1. Introduction
Bladder cancer (BlCa) is the 9th most common cancer type worldwide, with an estimated 400,000 new cases and 160,000 deaths per year, in both genders [
1]. Men are more susceptible and in more developed regions, BlCa represents has the 6th highest incidence among different cancers. BlCa may be categorized according to clinical, pathological, or molecular characteristics. Muscle-invasive bladder cancer (MIBC) which accounts for about 20% of all cases, represents the more aggressive form, being more likely to progress and metastasize, whereas non-muscle-invasive bladder cancer (NMIBC) is mostly characterized by multiple local recurrences, which, over time, also entail increased risk of invasion. Indeed, although most newly diagnosed patients present NMIBC (approximately 80%), there is variable risk of progression, with increased morbidity [
2,
3,
4].
Sirtuins (SIRTs) are a family of NAD
+-dependent deacetylases highly conserved among all living organisms. Seven different SIRTs (SIRT1–7) are described in mammals, also known as Class III histone deacetylases (HDACs), which differing among each other in substrate specificity and catalytic activity [
5]. Within the cell, these enzymes participate in control of important biological processes, including cell division, differentiation, metabolism, genomic stability, survival, senescence and organismal lifespan [
6]. In addition, SIRTs expression is deregulated in many cancer types [
7,
8,
9]. SIRT1 and SIRT3 may be up- or downregulated depending on the cancer type, acting either as oncogenes (e.g., colorectal or oral cancer) [
10,
11], or tumor suppressors (e.g., SIRT1 in bladder cancer, SIRT1 and SIRT3 in breast and prostate cancer) [
12,
13]. SIRT2 and SIRT4, on the other hand, are considered tumor suppressor, downregulated in glioma and hepatocellular carcinoma (SIRT2) [
14,
15], and bladder, gastric and lung cancer (SIRT4) [
16], among others. Although little is known about the role of SIRT5 in neoplastic transformation, it is overexpressed in non-small-cell lung cancer (NSCLC) [
17]. Concerning SIRT6, it is downregulated in several cancers, including hepatocellular carcinoma [
18,
19], but it is overexpressed in breast cancer and NSCLC [
20,
21]. Finally, an oncogenic function has been proposed for SIRT7 as it was found overexpressed in several epithelial cancers [
22,
23]. Moreover, SIRT7 is mostly localized in the nucleus and its deacetylase function needs to be disclosed, with only a few well characterized substrates reported [
24,
25]. SIRT7 deacetylase activity is related with histones, disclosing highly selective activity for lysine 18 of histone H3 (H3K18Ac), notwithstanding other protein targets involved in cell homeostasis and stress response [
24]. SIRT7 is also involved in ribosomes biogenesis and other mechanisms of cell proliferation [
26,
27].
Although sirtuins have been characterized in various neoplasms, their putative role in BlCa development and progression remains elusive with only a few published studies to date [
12,
16,
28]. Thus, we sought to comprehensively characterize SIRTs expression in BlCa tissues, comparing with normal bladder mucosa, assessing their potential as prognostic biomarkers. Furthermore, the phenotypic impact of SIRT7 deregulation in BlCa cells was also evaluated.
3. Discussion
Sirtuins, also known as Class III HDACs, are involved in many biological processes, including cell division, differentiation, metabolism, genomic stability, survival, senescence and organismal lifespan [
6], and variable SIRTs deregulated expression has been reported in many cancer types [
7,
8,
9]. Remarkably, sirtuins may act either as oncogenes or tumor suppressor genes in different tumor models [
12,
13,
14,
15,
16]. Thus, better understanding of the biological role of these unique enzymes in tumorigenesis might provide novel biomarkers for disease management as well as putative therapeutic targets.
Herein, we report, for the first time, a comprehensive evaluation of sirtuins (
SIRT1-7) mRNA expression in a series of 94 BlCa cases from a single institution and respective validation in TCGA dataset, comparing with normal bladder mucosa. Significant differences were depicted for all sirtuins, except for
SIRT3, with
SIRT1,
2,
4 and
5 downregulation and
SIRT6 and
7 overexpression. These findings were mostly validated in TCGA dataset, especially for
SIRT6 and
SIRT7. Previous studies on BlCa have mainly focused on SIRT1 and SIRT4 and were mostly based in publicly available datasets only [
12,
16], not providing a global picture of sirtuin deregulation in BlCa. Interestingly, besides significant differences between BlCa and urothelium, differential expression of some sirtuins was also disclosed between tumors with dissimilar clinical and biological behavior. Interestingly, although SIRT7 was overexpressed in BlCa, the more aggressive tumors (IHG) disclosed significantly lower expression levels compared to PLG and PHG, both in IPO Porto’s and TCGA cohorts. Furthermore, in MIBC both
SIRT7 transcript and protein disclosed a significant reduction compared to NMIBC. Remarkably, previous reports on SIRT7 in uterus, colon, kidney, ovary and prostate cancers revealed increased expression levels [
22,
24]. Nevertheless, in all those models, a strict oncogenic role was proposed for
SIRT7, whereas our findings suggest that, at the least in bladder carcinogenesis,
SIRT7 may play a dual role, eventually context-dependent. Furthermore, although we did not find significant differences in
SIRT6 transcript levels between PHG and IHG tumors (either in our and TCGA cohort), nor between different stages of MIBC, Wu et al. reported that SIRT6 protein levels declined significantly from T2 to T4 MIBC, which also suggests that the functional importance of sirtuins may change along cancer progression [
28]. The observed decreased overall survival in BlCa patients with lower
SIRT7 expression in TCGA cohort (eventually associated with higher grade and stage, as well as molecular BlCa subtype) further suggests that decreased SIRT7 impacts on neoplastic cell biology, promoting a more aggressive phenotype.
Taking in consideration
SIRT7 expression patterns in normal and neoplastic urothelium, we sought to characterize the effects of its deregulated expression at molecular level. Thus, after characterization of SIRT7 transcript and protein expression levels in neoplastic and benign urothelial cell lines, three cell lines were chosen for further experiments as their profile more closely replicated that of a spectrum of BlCa tissues. Interestingly, in vitro phenotypic assays demonstrated that although SIRT7 downregulation did not affect cell proliferation or apoptosis, with the exception of 5637 cell line, rather impairing cell motility, decreasing both cell migration and cell invasion capabilities in all modulated cell lines. These effects immediately suggested a putative association between SIRT7 and EMT, a process that is key for tumor invasion and metastization [
31,
32]. This hypothesis was confirmed as SIRT7 knockdown significantly associated with decreased E-Cadherin expression and augmented expression of a mesenchymal marker (N-Cadherin), and EMT-inducing transcription factors (SLUG and SNAIL), in the modulated BlCa cells. Although only a few studies investigated the relationship between sirtuins and EMT [
33], SIRT7 depletion in PC3 prostate cancer cell line was shown to impair migration and invasiveness, reprograming neoplastic cells towards epithelial gene expression [
22]. Our results indicate the opposite trend in BlCa cells, which might be due to the pleiotropic effects of sirtuins and/or the dissimilar molecular profile of prostate and urothelial cancer cells [
34].
Remarkably, we found that the mechanism by which SIRT7 affects CDH1 expression, and thus EMT, is probably linked to EZH2. EZH2 is a well-known member of the polycomb repressive complex 2 (PRC2), described as being involved in the transcription repression by catalyzing the repressive H3K27
me3 mark at several gene promotes, including
CDH1 [
35,
36,
37]. Previously, proteomic analyses demonstrated that among 250 candidate substrates, EZH2 was a SIRT7 substrate [
30,
38]. In our study, sh-SIRT7 cells showed increased total and acetylated EZH2 expression, followed by decreased ECAD protein. Concurrently, increased H3K27
me3 deposition at CDH1 promoter was also observed in the same cells. Thus, when SIRT7 is downregulated, EZH2 activity might be enhanced by acetylation, contributing to
CDH1 transcription repression through H3K27
me3 deposition in its promoter, as previously reported [
30].
CDH1 repression and concomitant EMT transcription factors’ upregulation (e.g., SNAIL and SLUG), might then lead to a shift from epithelial to mesenchymal phenotype, allowing for increased cancer cell motility. Indeed, upregulation of these specific EMT transcription factors, due to diverse upstream signals and post-transcriptional mechanisms, also corroborates our hypothetic mechanism. Indeed, both SNAIL and SLUG were shown to cooperate with PRC2, and specifically with EZH2, towards controlling the expression of several genes, relevant for neural crest development, including CDH1 [
39,
40]. Moreover, during EMT, Snail was proven to recruit EZH2 to specific genomic sites by the enrollment of the long non-coding RNA HOTAIR [
41]. Thus, our results suggest that EMT transcription factors’ upregulation in sh-SIRT7 BlCa cells might be due to the phenotypic shift in invasion and migration and, at the molecular level, by recruitment of EZH2 to specific targets.
Thus far, only a limited number of upstream SIRT7 transcription regulators, such as histone deacetylase 3 (HDAC3) and the X-box binding protein 1 (XBP1) molecules have been identified [
42,
43]. At post-transcriptional level, SIRT7 was shown be negatively regulated by microRNAs, such as those from microRNA-125 family [
44]. However, few reports deal with SIRT7 regulation by post-translational modifications [
45,
46,
47]. Hence, it would be important to further explore how regulation of SIRT7 occurs in BlCa, and unveil how SIRT7 expression shift occurs from non-invasive to invasive BlCa.
Moreover, although discovery of new prognostic biomarkers for BlCa is imperative for more effective disease management, the aim of our study was mostly to uncover how expression of all sirtuins was altered in BlCa, and to investigate whether they might be implicated in bladder carcinogenesis and/or disease progression and invasiveness. Indeed, we were able to demonstrate that for SIRT7. Nonetheless, the analyzed cohort was composed by patients diagnosed over a large time span (1991–2011) and the small number of events occurring in this cohort precluded a more robust and detailed statistical analysis.
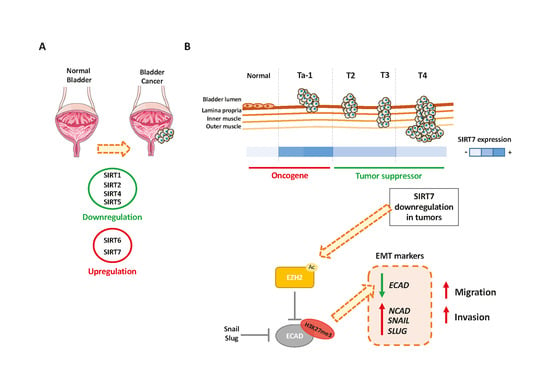
Overall, our results suggest that increased
SIRT7 expression occurs during urothelium neoplastic transformation, which usually results in the formation of non-invasive, papillary neoplasms or flat lesions like urothelial carcinoma in situ [
48]. At this stage, it is likely that SIRT7 is involved in promoting cell growth and survival, which are key to neoplastic development, eventually through deacetylation of H3K18 [
24]. Then, transition to an invasive phenotype might require
SIRT7 downregulation, involving EZH2 upregulation and acetylation, among other mechanisms, which promote EMT. Although the mechanism of SIRT7 deregulation in BlCa remains elusive, it is tempting to speculate whether it might be due to epigenetic mechanisms, which allow for the suggested plasticity of
SIRT7 gene expression during carcinogenesis and tumor progression.
4. Materials and Methods
4.1. Patients and Samples
Patients with primary bladder urothelial carcinoma (UCC), treated with transurethral resection (TUR) or radical cystectomy, between 1991 and 2011 at Portuguese Oncology Institute of Porto (IPO Porto), Portugal (
n = 94). A set of 19 morphologically normal bladder mucosa (NB) tissue samples was obtained from BlCa-free individuals (prostate cancer patients submitted to radical prostatectomy) and served as controls. All specimens were fresh-frozen at −80 °C and subsequently cut in a cryostat for confirmation of representativity and nucleic acid extraction. From each specimen, fragments were collected, formalin-fixed, and paraffin embedded for routine histopathological examination, including grading and pathological staging, by a dedicated uropathologist [
49]. Relevant clinical data was collected from clinical charts (
Table 1). Patients and controls were enrolled after informed consent. This study was approved by the institutional review board (Comissão de Ética para a Saúde) of IPO Porto (CES103-14).
4.2. Real-Time Quantitative PCR (RT-qPCR)
RNA was extracted from tissues and from MGHU3, 5637 and J82 sh-scramble and sh-SIRT7 cells using TRIzol
® (Invitrogen, Carlsbad, CA, USA), according to manufacturer’s instructions. For tissue RNA, cDNA synthesis was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems
®, Foster City, CA, USA), according to manufacturer’s instructions. Sirtuins transcript levels were quantified by RT-qPCR. Expression levels were evaluated using 4.5 µL of diluted cDNA, 5 µL of TaqMan
® Universal PCR Master Mix No AmpErase
® UNG (Applied Biosystems
®) and 0.5 µL of TaqMan
® Gene Expression Assay, specific for each sirtuin and reference genes, as described in
Table S1 (Applied Biosystems
®). Each sample was run in triplicate and the RT-qPCR conditions were: 2 min at 50 °C, followed by enzyme activation for 10 min at 95 °C, and 45 cycles which included a denaturation stage at 95 °C for 15 s and an extending stage at 60 °C for 60 s onds.
HPRT and
SDHA were both used as reference genes for normalization. Relative expression of target genes tested in each sample was determined as: [Gene Expression Level = (Gene Mean Quantity/(
HPRT1 &
SDHA) Mean Quantity) × 1000].
Concerning cell lines, 1000 ng of RNA were reverse transcribed using RevertAid RT kit (Thermo Fisher Scientific Inc., Waltham, MA, USA), according to manufacturer’s instructions. For 100 ng of cDNA, SIRT7 and NCAD transcript levels were quantified using TaqMan® Gene Expression Assay, as described above, in 396 well plates LightCycler480II (Roche, Basel, Switzerland). For ECAD and EZH2 genes, transcription levels were also evaluated in J82 sh-scramble and sh-SIRT7 cells in 396 well plates LightCycler480II (Roche) using Xpert Fast SYBER Mastermix Blue (GRiSP Research Solutions, Porto, Portugal) with specific primers (S2). Transcript levels for studied genes were then evaluated using ΔΔCt method, with HPRT and BGUS housekeeping genes as reference genes.
4.3. Immunohistochemistry
Immunohistochemistry was performed using the Novolink™ Max Polymer Detection System (Leica Biosystems, Wetzlar, Germany]. Three-μm thick tissues s tions from formalin-fixed and paraffin-embedded BlCa (corresponding to 88 of the 94 cases, for which there was archived tissue available) and controls (n = 25, consisting of normal urothelial mucosa collected from the urether of nephrectomy specimens with renal cell tumors) were cut, deparaffinized and rehydrated. Antigen retrieval was accomplished by microwaving the specimens at 800 W for 20 min in 10 mM sodium citrate buffer, pH = 6. Endogenous peroxidase activity was blocked by incubating the s tions in 0.6% hydrogen peroxide solution for 20 minutes. Primary monoclonal antibody for SIRT7 (HPA053669, Sigma-Aldrich™, St. Louis, MO, USA) was used in 1:500 dilution, and incubated at room-temperature (RT) for one hour. Then, 3,3′-diaminobenzidine (Sigma-Aldrich™) was used as chromogen for visualization and slides were mounted with Entellan® (Merck-Millipore, Burlington, MA, USA). Normal testicular tissue, showing intense SIRT7 immunoreactivity was used as positive control. SIRT7 immunoexpression was evaluated by a dedicated uropathologist and cases were classified using a semi-quantitative scale for both staining intensity (0—no staining; 1—intensity lower than normal urothelium; 2—intensity equal to normal urothelium; 3—intensity higher than normal urothelium) and percentage of positive cells (0—< 10%; 1—10–33%; 2—33–67%; 3— > 67%), in each tumor. Staining intensity and percentage of positive cell results were then combined in a single score (Score S = staining intensity x percentage of positive cells) assigned to each tumor, and further stratified into low expression (S < 4 = IHC−) and high expression (S ≥ 4 = IHC+ ) groups, which correspond to cases with less than 33% stained cells or staining intensity lower than normal urothelium, and cases with at least 33% stained cells with an intensity equal to or higher than normal urothelium.
4.4. TCGA Dataset Analysis in Bladder Urothelial Carcinoma Patients
The Cancer Genome Atlas (TCGA) dataset was interrogated for data on
SIRT1-7 expression and clinical information, when available, of 408 BlCa patients and 19 matched controls. All expression data from samples hybridized at the University of North Carolina, Lineberger Comprehensive Cancer Center, using Illumina HiSeq 2000 RNA Sequencing version 2 analysis, were downloaded from the GDC data portal (
https://portal.gdc.cancer.gov/). Biospecimen Core Resources (BCRs) provided the clinical data of each patient. This data is available for download through the GDC data portal (
https://portal.gdc.cancer.gov) (
Table 2).
4.5. Cell Lines Culture
5637, J82, T24 and TCCSUP BlCa cell lines and normal bladder cell line SV-HUC1 were selected for this study. All cell lines were purchased from ATCC and grown using recommended medium (Biochrom-Merck, Berlin, Germany) supplemented with 10% fetal bovine serum (FBS, Biochrom) and 1% penicillin/streptomycin (GIBCO, Invitrogen) at 37 °C and 5% CO2. Mycoplasma test was regularly performed for all cell lines using TaKaRa PCR Mycoplasma Detection Set (Clontech Laboratories, Mountain View, CA, USA).
4.6. Lentiviral Transduction
SIRT7 knockdown was performed through lentiviral transduction in J82 cell line using GIPZTM Lentiviral shRNA particles (DharmaconTM, Lafayette, CO, USA), and in MGHU3 and 5637 cell lines using SMARTvectorTM Inducible Lentiviral shRNA particles (target sequence: 5’-CCCTGCGTGCTGGTGAAGA-3’). All sh-SIRT7 vectors included the green fluorescent protein (GFP). Briefly, cells were seeded in 12 well/plate at density of 4 × 104 cells/well and incubated during 24 h in humidified chamber at 37 °C and 5% CO2. Then, culture medium was removed and 500 µL of completed medium with 8 µg/mL polybrene and lentiviral sh-SIRT7 particles with MOI 10 concentration were added. After 48 h, 1 µg/mL of puromycin dihydrochloride (Clontech Laboratories) was added to select stably transfected cells. For MGHU3 and 5637 cells, after puromycin selection, a treatment was performed with 100 ng/mL doxycycline in order to induce the Tet-On 3 G bipartite induction system. Additionally, J82, MGHU3 and 5637 control cells were generated using a non-target scramble shRNA under the same previously described conditions.
For clone selection, 10, 20 and 50 cells/well were seeded in 96 well plate after stable selection, and the isolated clones were grown until confluence for protein extraction, and subsequent western blot analysis for SIRT7 expression. Moreover, sh-SIRT7 cells were observed under the fluorescence microscope for GFP expression.
4.7. Protein Extraction
BlCa cell lines, sh-scramble/CTRL and sh-SIRT7 cells were grown until 80% confluence and homogenized in Kinexus lysis buffer supplemented with proteases inhibitors cocktail. Then, cells were sonicated for 5 cycles of 30 s ON and 30 s OFF (Bioruptor®, Diagenode, Liège, Belgium). After centrifugation, the supernatant was collected, and total protein was quantified according the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc.), according to the manufacture procedure.
For subcellular fractionation, Nuclear Extract kit (Active Motif, Carlsbad, CA, USA) was used. Briefly, bladder cancer cell lines, sh-scramble/CTRL and sh-SIRT7 cells were washed in 1 X PBS with phosphate inhibitors and scrapped. Subsequently, cells were suspended in hypotonic buffer and incubated on ice during 15 min. Additionally, a detergent was added, and samples were centrifuged at 14,000 rpm during 30 s at 4 °C. Supernatant (cytoplasmic fraction) was collected and stored at −80 °C until use. Then, cell pellets were resuspended and incubated in a complete lysis buffer solution (lysis buffer with proteases inhibitor cocktail and 10 mM DTT), following centrifugation and supernatant (nuclear fraction) collection and storage at −80 °C. Nuclear and cytoplasmic proteins were then quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc.), according to manufacture procedure.
4.8. Western Blot and Co-Immunoprecipitation
Aliquots of 30 µg total protein was separated in 10% polyacrylamide gel by SDS-PAGE and transferred onto an immunoblot PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA) in a 25 mM Tris-base/glycine buffer using a Trans-Blot Turbo Transfer system (Bio-Rad Laboratories). Membranes were blocked with 5% milk in TBS/0.1% Tween (TBS/T pH = 7.6) for 1 hour at RT. After incubation with primary antibodies for SIRT7 (1:350, HPA053669, Sigma-Aldrich) or EZH2 (1:500, NCL-L-EZH2, Leica Biosystems) for 1 h 30 min at RT, the membranes were washed in TBS/T and incubated with s ondary antibody coupled with horseradish peroxidase for 1 h at RT. The bound was visualized by chemiluminescence (Clarity WB ECL substrate, Bio-Rad) and quantification was performed using band densitometry analysis from the ImageJ software (version 1.6.1, National Institutes of Health). β-Actin (1:10,000, A1978, Sigma-Aldrich) for total protein and cytoplasmic protein analysis, and Laminin B1 (1:1000, D4Q42, Cell Signaling Technologies, Danvers, MA, USA) for nuclear protein, were used as loading controls. For co-immunoprecipitation assays, 200µg of total protein from cell lysates were incubated with anti-acetylated-lysine antibody (#9441, Cell Signaling Technology) and immunoprecipitated with Protein A/G magnetic beads (#16-663, Sigma-Aldrich) overnight at 4 °C. The final eluates were blotted with EZH2 primary antibody, as detailed above. Detailed information about western blot can be found at
Figure S8.
4.9. Immunofluorescence (IF)
Wild-type MGHU3, 5637 and J82, sh-scramble/CTRL and/or sh-SIRT7 cells were seeded on cover slips at 20,000 cells/well, overnight. Briefly, cells were fixed in methanol during 10 min and then blocked with 5% bovine serum albumin (BSA) during 30 min. After overnight SIRT7 (1:500, HPA053669, Sigma-Aldrich), ECAD (1:150, #3195, Cell Signaling Technology) and/or NCAD (1:50, #13116, Cell Signaling Technology) incubation at RT, cells were incubated with s ondary antibody anti-rabbit IgG-TRITC (1:500, T6778, Sigma-Aldrich) during 1 h at RT. Finally, after 1× PBS wash, cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) (AR1176, BOSTER Biological Technologies (Pleasanton, CA, USA) r in mounting medium. Pictures were taken on a IX51 fluorescence microscope (Olympus, Tokyo, Japan) equipped with an Olympus XM10digital camera using CellSens software.
4.10. Chromatin Immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) analysis was performed in sh-scramble/CTRL and sh-SIRT7 cells. For the crosslink step, formaldehyde solution (Sigma) was added to adherent cells (~1 × 107) media at 1% final concentration, and after an 8 minutes’ incubation at RT, reaction was immediately stopped by adding 1.5 mL of 2.5 M glycine and incubating for 5 min. Cells were then washed twice with ice-cold 1× PBS, scraped, harvested and centrifuged at 4 °C.
Cell pellets were homogenized with cell lysis buffer (10 mM Tris-HCL pH7.5, 10 mM NaCL, 0.5% NP-40) and left on ice for 1 h30, with intermittent vortexing, and then centrifuged at 4 °C. At this point, pellets were re-suspended in nuclei lysis buffer (50 mM Tris-HCL pH = 7.5, 10 mM EDTA pH = 8, 1% SDS) and incubated for 15 min on ice, followed by adding of 2× volumes of IP dilution buffer (16.7 mM Tris-HCL pH7.5, 167 mM NaCl, 1.2 mM EDTA pH = 8, 0.01% SDS). Chromatin was solubilized and sheared to 200–400 bp fragments using an ultra-sonicator (Bioruptor®, Diagenode) for 15 cycles of 30 s ON and 30 s OFF. Soluble chromatin was then centrifuged and stored at −80 °C until further use.
Before immunoprecipitation (IP), each 50 µL of chromatin was 1:10 diluted in dilution buffer (1.2 mM EDTA pH = 8, 16.7 mM Tris pH = 8, 167 mM NaCl, 1.1% Triton X-100, 0.01% SDS), and 5 µL of this solution was reserved in a new tube for the input control. After this, 20 µL of protein A+G magnetic beads (Millipore) were added to each IP sample, as well as ChIP-grade antibodies for Histone H3 (ab1791, Abcam, Cambridge, UK), tri-methylation of lysine 27 of histone H3 (H3K27me3, 07-449, Millipore), positive control (RNA polymerase II) and negative control (mouse IgG), at assay dependent concentration. IPs were incubated overnight at 4 °C with rotation. After incubation, magnetic beads were precipitated using 1.5 mL tubes magnet rack and washed with four different salt concentration buffers. At this point, elution buffer (50 mM Tris-HCL pH = 7.5, 10 mM EDTA pH = 8, 1% SDS) was added to all samples and input control, as well as 200 µg/mL of RNAse A, following an incubation of 30 min at 37 °C. After this, samples were incubated with proteinase K for 2 h at 62 °C, followed by an incubation of 10 min at 95 °C, for cross-link reversion.
DNA was extracted from samples using the Qiaquick gel extraction kit (Qiagen, Hilden, Germany), according to manufacture procedures, and stored at −20 °C until further use. For qPCR, two pairs of primers for CDH1 promoter were designed, both for ~325 bp before TSS (F—5′-TAACCCACCTAGACCCTAGCAA-3′, R–5′-GCTGATTGGCTGAGGGTTCA-3′) and for ~600 bp before TSS (F—5′-ACCTGTACTCCCAGCTACTAGA-3′, R—5′-GATGGGGTCTCACTCTTTCACC-3′). RT-qPCR was performed as mentioned above, and the relative amount of promoter DNA was normalized using Input Percent method.
4.11. Proximity Ligation Assay (PLA)
Sh-scramble/CTRL and sh-SIRT7 cells, were seeded in 1 cm2 coverslips and allowed to grow overnight. Then, cells were fixed in 4% formaldehyde (Sigma) for 10 min and permeabilized in 0.5% Triton X-100 (Sigma), for 5 min, at RT and gently stirred. PLA assay was performed using the commercial kit Duolink In Situ (OLINK Bioscience, Uppsala, Sweden), according to manufacturer’s instructions. The antibodies used were Histone H3 (ab1791, Abcam, Cambridge, UK), tri-methylation of Lysine 27 of Histone H3 (H3K27me3, 07-449, Millipore), SIRT7 (HPA053669, Sigma-Aldrich) and EZH2 (NCL-L-EZH2, Leica Biosystems). After the procedure, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (AR1176, BOSTER Biological Technology, Pleasanton, CA, USA) in mounting medium. Pictures were taken on an Olympus IX51 fluorescence microscope equipped with an Olympus XM10 digital camera using CellSens software.
4.12. Cell Viability Assay
To assess the role of SIRT7 in cell growth, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenultetrazolium (MTT) assay (Sigma-Aldrich) was performed. Briefly, sh-scramble/CTRL and sh-SIRT7 cells were seeded at 3000 cell/well density, overnight, in 96 well plate. Then, 5 µg/mL MTT solution in completed MEM medium was incubated during 1 h at 37 °C for 0 h, 24 h, 48 h and 72 h. Then, formazan crystals formed were dissolved in dimethyl sulfoxide (DMSO) and spectrophotometric measurement was done at 540 nm, using 655 nm as a reference absorbance (Fluostar Omega, BMG Labtech, Offenburg, Germany). The optical density (OD) obtained for 24 h, 48 h and 72 h was normalized for the 0 h time point. At least three independent experiments were performed.
4.13. Apoptosis Assay
Apoptosis was assessed using the APOPercentageTM kit (Biocolor Ltd., Belfast, Northern Ireland, UK). This assay uses a dye that is integrated by cells undergoing at early stage of apoptosis due to phosphatidylserine transmembrane movements, which results in APOPercentage dye incorporation by cells. Briefly, sh-scramble/CTRL and sh-SIRT7 cells were seeded in 24 well plate at density of 25,000 cell/well and incubated during 72 h in a humidified chamber at 37 °C and 5% CO2. At this time point, cells were incubated with 300 µL/well of APOPercentage dye solution at ratio 1:20 respectively, during 20 min at 37 °C. Then, cells were washed with PBS1 X and detached from well plate with TrypleTM Express (GBICO) during 10 min at 37 °C. After that, APOPercentage Dye Release reagent was added and plate were vigorously agitated during 15 min, following colorimetric measurement at 550 nm with 620 nm reference filter (Fluostar Omega). The H2O2 was used as a positive control. The OD obtained for apoptosis assay was normalized for the OD obtained by viability assay at the same time point. At least three independent experiments were performed.
4.14. Wound Healing Assay
Cells were seeded in 6 well plate at a density of 7.5 × 10
5 cell/well and allowed reach confluence at 37 °C, 5% CO
2. Then, a “wound” was made by manual scratching with a 200 µL pipette tip and cells were gently washed with 1× PBS. The “wounded” areas were photographed in specific wound sites (two sites for each wound) at 40× magnification using an Olympus IX51 inverted microscope equipped with an Olympus XM10 Digital Camera System every 24 h until wound closure. The relative migration distance (5 measures by wound) was calculated with the following formula: relative migration distance (%) = (A–B)/C × 100, where A is the width of cell wound at 0 h incubation, B is the width of cell wound after specific h of incubation, and C is the width mean of cell wound for 0 h of incubation. For relative migration distance, the results were analyzed using the beWound-Cell Migration Tool (Version 1.5) [
50]. At least three independent experiments were performed.
4.15. Invasion Assay
Invasion capacity of sh-scramble/CTRL and sh-SIRT7 cells was evaluated using a 24 well BD Biocoat Matrigel Invasion Chambers (BD Biosciences, San Jose, CA, USA). After rehydration of BD Matrigel Chambers during 2 h with MEM medium at 37 °C, cells at a density of 25,000 cells/ insert were seeded and incubated during 24 h at 37 °C in 5% CO2. Then, the non-invading cells were removed by with swab and invaded cells were fixed with methanol and staining with DAPI. Invaded cells were counted on an Olympus IX51 fluorescence microscope equipped with an Olympus XM10 digital camera using CellSens software. The % invasion normalized for total of amount cell seeded in BD Matrigel Chamber.
4.16. Statistical Analysis
All statistical analyses were performed using IBM® SPSS® Statistic software version 23 (IBM-SPSS Inc., Chicago, IL, USA) and graphs were built using GraphPad Prim 7.0 (GraphPad Software Inc., La Jolla, CA, USA). Significance level was set at p < 0.05, and Bonferroni’s correction was used when appropriate.
For both BlCa cohorts (IPO’s and TCGA), when applicable, Mann-Whitney U test (MW) was used to test for differences in sirtuins expression levels between NB and UCC tissue samples, pathological stages of cases divided in Ta-1 (NMIBC) and T2-4 (MIBC), and patients’ gender, and to assess differences in sh-scramble versus sh-SIRT7 conditions. Kruskall-Wallis test (KW) was performed to test for differences among UCC subtypes (papillary-low grade, papillary-high grade and invasive-high grade). Spearman’s rho was used to assess the correlation between SIRTs expression levels and age of the patients at diagnosis, and between SIRT7 and ECAD or NCAD expression levels. Associations between clinical grade or pathological stage and immunoexpression results were assessed by chi-square or Fisher’s exact test, and Somers’d directional measure was also computed.
Disease-specific and disease-free survival curves (Kaplan-Meier with log rank test) were computed for standard variables (tumor stage and grade) and for categorized SIRTs transcript levels. Moreover, the same analyses were also performed separately for NMIBC and MIBC cases. A Cox-regression model comprising all significant variables (univariable and multivariable model) was computed to assess the relative contribution of each variable to the follow-up status.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}