA proteins with NOTCH1 signaling, studies on mouse thymic lymphomas detected both the overexpression of HMGA1 and the activation of NOTCH1 signaling [203]. HMGA1 is a downstream target of Notch1 signaling, as shown by the presence of two NOTCH1/RBPJ cobinding sites of T/CTCCCACA in HMGA1 promoter regions and the consequent direct regulation of HMGA1 transcription by NOTCH1. Indeed, in human T leukemia cells, knockdown of HMGA1 provoked a significantly impaired cell growth and decreased expression of cyclin D and cyclin E. The observation of complexes between HMGA1 and retinoblastoma (RB) protein indicated the involvement of HMGA1 in cell cycle regulation and proliferation through NOTCH1 signaling, corroborating its leading role in leukemogenesis [203].
T-ALL frequently shows deletion within the CDKN2A/2B tumor suppressor locus that has been found in more than 70% of tested T-ALL samples [204]. As transgenic mice overexpressing HMGA1 develop aggressive T-ALL [205], the group by Di Cello et al. aimed to understand whether CDKN2A/2B genomic loss cooperates with HMGA1 in T-ALL development [65]. They reported that complete loss of CDKN2A accelerated leukemogenesis in HMGA1 transgenic mice that showed marked splenomegaly and significantly decreased OS compared with HMGA1 transgenic/CDKN2A WT mice. HMGA1a transgenic/CDKN2A null leukemias presented a T-cell origin immunophenotype with the expression of Th y 1.2, CD3, CD8, and α β T-Cell Receptor (TCR); a markedly accelerated development; and disease recapitulating the salient clinical and pathologic features of human T-ALL. Exploration of the publicly available databases showed that HMGA1 is one of the genes most frequently overexpressed in pediatric T-ALL compared with control BM samples, supporting the speculation that HMGA1 could play a causative role in T-ALL and could thus be a potential therapeutic target [65].
A recent study explored the correlation of HMGA1 expression with relapse in pediatric ALL [206]. Investigation of gene expression profiles generated from leukemic blasts from 86 children with relapsed B-ALL showed a significant increase in the mean expression of HMGA1 at relapse. Further consultation of published databases evidenced HMGA1 among the top 11% of genes as compared to B-cell progenitors in a prior study of pediatric B-ALL [207] and in the top 21% of overexpressed genes as compared to normal BM in another pediatric B-ALL study [208]. In this latter study, a high HMGA1 expression was noted in B-ALL samples, with a translocation in cMYC Proto-Oncogene (cMYC) t(8;14). HMGA1 is a cMYC transcriptional target [62], and HMGA1 high expression is maybe linked to cMYC translocations. As HMGA1 has a pivotal role in ES cells and cellular reprogramming [209] and has been identified as a leukemic stem cell signature in a murine model of AML [30], it could trigger refractory disease by inducing stem-like transcriptional programs [206].
A study on T-ALL with t(5;14)(q35;q32.2) highlights the involvement of HMGA1 in the mechanism of dysregulation of TLX3 or NKX2-5 homeobox genes as a consequence of 5q35 juxtaposition with 14q32.2 breakpoints dispersed across the BCL11B downstream genomic desert [210]. In vitro experiments with DNA inhibitory treatment in t(5;14) cells showed the presence of enhancers with multiple regulatory stigmata about 1 Mbp downstream of BCL11B. Further knockdown of either HMGA1 or PU.1 inhibited ectopic NKX2-5 and TLX3 expression, highlighting their implication in leukemic gene deregulation in t(5;14), likely through the generation of a putative t(5;14) enhanceosome in which HMGA1 is bound near enhancers, and PU.1 to promoters of homeobox genes [210]. As t(5;14) T-ALL is a major clinical entity, this information could be important to identify potential therapeutic targets such as DNA inhibitory treatments and histone deacetylase inhibition [210].
The HMGA1-STAT3 pathway also seems to play a crucial role in lymphoid neoplasias, such as T-ALL and Burkitts lymphoma, an aggressive lymphoid disease involving more mature B cells. HMGA1 regulates the expression of various tumor progression driver genes, such as STAT3, a signal transducer and activator of transcription [58,211] for which expression is associated to a refractory status in diverse tumors [212]. HMGA1 induces STAT3 expression that has a main role in inflammation, malignant transformation, and tumor progression [3,58,213,214]; STAT3 overexpression also leads to hematologic malignancies [3,58]. In vitro blocking of STAT3 function induces apoptosis in T-ALL cells derived from a HMGA1 transgenic model [58]. Indeed, the inhibition of HMGA1 or STAT3 function prevents Burkitt leukemia cells colony formation [58]. Blocking STAT3 function could be a new therapeutic approach in T-cell lymphoid tumors driven, at least in part, by HMGA1 altered expression, considering that HMGA1 has been demonstrated to drive leukemic transformation and refractory disease by inducing STAT3 and a stem-like transcriptional network [211] (Figure 4A).
As regards HMGA2, a cytogenetic analysis of abnormalities at the 12q12-q14 chromosomal locus conducted in 78 adult B- and T-cell ALL cases showed that, in 26% of cases, a submicroscopic deletion was present at the 12q14.3 locus targeting the region containing the HMGA2 gene [215].
Indeed, HMGA2 physiological function can also be indirectly altered. miRNA let-7b is frequently downregulated in KMT2A-rearranged ALL as a consequence of DNA hypermethylation of its promoter region, similar to what happens with the CDKN2A gene [216,217]. As a consequence, let-7b-regulated target genes are upregulated, such as HMGA2, that is normally negatively regulated by let-7b [74,79]. In 2015, Wu et al. demonstrated that, in infant leukemic cells, particularly in those positive to KMT2A-AFF1 (MLL-AF4), there was upregulation of HMGA2. As HMGA2 is a negative regulator of CDKN2A gene, a series of in vitro studies on KMT2A-AFF1-expressing cell lines showed that the use of the HMGA2 inhibitor netropsin in addition to the demethylating agent 5-azacytidine upregulated and sustained the expression of CDKN2A, resulting in a higher growth suppression as compared to treatment with 5-azacytidine alone (Figure 4B). These data highlighted that the let-7b-HMGA2-CDKN2A axis regulates cell proliferation of leukaemic cells and could be a possible molecular target for the treatment of infant ALL with KMT2A-AFF1 [218].
Supported by the findings regarding the key role of HMGA proteins in several important lymphoid pathways, it seems reasonable to suppose that, in the next years, HMGAs may be used as prognostic and/or targetable markers, even though further studies will be needed to corroborate the current evidence and to fully exploit them in clinical practice.
6. HMGA Proteins as Targetable Markers
In view of their role in carcinogenesis, HMGA proteins could be suitable candidates for molecular therapy against HMGA-overexpressing cancers. As they bind AT-specific minor grooves of DNA, their binding could be inhibited by other DNA minor groove binders/ligands, such as distamycin and netropsin [20,219], that have been studied as potential new therapies for several types of human cancer [2,220].
The search for HMGA1 inhibitors is ongoing. Despite toxicity issues that still need to be addressed, promising preliminary data have been obtained for flavopiridol and FR900482 [4,221].
A series of studies explored the use of STAT3 as a targetable marker in aggressive lymphoid malignancies overexpressing HMGA1. Nanoparticle delivery of STAT3 with G-quartet oligodeoxynucleotides that specifically inhibit STAT3 binding to DNA has been shown to have antileukemia effects in an HMGA1 transgenic model of aggressive T-ALL [211]. Indeed, BP-1-102, a salicylic acid-based inhibitor, was tested in preclinical models of ALL and Burkitts lymphoma with STAT3 activation [212], demonstrating that BP-1-102 inhibits STAT3 phosphorylation at tyrosine 705, preventing STAT3 dimerization, DNA binding, and the activation of downstream genes. It also decreases nuclear and cytoplasmic phosphorylated STAT3. Further treatment of cultured cells derived from B-lymphoid tumors previously shown to express high levels of HMGA1 and STAT3, including B-ALL, Burkitts leukemia, and T-ALL cells, showed a decreased proliferation [222]. However, there was no antitumor efficacy in murine xenograft models; the reason for this is still uncertain and may lie either in the limited exposure to the drug or in the induction of oncogenic pathways alternative to the one inhibited by BP-1-102 [222]. These experiments also helped to show that BP-1-102 treatment represses HMGA1 [222], highlighting that not only does HMGA1 induce STAT3 expression [58] but even STAT3 feeds forward to upregulate HMGA1, leading to an enhanced expression of both genes during tumor progression. In fact, a putative STAT3 consensus DNA binding site (TTN5AA) was found in the HMGA1 promoter region; it is conserved in humans and mice and could mediate STAT3 dimer binding and transactivation. In conjunction with direct transactivation, STAT3 could induce downstream factors that upregulate HMGA1 expression [222].
Furthermore, Bortezomib (BTZ) also seems to influence HMGA1 levels. This is a proteasome inhibitor used to suppress Diffuse large B-cell lymphoma (DLBCL) progression [223,224,225]. DLBCL is the most frequent non-Hodgkin lymphoma, showing variable clinical presentations as well as variable therapy responses [226,227,228]. In vitro studies on DLBCL CRL-2630 cells showed that BTZ treatment significantly inhibited the proliferation of DLBCL CRL-2630 cells [229]. After studies showing that exposure to BTZ induced an upregulation of miRNA 198 (miR-198) while depletion of the miR significantly reversed the inhibitory effect of BTZ on cell proliferation [230], further studies on miR-198 revealed the 3′-UTR of HMGA1 as a downstream target of miR-198, suppressing the expression of HMGA1 in DLBCL cells. This evidence links HMGA1 to BTZ treatment that decreased the level of HMAG1 and inhibited the migration of DLBCL cells [229,231,232,233].
Indeed, a recent screening of about 36,000 compounds indicated a class of phosphodiesterase inhibitors that help to suppress let-7 targets [234]. These potential drugs augment cAMP levels and elevate let-7 levels, inhibiting let-7 target genes such as HMGA2 and MYC and decreasing growth in multiple cancer cell lines [234].
In the treatment of CML patients, the introduction of specific tyrosine kinase inhibitors (TKI) has promoted great progress in patient management [235,236]. Several TKIs are currently enrolled in common practice (such as imatinib, nilotinib, and dasatinib), the use of which is strictly related to adverse events and to the observation of resistance phenomena developing during therapy [237,238]. These issues increase the need to devise alternative therapies; in recent years, studies on the use of epigenetic therapy associated with other therapies have seemed to offer valid therapeutic tools [239]. In this regard, the Vitkeviciene team investigated the potential role of two epigenetic modulators, EGCG (epigallocatechin-3-gallate) and BIX-01294 (1-benzylpiperidin-4-yl)-6,7-dimethoxy-2-(4-methyl-1,4-diazepan-1-yl) quinazolin-4-amine) in two AML and CML cell lines [240].
EGCG is a naturally occurring catechin in green tea. This molecule is known to have many functions, including antibacterial, antioxidant, and anti-inflammatory actions, but it also seems to have a potential antineoplastic role, inhibiting proliferation and inducing apoptosis in different tumors [241,242]. BIX-01294 is a synthetic molecule that allows the specific inhibition of EHMT2/G9a histone methyltransferase. EHMT2/G9a is an important protagonist of gene silencing mechanisms [243,244]. A study in vitro showed that EGCG and BIX-01294, acting as epigenetic modulators, cause cell cycle arrest in the G0/G1 phase; the lines treated with EGCG also showed an increased level of ATM, HMGA2, phosphorylation of ATM, and Senescence-associated (SA)-β-galactosidase staining, for which concerted action favors the cellular senescence phenomenon [240]. Although apoptosis as compared to the induction of senescence is believed to be more effective in the treatment of cancer, the use of substances capable of inducing senescence offers a great advantage in terms of therapy planning [245].
7. Conclusions
The studies conducted so far characterize HMGA proteins as master regulators of tumor progression, refractory disease, and cancer stem cell properties, providing strong evidence about their crucial function in malignant hematopoiesis.
The goal in the coming years could be to discover whether and how best this knowledge could be exploited in routine procedures now that high throughput genomic and transcriptomic technologies are available.
Indeed, the significance of a tailored therapy against tumors overexpressing HMGAs needs to be established in order to understand whether the differential expression of these proteins could be used in personalized therapeutic strategies, since several specific drugs for HMGA-involved pathways are available and others are being tested. Because HMGA proteins control gene transcription by remodeling chromatin and recruiting transcription factor complexes, an alternative approach to block their function could be by targeting downstream pathways induced by HMGAs. The differential function and involvement of HMGA proteins in tumor and normal cells could give such targeted therapy an advantage in terms of specificity and low toxicity.
Finally, the finding of HMGA1 involvement in reprogramming of somatic cells to fully pluripotent cells paves the way for its use in regenerative medicine to reprogram normal cells to stem-like cells [209].
Today, all these issues remain topics open to close research in this field, which will certainly need to continue, bearing in mind the potential of these proteins as personalized medicine targets.
Author Contributions
A.M., N.C., L.A., A.Z., G.S. and F.A. conceived and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to thank Mary Victoria Pragnell, B.A. for language revision of the manuscript. This work was supported by the “Associazione Italiana contro le Leucemie (AIL)-BARI”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Reeves, R. Molecular biology of HMGA proteins: Hubs of nuclear function. Gene 2001, 277, 63–81. [Google Scholar] [CrossRef]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Resar, L.M.S. The high mobility group A1 gene: Transforming inflammatory signals into cancer? Cancer Res. 2010, 70, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.N.; Resar, L.M.S. High mobility group A1 and cancer: Potential biomarker and therapeutic target. Histol. Histopathol. 2012, 27, 567–579. [Google Scholar]
- Friedmann, M.; Holth, L.T.; Zoghbi, H.Y.; Reeves, R. Organization, inducible-expression and chromosome localization of the human HMG-I(Y) nonhistone protein gene. Nucleic Acids Res. 1993, 21, 4259–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleynen, I.; Van de Ven, W.J.M. The HMGA proteins: A myriad of functions (Review). Int. J. Oncol. 2008, 32, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, S.; Ghosn, C.; DiSepio, D.; Molina, Y.; Sutter, M.; Klein, E.S.; Chandraratna, R.A. Retinoid-dependent recruitment of a histone H1 displacement activity by retinoic acid receptor. J. Biol. Chem. 1999, 274, 22563–22568. [Google Scholar] [CrossRef] [Green Version]
- Cesana, M.; Guo, M.H.; Cacchiarelli, D.; Wahlster, L.; Barragan, J.; Doulatov, S.; Vo, L.T.; Salvatori, B.; Trapnell, C.; Clement, K.; et al. A CLK3-HMGA2 Alternative Splicing Axis Impacts Human Hematopoietic Stem Cell Molecular Identity throughout Development. Cell Stem Cell 2018, 22, 575–588.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Mo, Q.; Wang, X. Oncological role of HMGA2 (Review). Int. J. Oncol. 2019, 55, 775–778. [Google Scholar] [CrossRef]
- Reeves, R.; Wolffe, A.P. Substrate structure influences binding of the non-histone protein HMG-I(Y) to free and nucleosomal DNA. Biochemistry 1996, 35, 5063–5074. [Google Scholar] [CrossRef]
- Nissen, M.S.; Reeves, R. Changes in superhelicity are introduced into closed circular DNA by binding of high mobility group protein I/Y. J. Biol. Chem. 1995, 270, 4355–4360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, D.A.; Pedulla, M.L.; Reeves, R. Directional binding of HMG-I(Y) on four-way junction DNA and the molecular basis for competitive binding with HMG-1 and histone H1. Nucleic Acids Res. 1999, 27, 2135–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, R.; Beckerbauer, L. HMGI/Y proteins: Flexible regulators of transcription and chromatin structure. Biochim. Biophys. Acta Gene Struct. Expr. 2001, 1519, 13–29. [Google Scholar] [CrossRef]
- Frank, O.; Schwanbeck, R.; Wiśniewski, J.R. Protein footprinting reveals specific binding modes of a high mobility group protein I to DNAs of different conformation. J. Biol. Chem. 1998, 273, 20015–20020. [Google Scholar] [CrossRef] [Green Version]
- Thanos, D.; Maniatis, T. Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 1995, 83, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Thanos, D.; Du, W.; Maniatis, T. The high mobility group protein HMG I(Y) is an essential structural component of a virus-inducible enhancer complex. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1993; Volume 58, pp. 73–81. [Google Scholar] [CrossRef] [PubMed]
- Thanos, D.; Maniatis, T. The High Mobility Group protein HMG I(Y) is required for NF-κB-dependent virus induction of the human IFN-β gene. Cell 1992, 71, 777–789. [Google Scholar] [CrossRef]
- Wolffe, A.P. Architectural transcription factors. Science 1994, 264, 1100–1101. [Google Scholar] [CrossRef]
- Carey, M. The enhanceosome and transcriptional synergy. Cell 1998, 92, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Radic, M.Z.; Saghbini, M.; Elton, T.S.; Reeves, R.; Hamkalo, B.A. Hoechst 33258, distamycin A, and high mobility group protein I (HMG-I) compete for binding to mouse satellite DNA. Chromosoma 1992, 101, 602–608. [Google Scholar] [CrossRef]
- Käs, E.; Poljak, L.; Adachi, Y.; Laemmli, U.K. A model for chromatin opening: Stimulation of topoisomerase II and restriction enzyme cleavage of chromatin by distamycin. EMBO J. 1993, 12, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, G.; Battista, S.; Belletti, B.; Thakur, S.; Pentimalli, F.; Trapasso, F.; Fedele, M.; Pierantoni, G.; Croce, C.M.; Fusco, A. Negative Regulation of BRCA1 Gene Expression by HMGA1 Proteins Accounts for the Reduced BRCA1 Protein Levels in Sporadic Breast Carcinoma. Mol. Cell. Biol. 2003, 23, 2225–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, S.M.; Sharpless, N.E. HMGA2, MicroRNAs, and Stem Cell Aging. Cell 2008, 135, 1013–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, J.; Kim, I.; Chada, K.; Morrison, S.J. Hmga2 promotes neural stem cell self-renewal in young but not olNishino, J., Kim, I., Chada, K., & Morrison, S. J. (2008). Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell 2008, 135, 227–239. [Google Scholar]
- Tzatsos, A.; Bardeesy, N. Ink4a/Arf regulation by let-7b and Hmga2: A genetic pathway governing stem cell aging. Cell Stem Cell 2008, 3, 469–470. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Benson, K.F.; Przybysz, K.; Liu, J.; Hou, Y.; Cherath, L.; Chada, K. Genomic structure and expression of the murine Hmgi-c gene. Nucleic Acids Res. 1996, 24, 4071–4077. [Google Scholar] [CrossRef]
- Rogalla, P.; Drechsler, K.; Frey, G.; Hennig, Y.; Helmke, B.; Bonk, U.; Bullerdiek, J. HMGI-C expression patterns in human tissues. Implications for the genesis of frequent mesenchymal tumors. Am. J. Pathol. 1996, 149, 775–779. [Google Scholar]
- Rommel, B.; Rogalla, P.; Jox, A.; Kalle, C.V.; Kazmierczak, B.; Wolf, J.; Bullerdiek, J. HMGI-C, a member of the high mobility group family of proteins, is expressed in hematopoietic stem cells and in leukemic cells. Leuk. Lymphoma 1997, 26, 603–607. [Google Scholar] [CrossRef]
- Gattas, G.J.; Quade, B.J.; Nowak, R.A.; Morton, C.C. HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes. Chromosomes Cancer 1999, 25, 316–322. [Google Scholar] [CrossRef]
- Somervaille, T.C.P.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Chiappetta, G.; Avantaggiato, V.; Visconti, R.; Fedele, M.; Battista, S.; Trapasso, F.; Merciai, B.M.; Fidanza, V.; Giancotti, V.; Santoro, M.; et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996, 13, 2439–2446. [Google Scholar] [PubMed]
- Zhou, X.; Benson, K.F.; Ashar, H.R.; Chada, K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 1995, 376, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Battista, S.; Fidanza, V.; Fedele, M.; Klein-Szanto, A.J.; Outwater, E.; Brunner, H.; Santoro, M.; Croce, C.M.; Fusco, A. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999, 59, 4793–4797. [Google Scholar] [PubMed]
- Melillo, R.M.; Pierantoni, G.M.; Scala, S.; Battista, S.; Fedele, M.; Stella, A.; De Biasio, M.C.; Chiappetta, G.; Fidanza, V.; Condorelli, G.; et al. Critical Role of the HMGI(Y) Proteins in Adipocytic Cell Growth and Differentiation. Mol. Cell. Biol. 2001, 21, 2485–2495. [Google Scholar] [CrossRef] [Green Version]
- Tallini, G.; Dal Cin, P. HMGI(Y) and HMGI-C dysregulation: A common occurrence in human tumors. Adv. Anat. Pathol. 1999, 6, 237–246. [Google Scholar] [CrossRef]
- Harada-Shirado, K.; Ikeda, K.; Ogawa, K.; Ohkawara, H.; Kimura, H.; Kai, T.; Noji, H.; Morishita, S.; Komatsu, N.; Takeishi, Y. Dysregulation of the MIRLET7/HMGA2 axis with methylation of the CDKN2A promoter in myeloproliferative neoplasms. Br. J. Haematol. 2015, 168, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.L.; Chen, C.C.; Chang, L.C. Gene expressions of HMGI-C and HMGI(Y) are associated with stage and metastasis in colorectal cancer. Int. J. Colorectal Dis. 2009, 24, 1281–1286. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Li, A.Y.-J.; Chen, L.; Lai, L.; Lin, H.H.; Hu, S.; Yao, L.; Peng, J.; Loera, S.; et al. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clin. Cancer Res. 2011, 17, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Pallante, P.; Sepe, R.; Puca, F.; Fusco, A. High mobility group a proteins as tumor markers. Front. Med. 2015, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Berlingieri, M.T.; Pierantoni, G.M.; Giancotti, V.; Santoro, M.; Fusco, A. Thyroid cell transformation requires the expression of the HMGA1 proteins. Oncogene 2002, 21, 2971–2980. [Google Scholar] [CrossRef] [Green Version]
- Scala, S.; Portella, G.; Fedele, M.; Chiappetta, G.; Fusco, A. Adenovirus-mediated suppression of HMGI(Y) protein synthesis as potential therapy of human malignant neoplasias. Proc. Natl. Acad. Sci. USA 2000, 97, 4256–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlingieri, M.T.; Manfioletti, G.; Santoro, M.; Bandiera, A.; Visconti, R.; Giancotti, V.; Fusco, A. Inhibition of HMGI-C protein synthesis suppresses retrovirally induced neoplastic transformation of rat thyroid cells. Mol. Cell. Biol. 1995, 15, 1545–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, M.; Berlingieri, M.T.; Scala, S.; Chiariotti, L.; Viglietto, G.; Rippel, V.; Bullerdiek, J.; Santoro, M.; Fusco, A. Truncated and chimeric HMGI-C genes induce neoplastic transformation of NIH3T3 murine fibroblasts. Oncogene 1998, 17, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, L.J.; Maher, J.F.; Bunton, T.E.; Resar, L.M. The oncogenic properties of the HMG-I gene family. Cancer Res. 2000, 60, 4256–4261. [Google Scholar]
- Baldassarre, G.; Fedele, M.; Battista, S.; Vecchione, A.; Klein-Szanto, A.J.P.; Santoro, M.; Waldmann, T.A.; Azimi, N.; Croce, C.M.; Fusco, A. Onset of natural killer cell lymphomas in transgenic mice carrying a truncated HMGI-C gene by the chronic stimulation of the IL-2 and IL-15 pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 7970–7975. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sumter, T.F.; Bhattacharya, R.; Tesfaye, A.; Fuchs, E.J.; Wood, L.J.; Huso, D.L.; Resar, L.M.S. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res. 2004, 64, 3371–3375. [Google Scholar] [CrossRef] [Green Version]
- Fedele, M.; Pentimalli, F.; Baldassarre, G.; Battista, S.; Klein-Szanto, A.J.P.; Kenyon, L.; Visone, R.; De Martino, I.; Ciarmiello, A.; Arra, C.; et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 2005, 24, 3427–3435. [Google Scholar] [CrossRef] [Green Version]
- Fedele, M.; Battista, S.; Kenyon, L.; Baldassarre, G.; Fidanza, V.; Klein-Szanto, A.J.P.; Parlow, A.F.; Visone, R.; Pierantoni, G.M.; Outwater, E.; et al. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene 2002, 21, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Chou, B.-K.; Mali, P.; Huang, X.; Ye, Z.; Dowey, S.N.; Resar, L.M.; Zou, C.; Zhang, Y.A.; Tong, J.; Cheng, L. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011, 21, 518–529. [Google Scholar] [CrossRef]
- Karp, J.E.; Smith, B.D.; Resar, L.S.; Greer, J.M.; Blackford, A.; Zhao, M.; Moton-Nelson, D.; Alino, K.; Levis, M.J.; Gore, S.D.; et al. Phase 1 and pharmacokinetic study of bolus-infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood 2011, 117, 3302–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.M.; Joseph, B.; Hillion, J.; Segal, J.; Karp, J.E.; Resar, L.M.S. Flavopiridol induces BCL-2 expression and represses oncogenic transcription factors in leukemic blasts from adults with refractory acute myeloid leukemia. Leuk. Lymphoma 2011, 52, 1999–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Chen, J.; Lee, S.; Clark, T.; Rowley, J.D.; Wang, S.M. The pattern of gene expression in human CD34(+) stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 13966–13971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolde, C.E.; Mukherjee, M.; Cho, C.; Resar, L.M.S. HMG-I/Y in human breast cancer cell lines. Breast Cancer Res. Treat. 2002, 71, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, A.; Di Cello, F.; Hillion, J.; Ronnett, B.M.; Elbahloul, O.; Ashfaq, R.; Dhara, S.; Prochownik, E.; Tworkoski, K.; Reeves, R.; et al. The high-mobility group A1 gene up-regulates cyclooxygenase 2 expression in uterine tumorigenesis. Cancer Res. 2007, 67, 3998–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cello, F.; Hillion, J.; Hristov, A.; Wood, L.J.; Mukherjee, M.; Schuldenfrei, A.; Kowalski, J.; Bhattacharya, R.; Ashfaq, R.; Resar, L.M.S. HMGA2 participates in transformation in human lung cancer. Mol. Cancer Res. 2008, 6, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Di Cello, F.; Hillion, J.; Kowalski, J.; Ronnett, B.M.; Aderinto, A.; Huso, D.L.; Resar, L.M.S. Cyclooxygenase inhibitors block uterine tumorigenesis in HMGA1a transgenic mice and human xenografts. Mol. Cancer Ther. 2008, 7, 2090–2095. [Google Scholar] [CrossRef] [Green Version]
- Hillion, J.; Dhara, S.; Sumter, T.F.; Mukherjee, M.; Di Cello, F.; Belton, A.; Turkson, J.; Jaganathan, S.; Cheng, L.; Ye, Z.; et al. The high-mobility group A1a/signal transducer and activator of transcription-3 axis: An achilles heel for hematopoietic malignancies? Cancer Res. 2008, 68, 10121–10127. [Google Scholar] [CrossRef] [Green Version]
- Hristov, A.C.; Cope, L.; Di Cello, F.; Reyes, M.D.; Singh, M.; Hillion, J.A.; Belton, A.; Joseph, B.; Schuldenfrei, A.; Iacobuzio-Donahue, C.A.; et al. HMGA1 correlates with advanced tumor grade and decreased survival in pancreatic ductal adenocarcinoma. Mod. Pathol. 2010, 23, 98–104. [Google Scholar] [CrossRef]
- Belton, A.; Gabrovsky, A.; Bae, Y.K.; Reeves, R.; Iacobuzio-Donahue, C.; Huso, D.L.; Resar, L.M.S. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS ONE 2012, 7, e30034. [Google Scholar] [CrossRef] [Green Version]
- Pomeroy, S.L.; Tamayo, P.; Gaasenbeek, M.; Sturla, L.M.; Angelo, M.; McLaughlin, M.E.; Kim, J.Y.H.; Goumnerova, L.C.; Black, P.M.; Lau, C.; et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 2002, 415, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.J.; Mukherjee, M.; Dolde, C.E.; Xu, Y.; Maher, J.F.; Bunton, T.E.; Williams, J.B.; Resar, L.M. HMG-I/Y, a new c-Myc target gene and potential oncogene. Mol. Cell. Biol. 2000, 20, 5490–5502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuldenfrei, A.; Belton, A.; Kowalski, J.; Talbot, C.C.; Di Cello, F.; Poh, W.; Tsai, H.-L.; Shah, S.N.; Huso, T.H.; Huso, D.L.; et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics 2011, 12, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efanov, A.; Zanesi, N.; Coppola, V.; Nuovo, G.; Bolon, B.; Wernicle-Jameson, D.; Lagana, A.; Hansjuerg, A.; Pichiorri, F.; Croce, C.M. Human HMGA2 protein overexpressed in mice induces precursor T-cell lymphoblastic leukemia. Blood Cancer J. 2014, 4, e227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cello, F.; Dhara, S.; Hristov, A.C.; Kowalski, J.; Elbahloul, O.; Hillion, J.; Roy, S.; Meijerink, J.P.P.; Winter, S.S.; Larson, R.S.; et al. Inactivation of the Cdkn2a locus cooperates with HMGA1 to drive T-cell leukemogenesis. Leuk. Lymphoma 2013, 54, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Petit, M.M.R.; Mols, R.; Schoenmakers, E.F.P.M.; Mandahl, N.; Van De Ven, W.J.M. LPP, the preferred fusion partner gene of HMGIC in lipomas, is a novel member of the LIM protein gene family. Genomics 1996, 36, 118–129. [Google Scholar] [CrossRef]
- Schoenmakers, E.F.; Huysmans, C.; Van de Ven, W.J. Allelic knockout of novel splice variants of human recombination repair gene RAD51B in t(12;14) uterine leiomyomas. Cancer Res. 1999, 59, 19–23. [Google Scholar]
- Schoenmakers, E.F.; Wanschura, S.; Mols, R.; Bullerdiek, J.; Van den Berghe, H.; Van de Ven, W.J. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat. Genet. 1995, 10, 436–444. [Google Scholar] [CrossRef]
- Kazmierczak, B.; Meyer-Bolte, K.; Tran, K.H.; Wöckel, W.; Breightman, I.; Rosigkeit, J.; Bartnitzke, S.; Bullerdiek, J. A high frequency of tumors with rearrangements of genes of the HMGI(Y) family in a series of 191 pulmonary chondroid hamartomas. Genes. Chromosomes Cancer 1999, 26, 125–133. [Google Scholar] [CrossRef]
- Odero, M.D.; Grand, F.H.; Iqbal, S.; Ross, F.; Roman, J.P.; Vizmanos, J.L.; Andrieux, J.; Laï, J.L.; Calasanz, M.J.; Cross, N.C.P. Disruption and aberrant expression of HMGA2 as a consequence of diverse chromosomal translocations in myeloid malignancies. Leukemia 2005, 19, 245–252. [Google Scholar] [CrossRef]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Geurts, J.M.W.; Schoenmakers, E.F.P.M.; Van De Ven, W.J.M. Molecular characterization of a complex chromosomal rearrangement in a pleomorphic salivary gland adenoma involving the 3′-UTR of HMGIC. Cancer Genet. Cytogenet. 1997, 95, 198–205. [Google Scholar] [CrossRef]
- Inoue, N.; Izui-Sarumaru, T.; Murakami, Y.; Endo, Y.; Nishimura, J.I.; Kurokawa, K.; Kuwayama, M.; Shime, H.; Machii, T.; Kanakura, Y.; et al. Molecular basis of clonal expansion of hematopoiesis in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood 2006, 108, 4232–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene Email alerting service The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 1025–1030. [Google Scholar] [CrossRef] [Green Version]
- Hebert, C.; Norris, K.; Scheper, M.A.; Nikitakis, N.; Sauk, J.J. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol. Cancer 2007, 6. [Google Scholar] [CrossRef] [Green Version]
- Baba, O.; Horie, T.; Nakao, T.; Hakuno, D.; Nakashima, Y.; Nishi, H.; Kuwabara, Y.; Nishiga, M.; Nishino, T.; Ide, Y.; et al. MicroRNA 33 Regulates the Population of Peripheral Inflammatory Ly6C high Monocytes through Dual Pathways. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquienelll, A.E.; Bettlnger, J.C.; Rougvle, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, X.; Obijuru, L.; Laser, J.; Aris, V.; Lee, P.; Mittal, K.; Soteropoulos, P.; Wei, J.-J. A micro-RNA signature associated with race, tumor size, and target gene activity in human uterine leiomyomas. Genes. Chromosomes Cancer 2007, 46, 336–347. [Google Scholar] [CrossRef]
- Grosveld, F.; van Assendelft, G.B.; Greaves, D.R.; Kollias, G. Position-independent, high-level expression of the human β-globin gene in transgenic mice. Cell 1987, 51, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Kottickal, L.V.; Sarada, B.; Ashar, H.; Chada, K.; Nagarajan, L. Preferential expression of HMGI-C isoforms lacking the acidic carboxy terminal in human leukemia. Biochem. Biophys. Res. Commun. 1998, 242, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Santulli, B.; Kazmierczak, B.; Napolitano, R.; Caliendo, I.; Chiappetta, G.; Rippe, V.; Bullerdiek, J.; Fusco, A. A 12q13 translocation involving the HMGI-C gene in Richter transformation of a chronic lymphocytic leukemia. Cancer Genet. Cytogenet. 2000, 119, 70–73. [Google Scholar] [CrossRef]
- Cirera-Salinas, D.; Pauta, M.; Allen, R.M.; Salerno, A.G.; Ramírez, C.M.; Chamorro-Jorganes, A.; Wanschel, A.C.; Lasuncion, M.A.; Morales-Ruiz, M.; Suarez, Y.; et al. Mir-33 regulates cell proliferation and cell cycle progression. Cell Cycle 2012, 11, 922–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Lange-Grünweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Rice, S.J.; Lai, S.-C.; Wood, L.W.; Helsley, K.R.; Runkle, E.A.; Winslow, M.M.; Mu, D. MicroRNA-33a mediates the regulation of high mobility group AT-hook 2 gene (HMGA2) by thyroid transcription factor 1 (TTF-1/NKX2-1). J. Biol. Chem. 2013, 288, 16348–16360. [Google Scholar] [CrossRef] [Green Version]
- Wiemer, E.A.C. The role of microRNAs in cancer: No small matter. Eur. J. Cancer 2007, 43, 1529–1544. [Google Scholar] [CrossRef]
- Godley, L.A. HMGA2 levels in CML: Reflective of miRNA gene regulation in a hematopoietic tumor? Leuk. Lymphoma 2007, 48, 1898–1899. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef]
- Sgarra, R.; Pegoraro, S.; D’Angelo, D.; Ros, G.; Zanin, R.; Sgubin, M.; Petrosino, S.; Battista, S.; Manfioletti, A.G. High Mobility Group A (HMGA): Chromatin Nodes Controlled by a Knotty miRNA Network. Int. J. Mol. Sci. 2020, 21, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyerinas, B.; Park, S.-M.; Shomron, N.; Hedegaard, M.M.; Vinther, J.; Andersen, J.S.; Feig, C.; Xu, J.; Burge, C.B.; Peter, M.E. Identification of let-7-regulated oncofetal genes. Cancer Res. 2008, 68, 2587–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copley, M.R.; Babovic, S.; Benz, C.; Knapp, D.J.H.F.; Beer, P.A.; Kent, D.G.; Wohrer, S.; Treloar, D.Q.; Day, C.; Rowe, K.; et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol. 2013, 15, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Wang, G.P.; Berry, C.C.; Malani, N.; Leboulch, P.; Fischer, A.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M.; Bushman, F.D. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood 2010, 115, 4356–4366. [Google Scholar] [CrossRef]
- De Vasconcellos, J.F.; Lee, Y.T.; Byrnes, C.; Tumburu, L.; Rabel, A.; Miller, J.L. HMGA2 Moderately Increases Fetal Hemoglobin Expression in Human Adult Erythroblasts. PLoS ONE 2016, 11, e0166928. [Google Scholar] [CrossRef]
- De Vasconcellos, J.F.; Byrnes, C.; Lee, Y.T.; Allwardt, J.M.; Kaushal, M.; Rabel, A.; Miller, J.L. Tough decoy targeting of predominant let-7 miRNA species in adult human hematopoietic cells. J. Transl. Med. 2017, 15, 169. [Google Scholar] [CrossRef]
- Bluteau, O.; Langlois, T.; Rivera-Munoz, P.; Favale, F.; Rameau, P.; Meurice, G.; Dessen, P.; Solary, E.; Raslova, H.; Mercher, T.; et al. Developmental changes in human megakaryopoiesis. J. Thromb. Haemost. 2013, 11, 1730–1741. [Google Scholar] [CrossRef]
- Stolla, M.C.; Catherman, S.C.; Kingsley, P.D.; Rowe, R.G.; Koniski, A.D.; Fegan, K.; Vit, L.; McGrath, K.E.; Daley, G.Q.; Palis, J. Lin28b regulates age-dependent differences in murine platelet function. Blood Adv. 2019, 3, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Battista, S.; Pentimalli, F.; Baldassarre, G.; Fedele, M.; Fidanza, V.; Croce, C.M.; Fusco, A. Loss of Hmga1 gene function affects embryonic stem cell lympho-hematopoietic differentiation. FASEB J. 2003, 17, 1496–1498. [Google Scholar] [CrossRef]
- Pierantoni, G.M.; Agosti, V.; Fedele, M.; Bond, H.; Caliendo, I.; Chiappetta, G.; Lo Coco, F.; Pane, F.; Turco, M.C.; Morrone, G.; et al. High-mobility group A1 proteins are overexpressed in human leukaemias. Biochem. J. 2003, 372, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berent-Maoz, B.; Montecino-Rodriguez, E.; Fice, M.; Casero, D.; Seet, C.S.; Crooks, G.M.; Lowry, W.; Dorshkind, K. The expansion of thymopoiesis in neonatal mice is dependent on expression of High mobility group A 2 protein (Hmga2). PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Watanabe, S.; Hino, Y.; Sakamoto, C.; Nakatsu, Y.; Okada, S.; Nakao, M. Hmga1 is differentially expressed and mediates silencing of the CD4/CD8 loci in T cell lineages and leukemic cells. Cancer Sci. 2012, 103, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Nikolajczyk, B.S.; Nelsen, B.; Sen, R. Precise alignment of sites required for mu enhancer activation in B cells. Mol. Cell. Biol. 1996, 16, 4544–4554. [Google Scholar] [CrossRef] [Green Version]
- Nikolajczyk, B.S.; Cortes, M.; Feinman, R.; Sen, R. Combinatorial determinants of tissue-specific transcription in B cells and macrophages. Mol. Cell. Biol. 1997, 17, 3527–3535. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, T.K.; Moore, M.W.; Yancopoulos, G.D.; Suh, H.; Lutzker, S.; Selsing, E.; Alt, F.W. Recombination between immunoglobulin variable region gene segments is enhanced by transcription. Nature 1986, 324, 585–589. [Google Scholar] [CrossRef]
- Sakai, E.; Bottaro, A.; Davidson, L.; Sleckman, B.P.; Alt, F.W. Recombination and transcription of the endogenous Ig heavy chain locus is effected by the Ig heavy chain intronic enhancer core region in the absence of the matrix attachment regions. Proc. Natl. Acad. Sci. USA 1999, 96, 1526–1531. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.T.; Andreucci, A.; Nikolajczyk, B.S. PU. 1-mediated transcription is enhanced by HMG-I(Y)-dependent structural mechanisms. J. Biol. Chem. 2001, 276, 9550–9557. [Google Scholar] [CrossRef] [Green Version]
- Nagulapalli, S.; Pongubala, J.M.; Atchison, M.L. Multiple proteins physically interact with PU.1. Transcriptional synergy with NF-IL6 beta (C/EBP delta, CRP3). J. Immunol. 1995, 155, 4330–4338. [Google Scholar]
- McCarthy, K.M.; McDevit, D.; Andreucci, A.; Reeves, R.; Nikolajczyk, B.S. HMGA1 co-activates transcription in B cells through indirect association with DNA. J. Biol. Chem. 2003, 278, 42106–42114. [Google Scholar] [CrossRef] [Green Version]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Dameshek, W. Some speculations on the myeloproliferative syndromes [editorial]. Blood. 1951;6(4):372-375. Blood 2016, 127, 663. [Google Scholar]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar]
- Rowley, J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Chen, C.C.; You, J.Y.; Lung, J.; Huang, C.E.; Chen, Y.Y.; Leu, Y.W.; Ho, H.Y.; Li, C.P.; Lu, C.H.; Lee, K.D.; et al. Aberrant let7a/HMGA2 signaling activity with unique clinical phenotype in JAK2-mutated myeloproliferative neoplasms. Haematologica 2017, 102, 509–518. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Vainchenker, W.; Delhommeau, F.; Constantinescu, S.N.; Bernard, O.A. New mutations and pathogenesis of myeloproliferative neoplasms. Blood 2011, 118, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Aliano, S.; Cirmena, G.; Garuti, A.; Fugazza, G.; Bruzzone, R.; Rocco, I.; Malacarne, M.; Ballestrero, A.; Sessarego, M. HMGA2 overexpression in polycythemia vera with t(12;21)(q14;q22). Cancer Genet. Cytogenet. 2007, 177, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Storlazzi, C.T.; Albano, F.; Locunsolo, C.; Lonoce, A.; Funes, S.; Guastadisegni, M.C.; Cimarosto, L.; Impera, L.; D’Addabbo, P.; Panagopoulos, I.; et al. t(3;12)(q26;q14) in polycythemia vera is associated with upregulation of the HMGA2 gene. Leukemia 2006, 20, 2190–2192. [Google Scholar] [CrossRef] [Green Version]
- Etienne, A.; Carbuccia, N.; Adélaïde, J.; Bekhouche, I.; Rémy, V.; Sohn, C.; Sainty, D.; Gastaut, J.A.; Olschwang, S.; Birnbaum, D.; et al. Rearrangements involving 12q in myeloproliferative disorders: Possible role of HMGA2 and SOCS2 genes. Cancer Genet. Cytogenet. 2007, 176, 80–88. [Google Scholar] [CrossRef]
- Andrieux, J.; Demory, J.L.; Dupriez, B.; Quief, S.; Plantier, I.; Roumier, C.; Bauters, F.; Laï, J.L.; Kerckaert, J.P. Dysregulation and Overexpression of HMGA2 in Myelofibrosis with Myeloid Metaplasia. Genes Chromosom. Cancer 2004, 39, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Zini, R.; Bogani, C.; Salati, S.; Pancrazzi, A.; Bianchi, E.; Mannelli, F.; Ferrari, S.; Le Bousse-Kerdilès, M.-C.; Bosi, A.; et al. Molecular Profiling of CD34 + Cells in Idiopathic Myelofibrosis Identifies a Set of Disease-Associated Genes and Reveals the Clinical Significance of Wilms’ Tumor Gene 1 ( WT1 ). Stem Cells 2007, 25, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Narita, M. Oncogenic HMGA2: Short or small? Genes Dev. 2007, 21, 1005–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, K.; Ogawa, K.; Takeishi, Y. The role of HMGA2 in the proliferation and expansion of a hematopoietic cell in myeloproliferative neoplasms. Fukushima J. Med. Sci. 2012, 58, 91–100. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Mazurier, F.; Dupont, S.; Chaligne, R.; Lamrissi-Garcia, I.; Tulliez, M.; Lippert, E.; Marion, F.X.; Pasquet, J.M.; Etienne, G.; et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood 2008, 112, 2429–2438. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Mason, P.J.; Bessler, M. 3′UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood 2011, 117, 5860–5869. [Google Scholar] [CrossRef] [Green Version]
- Li, O.; Li, J.; Dröge, P. DNA architectural factor and proto-oncogene HMGA2 regulates key developmental genes in pluripotent human embryonic stem cells. FEBS Lett. 2007, 581, 3533–3537. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Ikeda, K.; Ikezoe, T.; Harada-Shirado, K.; Ogawa, K.; Hashimoto, Y.; Sano, T.; Ohkawara, H.; Kimura, S.; Shichishima-Nakamura, A.; et al. Hmga2 collaborates with JAK2V617F in the development of myeloproliferative neoplasms. Blood Adv. 2017, 1, 1001–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguro, H.; Yuan, J.; Tanaka, S.; Miyagi, S.; Mochizuki-Kashio, M.; Ichikawa, H.; Yamazaki, S.; Koseki, H.; Nakauchi, H.; Iwama, A. Lethal myelofibrosis induced by Bmi1-deficient hematopoietic cells unveils a tumor suppressor function of the polycomb group genes. J. Exp. Med. 2012, 209, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Akada, H.; Nath, D.; Hutchison, R.E.; Mohi, G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood 2016, 127, 3410–3423. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.E.; Sausen, M.; Joseph, A.; Kingham, B.F.; Martin, E.S. Identification of a HMGA2-EFCAB6 gene rearrangement following next-generation sequencing in a patient with a t(12;22)(q14.3;q13.2) and JAK2V617F-positive myeloproliferative neoplasm. Cancer Genet. 2012, 205, 295–303. [Google Scholar] [CrossRef]
- Meyer, B.; Krisponeit, D.; Junghanss, C.; Escobar, H.M.; Bullerdiek, J. Quantitative expression analysis in peripheral blood of patients with chronic myeloid leukaemia: Correlation between HMGA2 expression and white blood cell count. Leuk. Lymphoma 2007, 48, 2008–2013. [Google Scholar] [CrossRef]
- Wei, J.; Li, H.; Wang, S.; Li, T.; Fan, J.; Liang, X.; Li, J.; Han, Q.; Zhu, L.; Fan, L.; et al. let-7 enhances osteogenesis and bone formation while repressing adipogenesis of human stromal/mesenchymal stem cells by regulating HMGA2. Stem Cells Dev. 2014, 23, 1452–1463. [Google Scholar] [CrossRef] [Green Version]
- Bruchova, H.; Merkerova, M.; Prchal, J.T. Aberrant expression of microRNA in polycythemia vera. Haematologica 2008, 93, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Tozzi, L.; Pancrazzi, A.; Bogani, C.; Antonioli, E.; Ponziani, V.; Poli, G.; Zini, R.; Ferrari, S.; Manfredini, R.; et al. MicroRNA expression profile in granulocytes from primary myelofibrosis patients. Exp. Hematol. 2007, 35, 1708.e1–1708.e12. [Google Scholar] [CrossRef]
- Zhan, H.; Cardozo, C.; Yu, W.; Wang, A.; Moliterno, A.R.; Dang, C.V.; Spivak, J.L. MicroRNA deregulation in polycythemia vera and essential thrombocythemia patients. Blood Cells Mol. Dis. 2013, 50, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; Casieri, P.; Minervini, A.; Specchia, G. SETBP1 and miR-4319 dysregulation in primary myelofibrosis progression to acute myeloid leukemia. J. Hematol. Oncol. 2012, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, J.; Caceres, N.; Constantinescu, S.N. Aberrant signal transduction pathways in myeloproliferative neoplasms. Leukemia 2008, 22, 1828–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, Y.; Inoue, N.; Shichishima, T.; Ohta, R.; Noji, H.; Maeda, Y.; Nishimura, J.I.; Kanakura, Y.; Kinoshita, T. Deregulated expression of HMGA2 is implicated in clonal expansion of PIGA deficient cells in paroxysmal nocturnal haemoglobinuria. Br. J. Haematol. 2012, 156, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Serio, B.; Selleri, C.; Maciejewski, J.P. Impact of immunogenetic polymorphisms in bone marrow failure syndromes. Mini Rev. Med. Chem. 2011, 11, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Shichishima, T.; Okamoto, M.; Ikeda, K.; Kaneshige, T.; Sugiyama, H.; Terasawa, T.; Osumi, K.; Maruyama, Y. HLA class II haplotype and quantitation of WT1 RNA in Japanese patients with paroxysmal nocturnal hemoglobinuria. Blood 2002, 100, 22–28. [Google Scholar] [CrossRef]
- Shichishima, T.; Noji, H.; Ikeda, K.; Akutsu, K.; Maruyama, Y. Bone marrow failure: The frequency of HLA class I alleles in Japanese patients with bone marrow failure. Haematologica 2006, 91, 856–857. [Google Scholar]
- Ikeda, K.; Shichishima, T.; Yasukawa, M.; Nakamura-Shichishima, A.; Noji, H.; Akutsu, K.; Osumi, K.; Maruyama, Y. The role of Wilms’ tumor gene peptide-specific cytotoxic T lymphocytes in immunologic selection of a paroxysmal nocturnal hemoglobinuria clone. Exp. Hematol. 2007, 35, 618–626. [Google Scholar] [CrossRef]
- Luzzatto, L.; Bessler, M. The dual pathogenesis of paroxysmal nocturnal hemoglobinuria. Curr. Opin. Hematol. 1996, 3, 101–110. [Google Scholar] [CrossRef]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA. Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrózek, K.; Heerema, N.A.; Bloomfield, C.D. Cytogenetics in acute leukemia. Blood Rev. 2004, 18, 115–136. [Google Scholar] [CrossRef]
- Rowley, J.D. Chromosomal translocations: Revisited yet again. Blood 2008, 112, 2183–2189. [Google Scholar] [CrossRef]
- Jin, J.; Yu, M.; Hu, C.; Ye, L.; Xie, L.; Jin, J.; Chen, F.; Tong, H. Pesticide exposure as a risk factor for myelodysplastic syndromes: A meta-analysis based on 1942 cases and 5359 controls. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumbo, C.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Impera, L.; Tota, G.; Parciante, E.; et al. Nanopore targeted sequencing for rapid gene mutations detection in acute myeloid leukemia. Genes (Basel) 2019, 10, 1026. [Google Scholar] [CrossRef] [Green Version]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Brunetti, C.; Anelli, L.; Zagaria, A.; Minervini, A.; Casieri, P.; Coccaro, N.; Tota, G.; et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn. Pathol. 2016, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Rockova, V.; Abbas, S.; Wouters, B.J.; Erpelinck, C.A.J.; Beverloo, H.B.; Delwel, R.; Van Putten, W.L.J.; Löwenberg, B.; Valk, P.J.M. Risk stratification of intermediate-risk acute myeloid leukemia: Integrative analysis of a multitude of gene mutation and gene expression markers. Blood 2011, 118, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Mroźek, K.; Marcucci, G.; Nicolet, D.; Maharry, K.S.; Becker, H.; Whitman, S.P.; Metzeler, K.H.; Schwind, S.; Wu, Y.Z.; Kohlschmidt, J.; et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 4515–4523. [Google Scholar] [CrossRef]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998, 92, 2322–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquis, M.; Beaubois, C.; Lavallée, V.P.; Abrahamowicz, M.; Danieli, C.; Lemieux, S.; Ahmad, I.; Wei, A.; Ting, S.B.; Fleming, S.; et al. High expression of HMGA2 independently predicts poor clinical outcomes in acute myeloid leukemia. Blood Cancer J. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Nyquist, K.B.; Panagopoulos, I.; Thorsen, J.; Roberto, R.; Wik, H.S.; Tierens, A.; Heim, S.; Micci, F. t(12;13)(q14;q31) leading to HMGA2 upregulation in acute myeloid leukaemia. Br. J. Haematol. 2012, 157, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Wei, X.; Zheng, L.; Zeng, J.; Liu, H.; Yang, S.; Tan, H. Amplified HMGA2 promotes cell growth by regulating Akt pathway in AML. J. Cancer Res. Clin. Oncol. 2016, 142, 389–399. [Google Scholar] [CrossRef]
- Ma, S.; Yang, L.L.; Niu, T.; Cheng, C.; Zhong, L.; Zheng, M.W.; Xiong, Y.; Li, L.L.; Xiang, R.; Chen, L.J.; et al. SKLB-677, an FLT3 and Wnt/β-catenin signaling inhibitor, displays potent activity in models of FLT3-driven AML. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Sokolowski, K.M.; Koprowski, S.; Kunnimalaiyaan, S.; Balamurugan, M.; Gamblin, T.C.; Kunnimalaiyaan, M. Potential Molecular Targeted Therapeutics: Role of PI3-K/Akt/mTOR Inhibition in Cancer. Anticancer Agents Med. Chem. 2016, 16, 29–37. [Google Scholar] [CrossRef]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing leukemia stem cells to NF-κB inhibitor treatment in vivo by inactivation of both TNF and IL-1 signaling. Oncotarget 2017, 8, 8420–8435. [Google Scholar]
- Cook, A.M.; Li, L.; Ho, Y.; Lin, A.; Li, L.; Stein, A.; Forman, S.; Perrotti, D.; Jove, R.; Bhatia, R. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood 2014, 123, 2826–2837. [Google Scholar] [CrossRef] [Green Version]
- Arcaro, A.; Guerreiro, A. The Phosphoinositide 3-Kinase Pathway in Human Cancer: Genetic Alterations and Therapeutic Implications. Curr. Genomics 2007, 8, 271–306. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Xu, H.; Chen, G.; Wei, X.; Yu, B.; Ye, J.; Xu, L.; Tan, H. Silencing of HMGA2 reverses retardance of cell differentiation in human myeloid leukaemia. Br. J. Cancer 2018, 118, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Gu, Y.; Wang, G.; Hu, Q.; Chen, S.; Wang, Y.; Zhao, M. HMGA2 regulates acute myeloid leukemia progression and sensitivity to daunorubicin via Wnt/β-catenin signaling. Int. J. Mol. Med. 2019, 44, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, L.; Zhang, J.; Tang, W.; Zhang, N.; He, M.; Guo, Y.; Wang, Z. HMGA2 elicits EMT by activating the Wnt/β-catenin pathway in gastric cancer. Dig. Dis. Sci. 2013, 58, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Wend, P.; Runke, S.; Wend, K.; Anchondo, B.; Yesayan, M.; Jardon, M.; Hardie, N.; Loddenkemper, C.; Ulasov, I.; Lesniak, M.S.; et al. WNT10B/β-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol. Med. 2013, 5, 264–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.R.; Abboud, C.N.; Altman, J.; Appelbaum, F.R.; Arber, D.A.; Attar, E.; Borate, U.; Coutre, S.E.; Damon, L.E.; Goorha, S.; et al. NCCN Clinical Practice Guidelines Acute myeloid leukemia. J. Natl. Compr. Canc. Netw. 2012, 10, 984–1021. [Google Scholar] [CrossRef] [Green Version]
- Quiney, C.; Billard, C.; Faussat, A.M.; Salanoubat, C.; Kolb, J.P. Hyperforin inhibits P-gp and BCRP activities in chronic lymphocytic leukaemia cells and myeloid cells. Leuk. Lymphoma 2007, 48, 1587–1599. [Google Scholar] [CrossRef]
- Andrieux, J.; Bilhou-Nabera, C.; Lippert, E.; Le Bousse-Kerdiles, M.C.; Dupriez, B.; Grardel, N.; Pierre-Louis, O.; Desterke, C.; Praloran, V.; Luc Laï, J.; et al. Expression of HMGA2 in PB leukocytes and purified CD34+ cells from controls and patients with Myelofibrosis and myeloid metaplasia. Leuk. Lymphoma 2006, 47, 1956–1959. [Google Scholar] [CrossRef]
- Thorsteinsdottir, U.; Mamo, A.; Kroon, E.; Jerome, L.; Bijl, J.; Lawrence, H.J.; Humphries, K.; Sauvageau, G. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 2002, 99, 121–129. [Google Scholar] [CrossRef]
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; Van Den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009, 113, 2375–2385. [Google Scholar] [CrossRef] [Green Version]
- Sauvageau, G.; Lansdorp, P.M.; Eaves, C.J.; Hogge, D.E.; Dragowska, W.H.; Reid, D.S.; Largman, C.; Lawrence, H.J.; Humphries, R.K. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl. Acad. Sci. USA 1994, 91, 12223–12227. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Song, C.-X.; Huang, H.; Frankenberger, C.A.; Sankarasharma, D.; Gomes, S.; Chen, P.; Chen, J.; Chada, K.K.; He, C.; et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 9920–9925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsteinsdottir, U.; Kroon, E.; Jerome, L.; Blasi, F.; Sauvageau, G. Defining Roles for HOX and MEIS1 Genes in Induction of Acute Myeloid Leukemia. Mol. Cell. Biol. 2001, 21, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmrich, S.; Katsman-Kuipers, J.E.; Henke, K.; Khatib, M.E.; Jammal, R.; Engeland, F.; Dasci, F.; Zwaan, C.M.; Den Boer, M.L.; Verboon, L.; et al. MiR-9 is a tumor suppressor in pediatric AML with t(8;21). Leukemia 2014, 28, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; Lapierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and Lin28B inhibit let-7 MicroRNA biogenesis by distinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Heo, I.; Joo, C.; Cho, J.; Ha, M.; Han, J.; Kim, V.N. Lin28 Mediates the Terminal Uridylation of let-7 Precursor MicroRNA. Mol. Cell 2008, 32, 276–284. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective blockade of microRNA processing by Lin28. Science 2008, 320, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Rybak, A.; Fuchs, H.; Smirnova, L.; Brandt, C.; Pohl, E.E.; Nitsch, R.; Wulczyn, F.G. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat. Cell Biol. 2008, 10, 987–993. [Google Scholar] [CrossRef]
- Newman, M.A.; Thomson, J.M.; Hammond, S.M. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 2008, 14, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Büssing, I.; Slack, F.J.; Großhans, H. let-7 microRNAs in development, stem cells and cancer. Trends Mol. Med. 2008, 14, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qin, Y.Z.; Yang, S.; Wang, Y.; Chang, Y.J.; Zhao, T.; Jiang, Q.; Huang, X.J. Meis1 is critical to the maintenance of human acute myeloid leukemia cells independent of MLL rearrangements. Ann. Hematol. 2017, 96, 567–574. [Google Scholar] [CrossRef] [PubMed]
- In’t Hout, F.E.M.; van der Reijden, B.A.; Monteferrario, D.; Jansen, J.H.; Huls, G. High expression of transcription factor 4 (TCF4) is an independent adverse prognostic factor in acute myeloid leukemia that could guide treatment decisions. Haematologica 2014, 99, e257–e259. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.; Muselman, A.; Du, R.; Harada, Y.; Scholl, A.G.; Yan, M.; Matsuura, S.; Weng, S.; Harada, H.; Zhang, D.E. Hmga2 is a direct target gene of RUNX1 and regulates expansion of myeloid progenitors in mice. Blood 2014, 124, 2203–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerritsen, M.; Yi, G.; Tijchon, E.; Kuster, J.; Schuringa, J.J.; Martens, J.H.A.; Vellenga, E. RUNX1 mutations enhance self-renewal and block granulocytic differentiation in human in vitro models and primary AMLs. Blood Adv. 2019, 3, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, G.; Rousseau, B.; Marquis, M.; Beaubois, C.; Sauvageau, G.; Hébert, J. Cost-Effectiveness Analysis of a HMGA2 Prognostic Test for Acute Myeloid Leukemia in a Canadian Setting. Appl. Health Econ. Health Policy 2019, 17, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Hunger, S.P.; Loh, M.L.; Whitlock, J.A.; Winick, N.J.; Carroll, W.L.; Devidas, M.; Raetz, E.A. Children’s oncology Group’s 2013 blueprint for research: Acute lymphoblastic leukemia. Pediatr. Blood Cancer 2013, 60, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Next-generation sequencing in acute lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 2929. [Google Scholar] [CrossRef] [Green Version]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris IV, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, M.; Fidanza, V.; Battista, S.; Pentimalli, F.; Klein-Szanto, A.J.P.; Visone, R.; De Martino, I.; Curcio, A.; Morisco, C.; Del Vecchio, L.; et al. Haploinsufficiency of the Hmga1 gene causes cardiac hypertrophy and myelo-lymphoproliferative disorders in mice. Cancer Res. 2006, 66, 2536–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Li, Y.-S.; Tang, H.-B. High mobility group A1 protein acts as a new target of Notch1 signaling and regulates cell proliferation in T leukemia cells. Mol. Cell. Biochem. 2013, 374, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Bartram, C.R.; Seriu, T.; Miller, C.W.; Tobler, A.; Janssen, J.W.; Reiter, A.; Ludwig, W.D.; Zimmermann, M.; Schwaller, J. Analysis of a family of cyclin-dependent kinase inhibitors: P15/MTS2/INK4B, p16/MTS1/INK4A, and p18 genes in acute lymphoblastic leukemia of childhood. Blood 1995, 86, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, S.W.; Sherr, C.J. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr. Opin. Genet. Dev. 2003, 13, 77–83. [Google Scholar] [CrossRef]
- Roy, S.; Di Cello, F.; Kowalski, J.; Hristov, A.C.; Tsai, H.L.; Bhojwani, D.; Meyer, J.A.; Carroll, W.L.; Belton, A.; Resar, L.M.S. HMGA1 overexpression correlates with relapse in childhood B-lineage acute lymphoblastic leukemia. Leuk. Lymphoma 2013, 54, 2565–2567. [Google Scholar] [CrossRef] [Green Version]
- Maia, S.; Haining, W.N.; Ansén, S.; Xia, Z.; Armstrong, S.A.; Seth, N.P.; Ghia, P.; den Boer, M.L.; Pieters, R.; Sallan, S.E.; et al. Gene expression profiling identifies BAX-delta as a novel tumor antigen in acute lymphoblastic leukemia. Cancer Res. 2005, 65, 10050–10058. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.; Olofsson, T.; Lindgren, D.; Nilsson, B.; Ritz, C.; Edén, P.; Lassen, C.; Råde, J.; Fontes, M.; Mörse, H.; et al. Molecular signatures in childhood acute leukemia and their correlations to expression patterns in normal hematopoietic subpopulations. Proc. Natl. Acad. Sci. USA 2005, 102, 19069–19074. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.N.; Kerr, C.; Cope, L.; Zambidis, E.; Liu, C.; Hillion, J.; Belton, A.; Huso, D.L.; Resar, L.M.S. HMGA1 Reprograms Somatic Cells into Pluripotent Stem Cells by Inducing Stem Cell Transcriptional Networks. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Nagel, S.; Scherr, M.; Kel, A.; Hornischer, K.; Crawford, G.E.; Kaufmann, M.; Meyer, C.; Drexler, H.G.; MacLeod, R.A.F. Activation of TLX3 and NKX2-5 in t(5;14)(q35;q32) T-cell acute lymphoblastic leukemia by remote 3′-BCL11B enhancers and coregulation by PU.1 and HMGA1. Cancer Res. 2007, 67, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Hillion, J.; Belton, A.M.; Shah, S.N.; Turkson, J.; Jing, N.; Tweardy, D.J.; Di Cello, F.; Huso, D.L.; Resar, L.M.S. Nanoparticle delivery of inhibitory signal transducer and activator of transcription 3 G-quartet oligonucleotides blocks tumor growth in HMGA1 transgenic model of T-cell leukemia. Leuk. Lymphoma 2014, 55, 1194–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yue, P.; Page, B.D.G.; Li, T.; Zhao, W.; Namanja, A.T.; Paladino, D.; Zhao, J.; Chen, Y.; Gunning, P.T.; et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. USA 2012, 109, 9623–9628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, N.; Li, Y.; Xiong, W.; Sha, W.; Jing, L.; Tweardy, D.J. G-quartet oligonucleotides: A new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004, 64, 6603–6609. [Google Scholar] [CrossRef] [Green Version]
- Jing, N.; Zhu, Q.; Yuan, P.; Li, Y.; Mao, L.; Tweardy, D.J. Targeting signal transducer and activator of transcription 3 with G-quartet oligonucleotides: A potential novel therapy for head and neck cancer. Mol. Cancer Ther. 2006, 5, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.S.; Kantarjian, H.M.; Bueso-Ramos, C.E.; Medeiros, L.J.; Haidar, M.A. Frequent deletions at 12q14.3 chromosomal locus in adult acute lymphoblastic leukemia. Genes. Chromosomes Cancer 2005, 42, 87–94. [Google Scholar] [CrossRef]
- Nishi, M.; Eguchi-Ishimae, M.; Wu, Z.; Gao, W.; Iwabuki, H.; Kawakami, S.; Tauchi, H.; Inukai, T.; Sugita, K.; Hamasaki, Y.; et al. Suppression of the let-7b microRNA pathway by DNA hypermethylation in infant acute lymphoblastic leukemia with MLL gene rearrangements. Leukemia 2013, 27, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Sugita, K.; Inukai, T.; Goi, K.; Iijima, K.; Tezuka, T.; Kojika, S.; Shiraishi, K.; Miyamoto, N.; Karakida, N.; et al. P16/MTS1/INK4A gene is frequently inactivated by hypermethylation in childhood acute lymphoblastic leukemia with 11q23 translocation. Leukemia 1999, 13, 884–890. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Eguchi-Ishimae, M.; Yagi, C.; Iwabuki, H.; Gao, W.; Tauchi, H.; Inukai, T.; Sugita, K.; Ishii, E.; Eguchi, M. HMGA2 as a potential molecular target in KMT2A-AFF1-positive infant acute lymphoblastic leukaemia. Br. J. Haematol. 2015, 171, 818–829. [Google Scholar] [CrossRef]
- Miao, Y.; Cui, T.; Leng, F.; Wilson, W.D. Inhibition of high-mobility-group A2 protein binding to DNA by netropsin: A biosensor-surface plasmon resonance assay. Anal. Biochem. 2008, 374, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Fedele, M.; Fusco, A. HMGA and Cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 48–54. [Google Scholar] [CrossRef]
- Beckerbauer, L.; Tepe, J.J.; Cullison, J.; Reeves, R.; Williams, R.M. FR900482 class of anti-tumor drugs cross-links oncoprotein HMG I/Y to DNA in vivo. Chem. Biol. 2000, 7, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Belton, A.; Xian, L.; Huso, T.; Koo, M.; Luo, L.Z.; Turkson, J.; Page, B.D.G.; Gunning, P.T.; Liu, G.; Huso, D.L.; et al. STAT3 inhibitor has potent antitumor activity in B-lineage acute lymphoblastic leukemia cells overexpressing the high mobility group A1 (HMGA1)–STAT3 pathway. Leuk. Lymphoma 2016, 57, 2681–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böll, B.; Hansen, H.; Heuck, F.; Reiners, K.; Borchmann, P.; Rothe, A.; Engert, A.; Pogge von Strandmann, E. The fully human anti-CD30 antibody 5F11 activates NF-{kappa}B and sensitizes lymphoma cells to bortezomib-induced apoptosis. Blood 2005, 106, 1839–1842. [Google Scholar]
- Jia, L.; Gopinathan, G.; Sukumar, J.T.; Gribben, J.G. Blocking autophagy prevents bortezomib-induced NF-κB activation by reducing I-κBα degradation in lymphoma cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-P.; Ding, D.-Z.; Shi, B.; Zhang, S.-Q.; Gu, L.-L.; Wang, Y.-C.; Cheng, C. Expression of TRIM28 correlates with proliferation and Bortezomib-induced apoptosis in B-cell non-Hodgkin lymphoma. Leuk. Lymphoma 2018, 59, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Pongas, G.N.; Annunziata, C.M.; Staudt, L.M. PI3Kδ inhibition causes feedback activation of PI3Kα in the ABC subtype of diffuse large B-cell lymphoma. Oncotarget 2017, 8, 81794–81802. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Shao, Q.; Li, C.; Liu, H.; Li, J.; Wang, Y.; Song, W.; Li, L.; Wang, G.; Shao, Z.; et al. Effects of microRNA-21 on apoptosis by regulating the expression of PTEN in diffuse large B-cell lymphoma. Medicine (Baltimore) 2017, 96, e7952. [Google Scholar] [CrossRef]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Perrone, T.; Specchia, G.; Albano, F. Molecular Complexity of Diffuse Large B-Cell Lymphoma: Can It Be a Roadmap for Precision Medicine? Cancers (Basel) 2020, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Shen, J.; Wang, Q.; Zhang, M. Bortezomib inhibited the progression of diffuse large B-cell lymphoma via targeting miR-198. Biomed. Pharmacother. 2018, 108, 43–49. [Google Scholar] [CrossRef]
- Shen, Y.; Lu, L.; Xu, J.; Meng, W.; Qing, Y.; Liu, Y.; Zhang, B.; Hu, H. Bortezomib induces apoptosis of endometrial cancer cells through microRNA-17-5p by targeting p21. Cell Biol. Int. 2013, 37, 1114–1121. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, H.; Hu, Z.; Mao, Y.; Xu, X.; Zhu, Y.; Xu, X.; Wu, J.; Li, S.; Mao, Q.; et al. miR-26a inhibits proliferation and motility in bladder cancer by targeting HMGA1. FEBS Lett. 2013, 587, 2467–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Tao, T.; Liu, C.; Guan, H.; Huang, Y.; Xu, B.; Chen, M. Downregulation of miR-195 promotes prostate cancer progression by targeting HMGA1. Oncol. Rep. 2016, 36, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wang, J.; Jia, Y.; Shen, F.; Han, W.; Kang, Y. MiR-142-3p functions as a potential tumor suppressor in human osteosarcoma by targeting HMGA1. Cell. Physiol. Biochem. 2014, 33, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Cinkornpumin, J.; Roos, M.; Nguyen, L.; Liu, X.; Gaeta, X.; Lin, S.; Chan, D.N.; Liu, A.; Gregory, R.I.; Jung, M.; et al. A small molecule screen to identify regulators of let-7 targets. Sci. Rep. 2017, 7, 15973. [Google Scholar] [CrossRef]
- Manley, P.W.; Cowan-Jacob, S.W.; Mestan, J. Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochim. Biophys. Acta 2005, 1754, 3–13. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Rossi, A.R.; Breccia, M.; Abruzzese, E.; Castagnetti, F.; Luciano, L.; Gozzini, A.; Annunziata, M.; Martino, B.; Stagno, F.; Cavazzini, F.; et al. Outcome of 82 chronic myeloid leukemia patients treated with nilotinib or dasatinib after failure of two prior tyrosine kinase inhibitors. Haematologica 2013, 98, 399–403. [Google Scholar] [CrossRef]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Vitkeviciene, A.; Baksiene, S.; Borutinskaite, V.; Navakauskiene, R. Epigallocatechin-3-gallate and BIX-01294 have different impact on epigenetics and senescence modulation in acute and chronic myeloid leukemia cells. Eur. J. Pharmacol. 2018, 838, 32–40. [Google Scholar] [CrossRef]
- Gan, R.-Y.; Li, H.-B.; Sui, Z.-Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit. Rev. Food Sci. Nutr. 2018, 58, 924–941. [Google Scholar] [CrossRef]
- Granja, A.; Pinheiro, M.; Reis, S. Epigallocatechin gallate nanodelivery systems for cancer therapy. Nutrients 2016, 8, 307. [Google Scholar] [CrossRef]
- Huang, J.; Dorsey, J.; Chuikov, S.; Pérez-Burgos, L.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [Green Version]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
Figure 1.
HMGA2 development-specific isoforms: HSCs feature distinct alternative splicing patterns in various key hematopoietic regulators, one of which is HMGA2. In humans, two alternative splicing isoforms have recently been discovered: the HMGA2-L isoform, which is sensitive to degradation by let-7 and is dominant in fetal HSCs, and the HMGA2-S isoform, that is resistant to degradation by let-7 and is prevalent in newborn HSCs. The two isoforms reveal alternative 3’UTR usage; they share the first three exons and differ in their terminal exon usage (including C-terminal domains and 3’UTRs). The HMGA2-S 3’UTR is one third the length of the HMGA2-L 3’UTR and lacks the seven let-7 sites as well as the other most conserved miRNA sites. The balance between alternative isoforms and the escape from miRNA-mediated control seems to depend on the activity of the splicing kinase CLK3, which promotes exon skipping and thus influences the HMGA2 splicing pattern [8]. HSCs-hematopoietic stem cells; 3’UTR-3′-Untranslated Region.
Figure 1.
HMGA2 development-specific isoforms: HSCs feature distinct alternative splicing patterns in various key hematopoietic regulators, one of which is HMGA2. In humans, two alternative splicing isoforms have recently been discovered: the HMGA2-L isoform, which is sensitive to degradation by let-7 and is dominant in fetal HSCs, and the HMGA2-S isoform, that is resistant to degradation by let-7 and is prevalent in newborn HSCs. The two isoforms reveal alternative 3’UTR usage; they share the first three exons and differ in their terminal exon usage (including C-terminal domains and 3’UTRs). The HMGA2-S 3’UTR is one third the length of the HMGA2-L 3’UTR and lacks the seven let-7 sites as well as the other most conserved miRNA sites. The balance between alternative isoforms and the escape from miRNA-mediated control seems to depend on the activity of the splicing kinase CLK3, which promotes exon skipping and thus influences the HMGA2 splicing pattern [8]. HSCs-hematopoietic stem cells; 3’UTR-3′-Untranslated Region.

Figure 2.
Different expression of HMGA2 in hematological diseases: Following the truncation of the 3′-UTR, the binding sites for let-7 are lost and the negative regulation of miRNA on the expression of HMGA2 no longer occurs. Overexpression of HMGA2 leads to a clonal growth advantage of the hematopoietic cell, as observed in MPN, PNH, and MDS patients. HMGA2-High mobility group AT-Hook 2; 3′-UTR-3′-Untranslated Region; MPN-Myeloproliferative neoplasms; PNH-paroxysmal nocturnal hemoglobinuria; MDS-myelodyslastic syndrome.
Figure 2.
Different expression of HMGA2 in hematological diseases: Following the truncation of the 3′-UTR, the binding sites for let-7 are lost and the negative regulation of miRNA on the expression of HMGA2 no longer occurs. Overexpression of HMGA2 leads to a clonal growth advantage of the hematopoietic cell, as observed in MPN, PNH, and MDS patients. HMGA2-High mobility group AT-Hook 2; 3′-UTR-3′-Untranslated Region; MPN-Myeloproliferative neoplasms; PNH-paroxysmal nocturnal hemoglobinuria; MDS-myelodyslastic syndrome.

Figure 3.
The role of HOXA9 regulation in normal myeloid differentiation: (A) HOXA9 overexpression maintains the self-renewal capacity of leukemic stem cells and blocks the differentiation process. (B) In some leukemia cell lines, HMGA2 knockdown affects HOXA9 expression, and this influences the differentiation processes in myeloid cells. HOXA9-Homeobox A9; HMGA2-High mobility group AT-Hook 2.
Figure 3.
The role of HOXA9 regulation in normal myeloid differentiation: (A) HOXA9 overexpression maintains the self-renewal capacity of leukemic stem cells and blocks the differentiation process. (B) In some leukemia cell lines, HMGA2 knockdown affects HOXA9 expression, and this influences the differentiation processes in myeloid cells. HOXA9-Homeobox A9; HMGA2-High mobility group AT-Hook 2.

Figure 4.
HMGA involvement in lymphoid malignancies: Graph representing some HMGA-controlled pathways involved in lymphoid tumorigenesis. (A) The HMGA1–STAT3 pathway plays a crucial role in lymphoid neoplasias, such as T-ALL and Burkitts lymphoma. HMGA1 induces STAT3 expression [58,211] that has a main role in malignant transformation and tumor progression and in determining a treatment refractory status [212]. In vitro blocking of HMGA1 or STAT3 function seems to be a promising new therapeutic approach in T-cell lymphoid tumors to induce apoptosis and to inhibit cell growth [211]. (B) KMT2A-rearranged ALLs frequently show miRNA let-7b downregulation [216,217]. As a consequence, let-7b-regulated target genes are upregulated, including HMGA2. HMGA2 overexpression has an impact on its downstream targets, such as the CDKN2A gene, that is negatively regulated by HMGA2. This circumstance triggers leukemic proliferation. The use of the HMGA2 inhibitor netropsin in addition to the demethylating agent 5-azacytidine has been demonstrated to upregulate and sustain the expression of CDKN2A, resulting in a higher growth suppression compared to treatment with 5-azacytidine alone [218]. HMGA-High mobility group AT-Hook; T-ALL-T-cell Acute lymphoblastic leukemia.
Figure 4.
HMGA involvement in lymphoid malignancies: Graph representing some HMGA-controlled pathways involved in lymphoid tumorigenesis. (A) The HMGA1–STAT3 pathway plays a crucial role in lymphoid neoplasias, such as T-ALL and Burkitts lymphoma. HMGA1 induces STAT3 expression [58,211] that has a main role in malignant transformation and tumor progression and in determining a treatment refractory status [212]. In vitro blocking of HMGA1 or STAT3 function seems to be a promising new therapeutic approach in T-cell lymphoid tumors to induce apoptosis and to inhibit cell growth [211]. (B) KMT2A-rearranged ALLs frequently show miRNA let-7b downregulation [216,217]. As a consequence, let-7b-regulated target genes are upregulated, including HMGA2. HMGA2 overexpression has an impact on its downstream targets, such as the CDKN2A gene, that is negatively regulated by HMGA2. This circumstance triggers leukemic proliferation. The use of the HMGA2 inhibitor netropsin in addition to the demethylating agent 5-azacytidine has been demonstrated to upregulate and sustain the expression of CDKN2A, resulting in a higher growth suppression compared to treatment with 5-azacytidine alone [218]. HMGA-High mobility group AT-Hook; T-ALL-T-cell Acute lymphoblastic leukemia.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Minervini, A.; Coccaro, N.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. HMGA Proteins in Hematological Malignancies. Cancers 2020, 12, 1456. https://doi.org/10.3390/cancers12061456
AMA Style
Minervini A, Coccaro N, Anelli L, Zagaria A, Specchia G, Albano F. HMGA Proteins in Hematological Malignancies. Cancers. 2020; 12(6):1456. https://doi.org/10.3390/cancers12061456
Chicago/Turabian StyleMinervini, Angela, Nicoletta Coccaro, Luisa Anelli, Antonella Zagaria, Giorgina Specchia, and Francesco Albano. 2020. "HMGA Proteins in Hematological Malignancies" Cancers 12, no. 6: 1456. https://doi.org/10.3390/cancers12061456
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.