GRK2-Dependent HuR Phosphorylation Regulates HIF1α Activation under Hypoxia or Adrenergic Stress

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
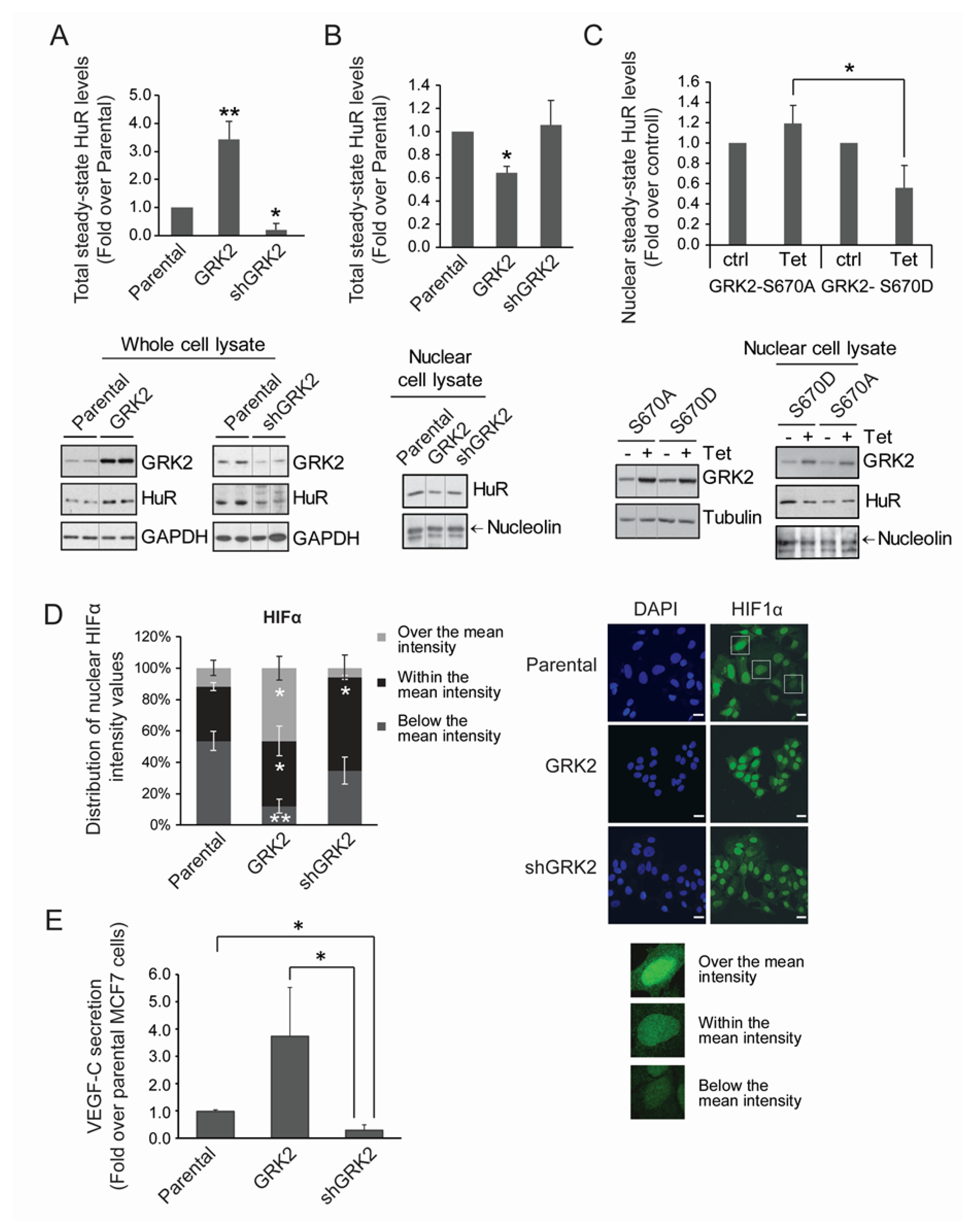
2.1. HuR Protein Levels Positively Correlate with GRK2 Activity
2.2. HuR Is a GRK2 Phosphorylation Substrate
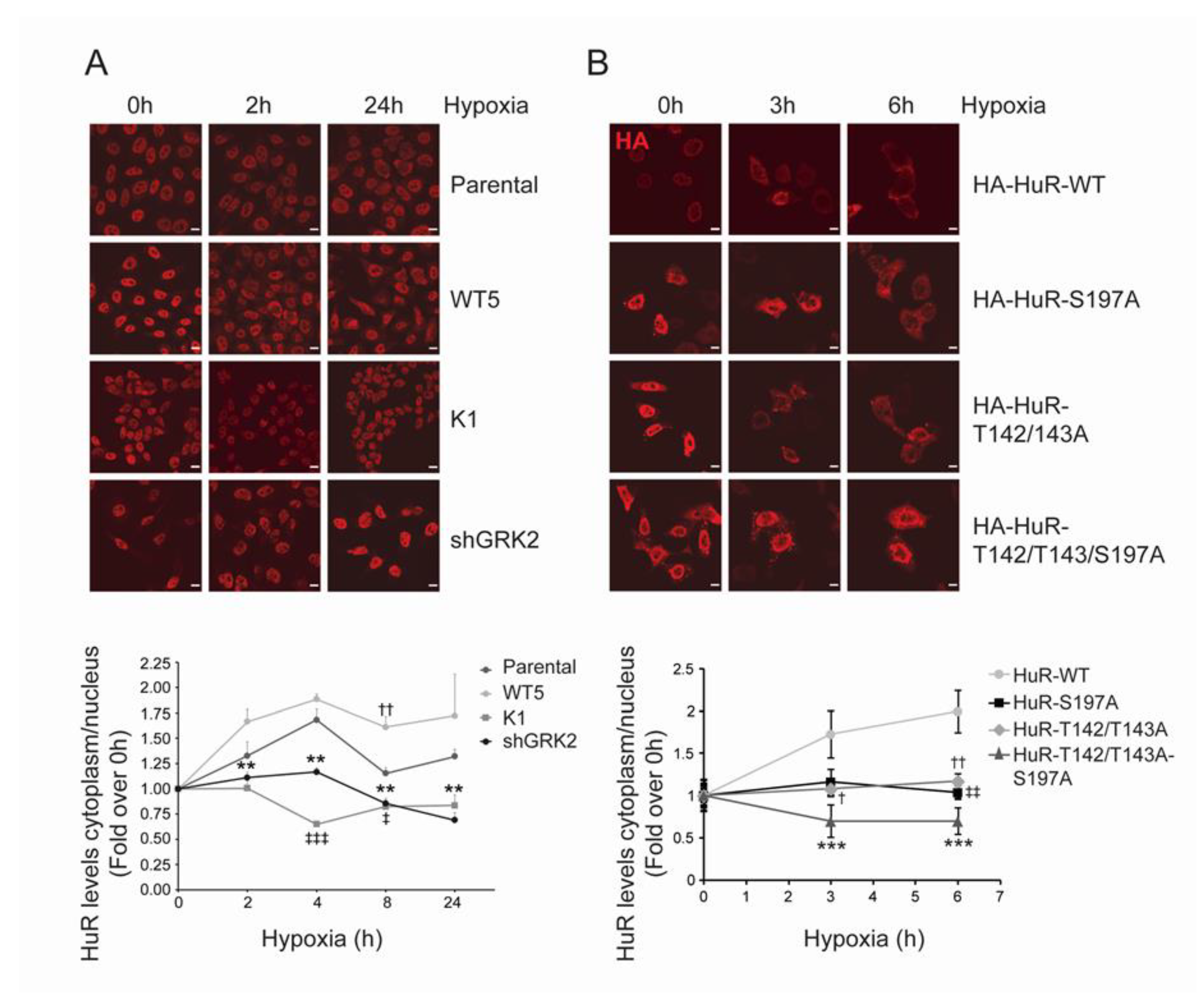
2.3. GRK2 Activity Modulates the Hypoxia-Induced Modulation of HuR Cellular Levels and Cytoplasmic Shuttling
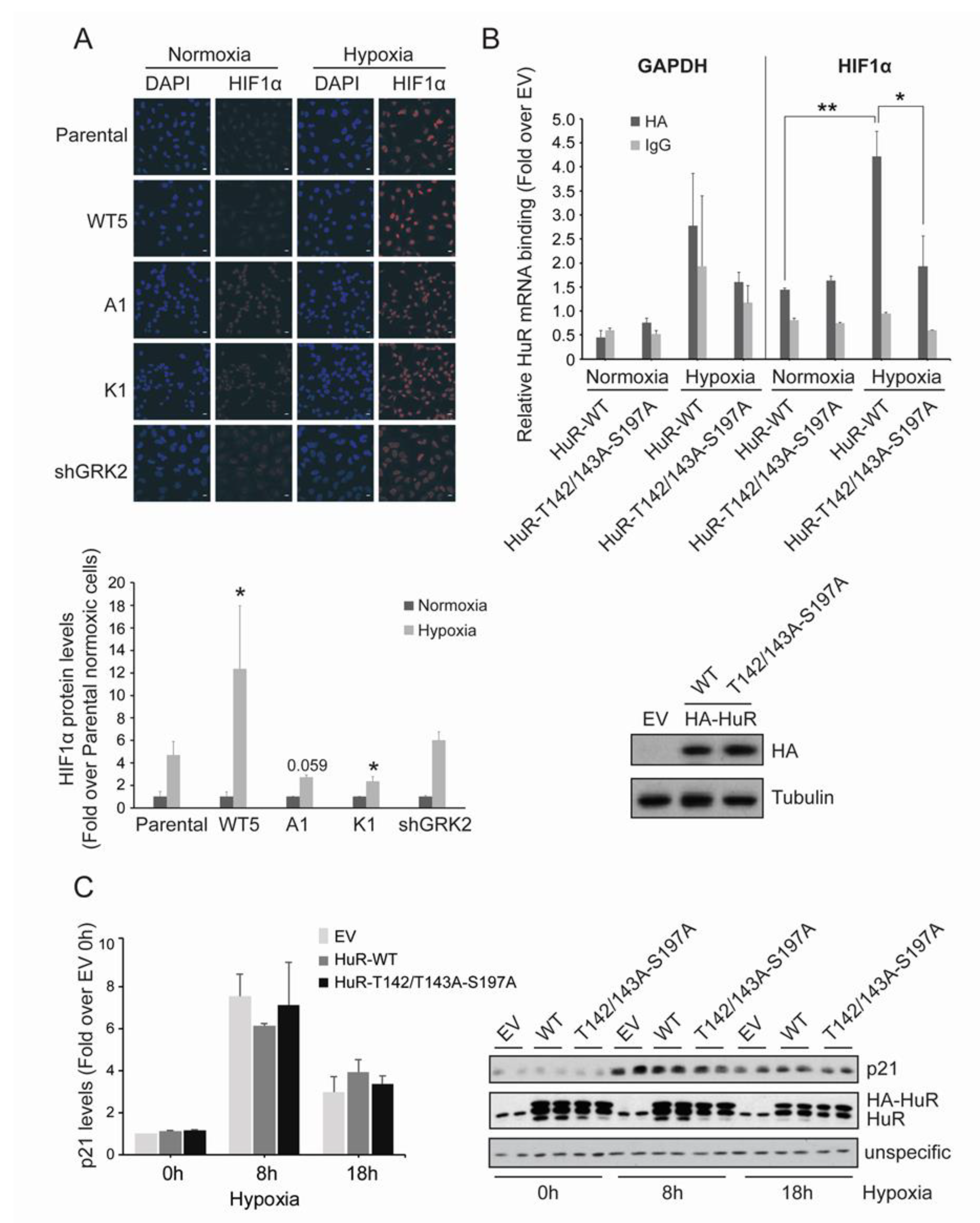
2.4. GRK2 Phosphorylation of HuR Is Required for HIF-1α Upregulation in Response to Hypoxia
2.5. GRK2 Fosters the HuR/HIF-1α Axis and a Pro-Lymphangiogenic Response in the Normoxic Breast Luminal Cancer Cells
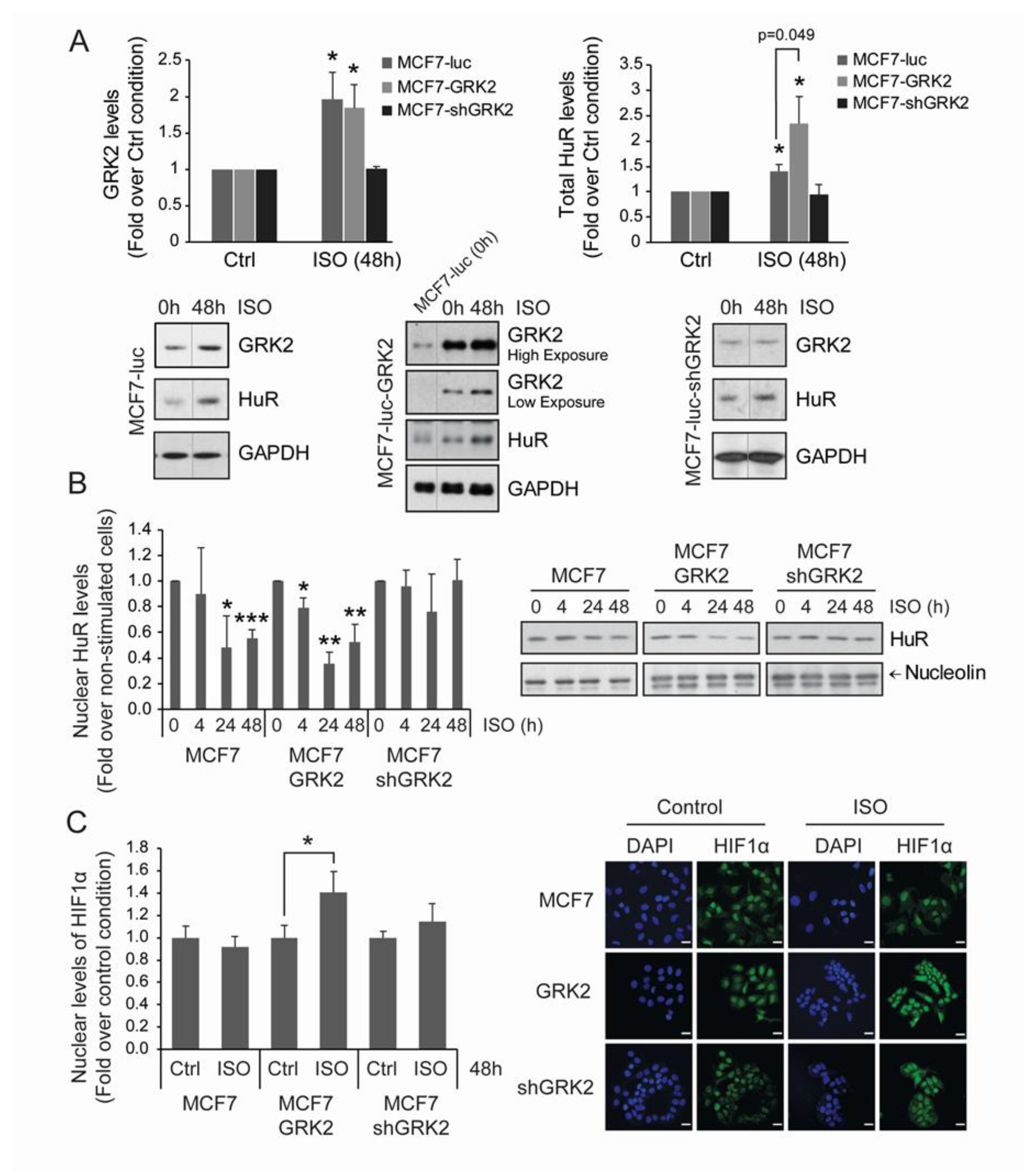
2.6. Adrenergic Stress Induces a HuR/HIF1-α Hypoxia-Like Response in MCF7 Breast Cancer Cells in a GRK2-Mediated Manner
3. Discussion
4. Methods
4.1. Cell Culture and Treatments
4.2. Plasmids and Cell Transfection
4.3. Immunoprecipitation, Western Blot, Dot Blot, and ELISA
4.4. Immunofluorescence and Confocal Microscopy
4.5. Kinase Activity Assays
4.6. Mass Spectrometry Analysis
4.7. RNA Immunoprecipitation
4.8. Bioinformatics Analysis of HuR and GRK2 Expression in Human Breast Tumors
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliff, P.J. New horizons in hypoxia signaling pathways. Exp. Cell Res. 2017, 356, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Laitala, A.; Erler, J.T. Hypoxic Signalling in Tumour Stroma. Front. Oncol. 2018, 29, 189. [Google Scholar] [CrossRef]
- Velaei, K.; Samadi, N.; Barazvan, B.; Soleimani, A.; Rad, J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast 2016, 30, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines 2018, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Masson, N.; Ratcliffe, P.J. Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) Signaling Increases the Rate of Hypoxia-Inducible Factor 1α (HIF-1α) Synthesis: Novel Mechanism for HIF-1-Mediated Vascular Endothelial Growth Factor Expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, H.I.; Asosingh, K.; Stephens, O.R.; Queisser, K.A.; Xu, W.; Willard, B.; Erzurum, S.C. Hypoxia sensing through β-adrenergic receptors. JCI Insight 2016, 1, e90240. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.X.; Li, W.; Xu, T.R. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Gras, E.; Belaidi, E.; Briançon-Marjollet, A.; Pépin, J.L.; Arnaud, C.; Godin-Ribuot, D. Endothelin-1 mediates intermittent hypoxia-induced inflammatory vascularremodeling through HIF-1 activation. J. Appl. Physiol. 2016, 120, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Ribas, C.; Sánchez-Madrid, F.; Mayor, F., Jr. G protein-coupled receptor kinase 2 (GRK2) as a multifunctional signaling hub. Cell. Mol. Life Sci. 2019, 76, 4423–4446. [Google Scholar] [CrossRef] [Green Version]
- Evron, T.; Daigle, T.L.; Caron, M.G. GRK2: Multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 2012, 33, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Han, C.C.; Ma, Y.; Li, Y.; Wang, Y.; Wei, W. Regulatory effects of GRK2 on GPCRs and non-GPCRs and possible use as a drug target. Int. J. Mol. Med. 2016, 38, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Nogués, L.; Reglero, C.; Rivas, V.; Salcedo, A.; Lafarga, V.; Neves, M.; Zhou, X.Z. G Protein-coupled Receptor Kinase 2 (GRK2) Promotes Breast Tumorigenesis through a HDAC6-Pin1 Axis. EBioMedicine 2016, 13, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogués, L.; Palacios-García, J.; Reglero, C.; Rivas, V.; Neves, M.; Ribas, C.; Mayor, F., Jr. G protein-coupled receptor kinases (GRKs) in tumorigenesis and cancer progression: GPCR regulators and signaling hubs. Semin. Cancer Biol. 2018, 48, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Rivas, V.; Carmona, R.; Muñoz-Chápuli, R.; Mendiola, M.; Nogués, L.; Reglero, C.; Mayor, F. Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2. J. Clin. Investig. 2013, 123, 4714–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. WIREs RNA 2010, 1, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Kotta-Loizou, I.; Vasilopoulos, S.N.; Coutts, R.H.; Theocharis, S. Current Evidence and Future Perspectives on HuR and Breast Cancer Development, Prognosis, and Treatment. Neoplasia 2016, 18, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, K.M.; Daly, J.M.; Beveridge, D.J.; Thomson, A.M.; Voon, D.C.; Furneaux, H.M.; Leedman, P.J. The 3′-untranslated region of p21WAF1 mRNA is a composite cis-acting sequence bound by RNA-binding proteins from breast cancer cells, including HuR and poly(C)-binding protein. J. Biol. Chem. 2003, 278, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López de Silanes, I.; Zhan, M.; Lal, A.; Yang, X.; Gorospe, M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA 2004, 101, 2987–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, F., Jr.; Penela, P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2012, 31, 856–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peregrin, S.; Jurado-Pueyo, M.; Campos, P.M.; Sanz-Moreno, V.; Ruiz-Gomez, A.; Crespo, P.; Murga, C. Phosphorylation of p38 by GRK2 at the docking groove unveils a novel mechanism for inactivating p38MAPK. Curr. Biol. 2006, 16, 2042–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.H.; Abdelmohsen, K.; Lal, A.; Pullmann, R., Jr.; Yang, X.; Galban, S.; Gorospe, M. Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 2008, 22, 1804–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.H.; Yang, X.; Kuwano, Y.; Gorospe, M. Modification at HuR(S242) alters HuR localization and proliferative influence. Cell Cycle 2008, 7, 3371–3377. [Google Scholar] [CrossRef]
- Doller, A.; Pfeilschifter, J.; Eberhardt, W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 2008, 20, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Doller, A.; Schlepckow, K.; Schwalbe, H.; Pfeilschifter, J.; Eberhardt, W. Tandem phosphorylation of serines 221 and 318 by protein kinase Cdelta coordinates mRNA binding and nucleocytoplasmic shuttling of HuR. Mol. Cell. Biol. 2010, 30, 1397–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafarga, V.; Cuadrado, A.; Lopez de Silanes, I.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell. Biol. 2009, 29, 4341–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talwar, S.; Jin, J.; Carroll, B.; Liu, A.; Gillespie, M.B.; Palanisamy, V. Caspase-mediated cleavage of RNA-binding protein HuR regulates c-Myc protein expression after hypoxic stress. J. Biol. Chem. 2011, 286, 32333–32343. [Google Scholar] [CrossRef] [Green Version]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. WIREs RNA 2017, 8, e1372. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Furneaux, H.; Cheng, H.; Caldwell, M.C.; Hutter, D.; Liu, Y.; Gorospe, M. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 2000, 20, 760–769. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Das, S.; Sengupta, S. Involvement of HuR in the serum starvation induced autophagy through regulation of Beclin1 in breast cancer cell-line, MCF-7. Cell Signal 2019, 61, 78–85. [Google Scholar] [CrossRef]
- Gallouzi, I.E.; Brennan, C.M.; Steitz, J.A. Protein ligands mediate the CRM1-dependent export of HuR in response to heat shock. RNA 2001, 7, 1348–1361. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.F.; Jimbo, M.; Wulfkuhle, J.; Gallagher, I.; Deng, J.; Enyenihi, L.; Risbud, M.V. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene 2016, 35, 2529–2541. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.S.; Bauer, A.L.; Striet, J.B.; Schnell, P.O.; Czyzyk-Krzeska, M.F. Calcium signaling stimulates translation of HIF-alpha during hypoxia. FASEB J. 2006, 20, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, K.; Milke, L.; Rübsamen, D.; Menrad, H.; Schmid, T.; Brüne, B. HIF-1α protein is upregulated in HIF-2α depleted cells via enhanced translation. FEBS Lett. 2012, 586, 1652–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alitalo, K. The lymphatic vasculature in disease. Nat. Med. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Pérez, D.; Rohde, A.; Callejón, G.; Pérez-Ruiz, E.; Rodrigo, I.; Rivas-Ruiz, F.; Zarcos, I. Correlation between serum levels of vascular endothelial growth factor-C and sentinel lymph node status in early breast cancer. Tumour. Biol. 2015, 36, 9285–9293. [Google Scholar] [CrossRef]
- Ni, X.; Zhao, Y.; Ma, J.; Xia, T.; Liu, X.; Ding, Q.; Zha, X.; Wang, S. Hypoxia-induced factor-1 alpha upregulates vascular endothelial growth factor C to promote lymphangiogenesis and angiogenesis in breast cancer patients. J. Biomed. Res. 2013, 27, 478–485. [Google Scholar]
- Penela, P. Chapter Three—Ubiquitination and Protein Turnover of G-Protein-Coupled Receptor Kinases in GPCR Signaling and Cellular Regulation. Prog. Mol. Biol. Transl. Sci. 2016, 141, 85–140. [Google Scholar]
- Obeid, E.I.; Conzen, S.D. The role of adrenergic signaling in breast cancer biology. Cancer Biomark. 2013, 13, 161–169. [Google Scholar] [CrossRef]
- Childers, W.K.; Hollenbeak, C.S.; Cheriyath, P. β-Blockers Reduce Breast Cancer Recurrence and Breast Cancer Death: A Meta-Analysis. Clin. Breast Cancer 2015, 15, 426–431. [Google Scholar] [CrossRef]
- Penela, P.; Ribas, C.; Aymerich, I.; Eijkelkamp, N.; Barreiro, O.; Heijnen, C.J.; Mayor, F. G protein-coupled receptor kinase 2 positively regulates epithelial cell migration. EMBO J. 2008, 27, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Sato, P.Y.; Chuprun, J.K.; Peroutk, R.J.; Otis, N.J.; Ibetti, J.; Koch, W.J. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ. Res. 2013, 112, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Elorza, A.; Sarnago, S.; Mayor, F., Jr. Beta-arrestin- and c-Src-dependent degradation of G-protein-coupled receptor kinase 2. EMBO J. 2001, 20, 5129–5138. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Inserte, J.; Ramos, P.; Rodriguez-Sinovas, A.; Garcia-Dorado, D.; Mayor, F., Jr. Degradation of GRK2 and AKT is an early and detrimental event in myocardial ischemia/reperfusion. EBioMedicine 2019, 48, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Pyen, J.; Lee, I.; Lee, H.; Kim, Y.; Kim, T. Cobalt chloride-induced apoptosis and extracellular signal-regulated protein kinase 1/2 activation in rat C6 glioma cells. J. Biochem. Mol. Biol. 2004, 37, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güttinger, S.; Mühlhäusser, P.; Koller-Eichhorn, R.; Brennecke, J.; Kutay, U. Transportin2 functions as importin and mediates nuclear import of HuR. Proc. Natl. Acad. Sci. USA 2004, 101, 2918–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.C.; Steitz, J.A. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc. Natl. Acad. Sci. USA 1998, 95, 15293–15298. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.M.; Gallouzi, I.E.; Steitz, J.A. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J. Cell Biol. 2000, 151, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Rebane, A.; Aab, A.; Steitz, J.A. Transportins 1 and 2 are redundant nuclear import factors for hnRNP A1 and HuR. RNA 2004, 10, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Yang, X.; Kawai, T.; López de Silanes, I.; Mazan-Mamczarz, K.; Chen, P.; Gorospe, M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1: Involvement in the nuclear import of RNA-binding protein HuR. J. Biol. Chem. 2004, 279, 48376–48388. [Google Scholar] [CrossRef] [Green Version]
- Bychkov, E.; Zurkovsky, L.; Garret, M.B.; Ahmed, M.R.; Gurevich, E.V. Distinct cellular and subcellular distributions of G protein-coupled receptor kinase and arrestin isoforms in the striatum. PLoS ONE 2012, 7, e48912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zeng, F.; Liu, Q.; Liu, H.; Liu, Z.; Niu, L.; Li, X. The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Lal, P.; Cerofolini, L.; D’Agostino, V.G.; Zucal, C.; Fuccio, C.; Bonomo, I.; Preet, R. Regulation of HuR structure and function by dihydrotanshinone-I. Nucleic Acids Res. 2017, 45, 9514–9527. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Srikantan, S.; Yang, X.; Lal, A.; Kim, H.H.; Kuwano, Y.; Gorospe, M. Ubiquitin-mediated proteolysis of HuR by heat shock. EMBO J. 2009, 28, 1271–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Huang, A.; Zhang, A.; Zhou, C. HuR promotes breast cancer cell proliferation and survival via binding to CDK3 mRNA. Biomed. Pharmacother. 2017, 91, 788–795. [Google Scholar] [CrossRef]
- Calaluce, R.; Gubin, M.M.; Davis, J.W.; Magee, J.D.; Chen, J.; Kuwano, Y.; Atasoy, U. The RNA binding protein HuR differentially regulates unique subsets of mRNAs in estrogen receptor negative and estrogen receptor positive breast cancer. BMC Cancer 2010, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Galbán, S.; Kuwano, Y.; Pullmann, R., Jr.; Martindale, J.L.; Kim, H.H.; Lal, A.; Lewis, S.M. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1alpha. Mol. Cell. Biol. 2008, 28, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Sheflin, L.G.; Zou, A.P.; Spaulding, S.W. Androgens regulate the binding of endogenous HuR to the AU-rich 3’UTRs of HIF-1alpha and EGF mRNA. Biochem. Biophys. Res. Commun. 2004, 322, 644–651. [Google Scholar] [CrossRef]
- Yasuda, M.; Hatanaka, T.; Shirato, H.; Nishioka, T. Cell type-specific reciprocal regulation of HIF1A gene expression is dependent on 5′- and 3′-UTRs. Biochem. Biophys. Res. Commun. 2014, 447, 638–643. [Google Scholar] [CrossRef]
- Mitsunari, K.; Miyata, Y.; Asai, A.; Matsuo, T.; Shida, Y.; Hakariya, T.; Sakai, H. Human antigen R is positively associated with malignant aggressiveness via upregulation of cell proliferation, migration, and vascular endothelial growth factors and cyclooxygenase-2 in prostate cancer. Transl. Res. 2016, 175, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Morfoisse, F.; Kuchnio, A.; Frainay, C.; Gomez-Brouchet, A.; Delisle, M.B.; Marzi, S.; Bousquet, C. Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1α-independent translation-mediated mechanism. Cell Rep. 2014, 6, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Choy, C.; Raytis, J.L.; Smit, D.D.; Duenas, M.; Neman, J.; Jandial, R.; Lew, M.W. Inhibition of β2-adrenergic receptor reduces triple-negative breast cancer brain metastases: The potential benefit of perioperative β-blockade. Oncol. Rep. 2016, 35, 3135–3142. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; Cole, S.; Costanzo, E.; Bradley, S.; Coffin, J.; Jabbari, S.; Sood, A.K. Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin. Cancer Res. 2003, 9, 4514–4521. [Google Scholar]
- Jafferjee, M.; Reyes Valero, T.; Marrero, C.; McCrink, K.A.; Brill, A.; Lymperopoulos, A. GRK2 Up-Regulation Creates a Positive Feedback Loop for Catecholamine Production in Chromaffin. Cells Mol. Endocrinol. 2016, 30, 372–381. [Google Scholar]
- Aluja, D.; Inserte, J.; Penela, P.; Ramos, P.; Ribas, C.; Iñiguez, M.Á.; Garcia-Dorado, D. Calpains mediate isoproterenol-induced hypertrophy through modulation of GRK2. Basic Res. Cardiol. 2019, 114, 21. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Yang, C.; Wu, J.; Lu, H.; Zheng, X.; Zhang, Y.; Li, Z. Downregulation of β-Adrenoceptors in Isoproterenol-Induced Cardiac Remodeling through HuR. PLoS ONE 2016, 11, e0152005. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Lefkowitz, R.J. β-Arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 2006, 281, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hayre, M.; Eichel, K.; Avino, S.; Zhao, X.; Steffen, D.J.; Feng, X.; Inoue, A. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal 2017, 10, eaal3395. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Huang, J.; Xiang, Y.; Bastepe, M.; Jüppner, H.; Kobilka, B.K.; Huang, X.Y. Dosage-dependent switch from G protein-coupled to G protein-independent signaling by a GPCR. EMBO J. 2007, 26, 53–64. [Google Scholar] [CrossRef]
- Keene, D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 2006, 1, 302. [Google Scholar] [CrossRef]
- Cortazar, A.R.; Torrano, V.; Martín-Martín, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Apaolaza, I. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reglero, C.; Lafarga, V.; Rivas, V.; Albitre, Á.; Ramos, P.; Berciano, S.R.; Tapia, O.; Martínez-Chantar, M.L.; Mayor Jr, F.; Penela, P. GRK2-Dependent HuR Phosphorylation Regulates HIF1α Activation under Hypoxia or Adrenergic Stress. Cancers 2020, 12, 1216. https://doi.org/10.3390/cancers12051216
Reglero C, Lafarga V, Rivas V, Albitre Á, Ramos P, Berciano SR, Tapia O, Martínez-Chantar ML, Mayor Jr F, Penela P. GRK2-Dependent HuR Phosphorylation Regulates HIF1α Activation under Hypoxia or Adrenergic Stress. Cancers. 2020; 12(5):1216. https://doi.org/10.3390/cancers12051216
Chicago/Turabian StyleReglero, Clara, Vanesa Lafarga, Verónica Rivas, Ángela Albitre, Paula Ramos, Susana R. Berciano, Olga Tapia, María L. Martínez-Chantar, Federico Mayor Jr, and Petronila Penela. 2020. "GRK2-Dependent HuR Phosphorylation Regulates HIF1α Activation under Hypoxia or Adrenergic Stress" Cancers 12, no. 5: 1216. https://doi.org/10.3390/cancers12051216