ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 While ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
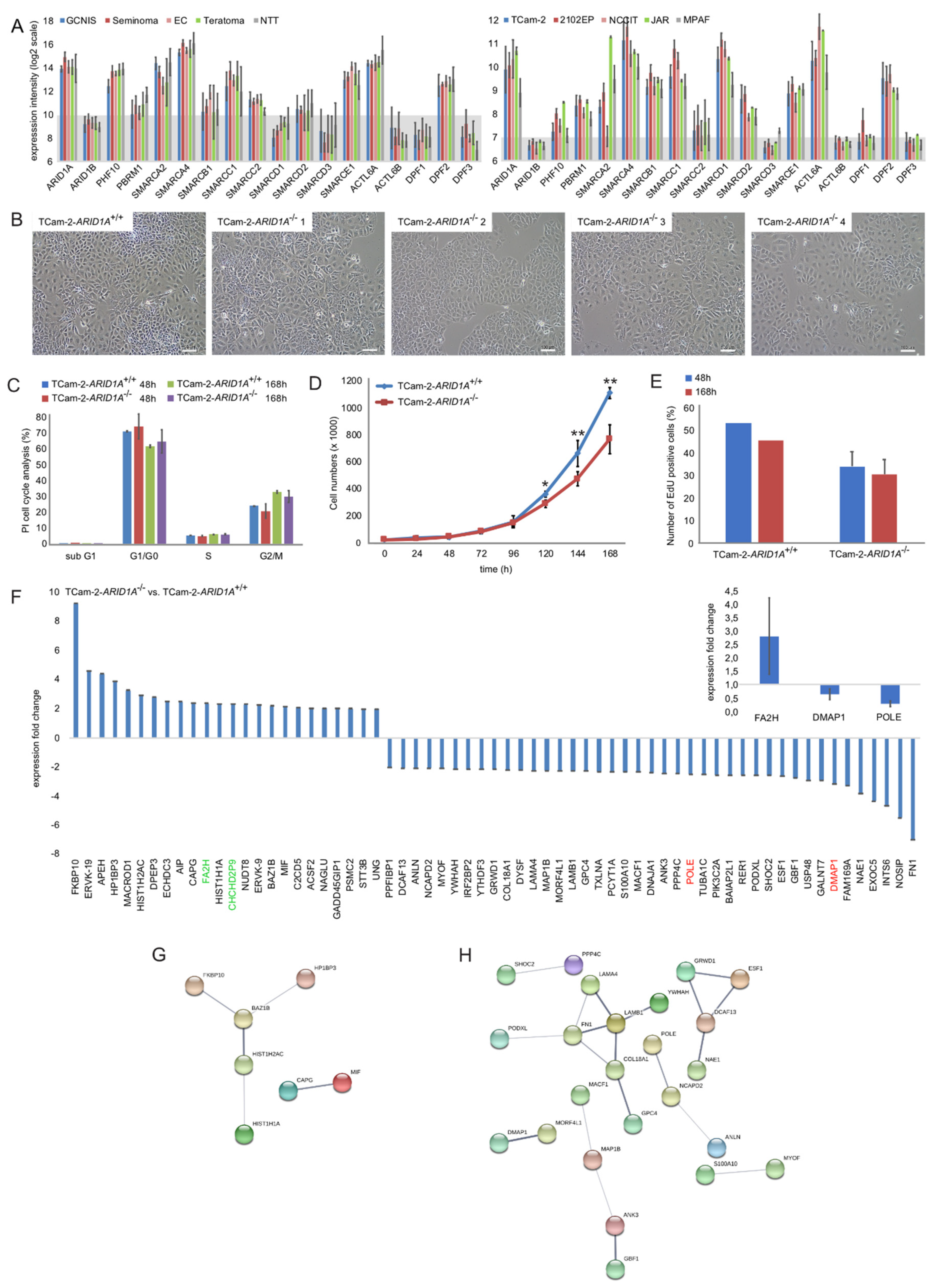
2.1. Genomic and Molecular Characterization of ARID1A and the SWI/SNF Complex
2.2. Deciphering the Molecular Function of ARID1A in GCTs
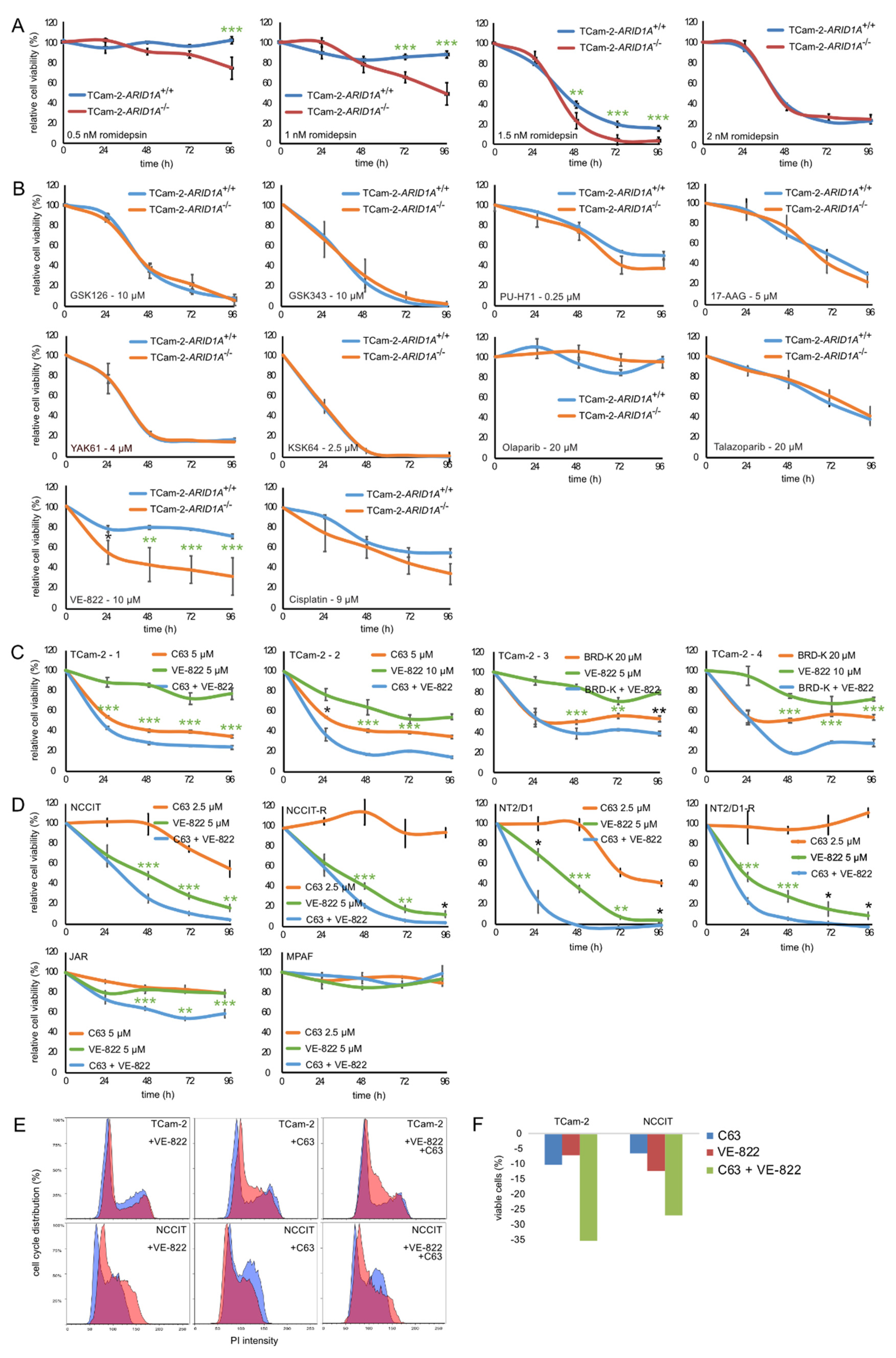
2.3. Targeting ARID1A as a Therapeutic Option
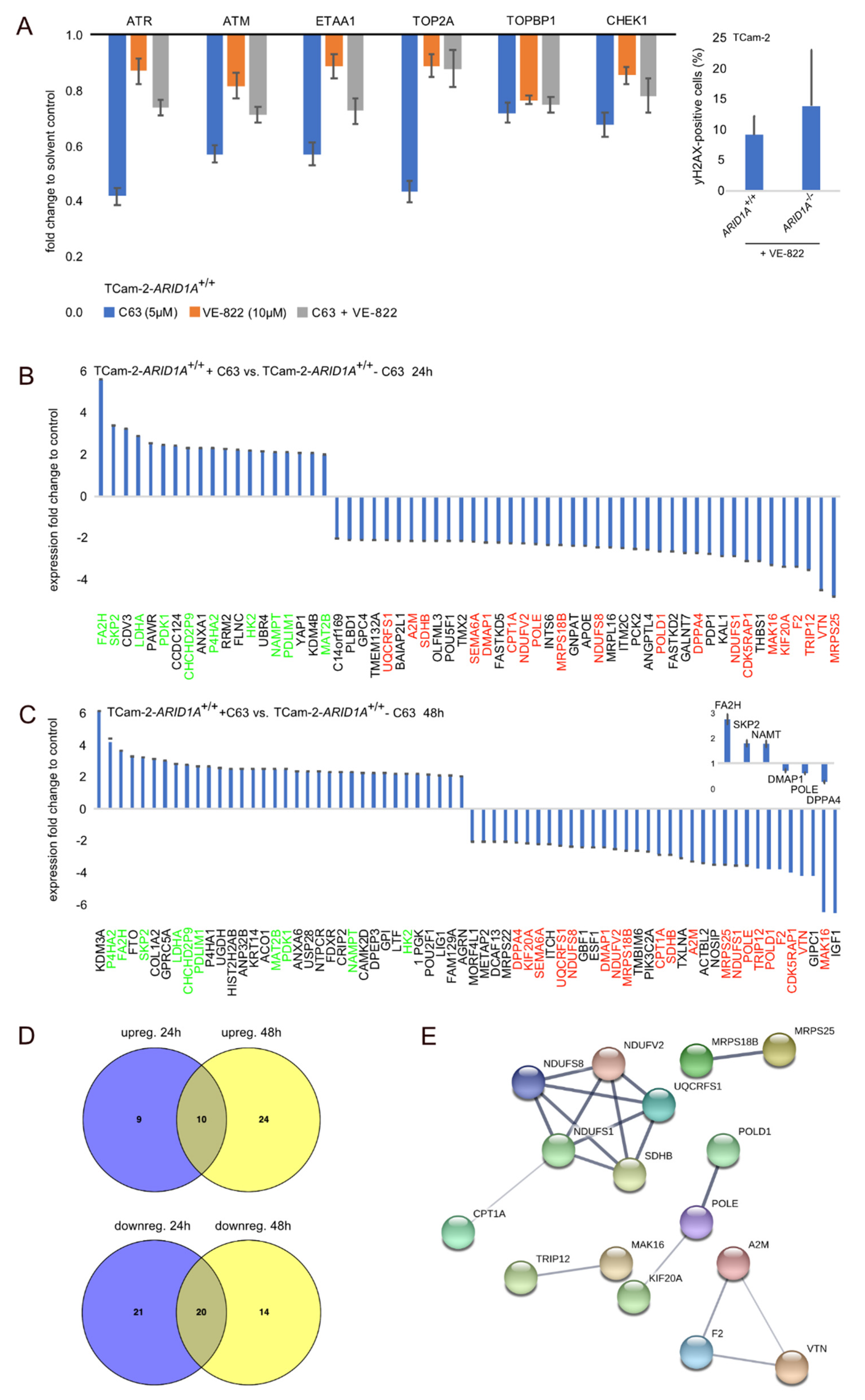
2.4. Deciphering the Molecular Function of the ARID1A-Inhibitor C63
2.5. The Effect of ARID1A-Deficiency on the Pluripotency Program
3. Discussion
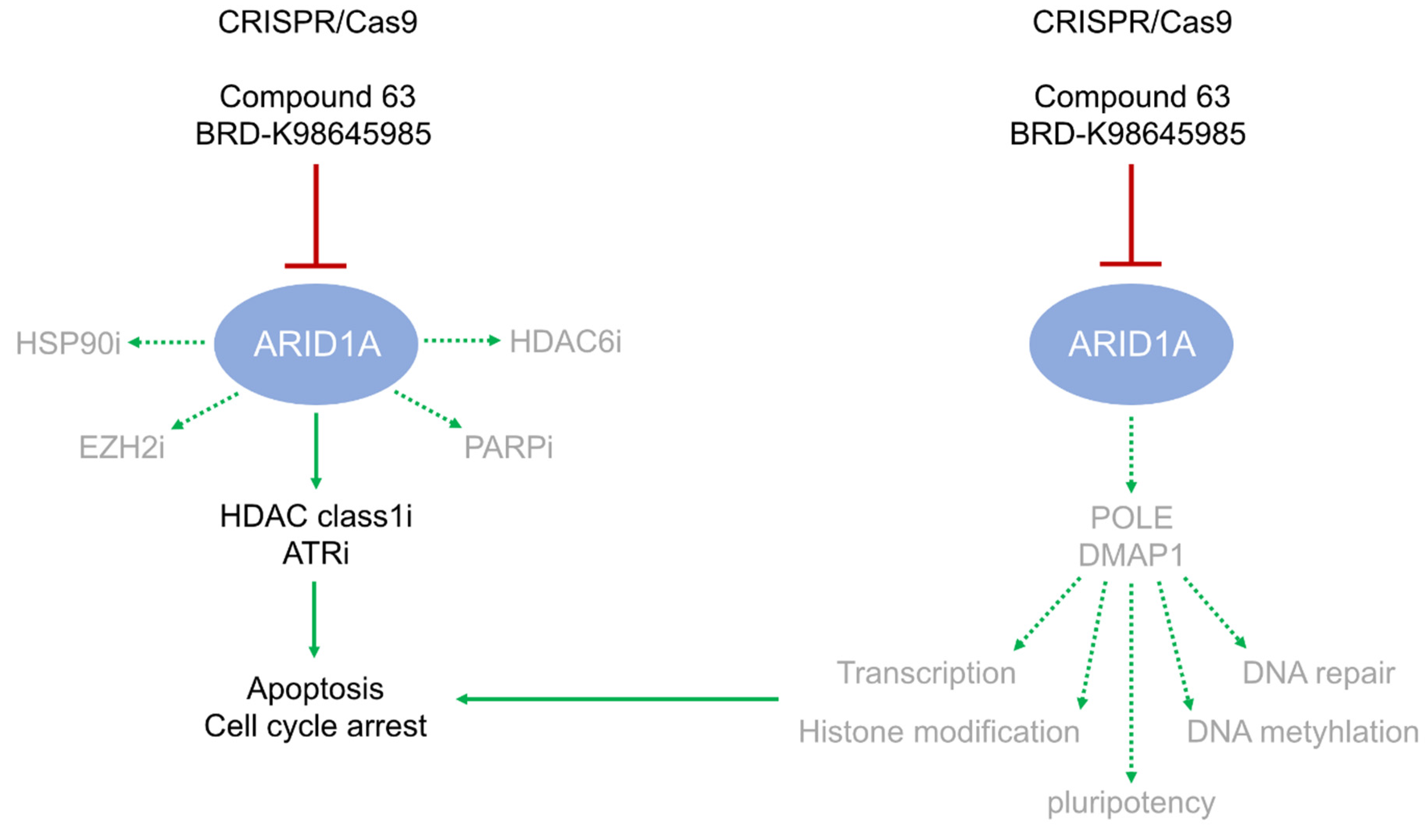
3.1. ARID1A Deficiency or Inhibition Sensitizes GCT Cells to ATR Inhibition
3.2. POLE and DMPA1 are Putative ARID1A Effectors
3.3. ARID1A Transiently Affects the Pluripotency Program in Seminoma and EC Cells
4. Material and Methods
4.1. Ethics Statement
4.2. Cell Culture
4.3. Inhibitor Preparation and Application
4.4. Gene Editing
4.5. Measurement of Cell Viability
4.6. Flow Cytometry
4.7. Immunofluorescence
4.8. Measurement of Cell Growth
4.9. DNA, RNA and Protein Isolation
4.10. Western Blot
4.11. Sanger Sequencing
4.12. Mass Spectrometry: Sample Preparation
4.13. Mass Spectrometry: LC-MS Analysis
4.14. Mass Spectrometry: Computational Mass Spectrometric Data Analysis
4.15. Quantitative RT-PCR
4.16. Illumina HT-12v4/Affymetrix Expression Arrays and Illumina 450k DNA Methylation Array
4.17. Online Analyses Tools
4.18. Statistical Analysis
5. Conclusions
6. Limitations of This Study
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oosterhuis, J.W.; Looijenga, L.H.J. Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 2005, 5, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Berney, D.; Looijenga, L.H.J.; Idrees, M.; Oosterhuis, J.W.; Meyts, E.R.-D.; Ulbright, T.M.; Skakkebaek, N.E. Germ cell neoplasiain situ (GCNIS): Evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 2016, 69, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, J.; Elghiaty, A.; Ham, W.S. Recent global trends in testicular cancer incidence and mortality. Med. (U.S.) 2018, 97, e12390. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Seidel, C.; Bokemeyer, C. Therapeutic approaches for refractory germ cell cancer. Expert Rev. Anticancer. Ther. 2018, 18, 389–397. [Google Scholar] [CrossRef]
- Oing, C.; Giannatempo, P.; Honecker, F.U.; Oechsle, K.; Bokemeyer, C.; Beyer, J. Palliative treatment of germ cell cancer. Cancer Treat. Rev. 2018, 71, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Oing, C.; Lorch, A. The Role of Salvage High-Dose Chemotherapy in Relapsed Male Germ Cell Tumors. Oncol. Res. Treat. 2018, 41, 365–369. [Google Scholar] [CrossRef]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J. Cell Mol. Med. 2017, 21, 1300–1314. [Google Scholar] [CrossRef]
- Reisman, D.N.; Glaros, S.; Thompson, A.E. The SWI/SNF complex and cancer. Oncogene 2009, 28, 1653–1668. [Google Scholar] [CrossRef] [Green Version]
- Dykhuizen, E.C.; Hargreaves, D.C.; Miller, E.L.; Cui, K.; Korshunov, A.; Kool, M.; Pfister, S.; Cho, Y.-J.; Zhao, K.; Crabtree, G.R. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 2013, 497, 624–627. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.-M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.Y.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Iii, F.J.R.; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nature 2017, 19, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [Green Version]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, K.; Hamacher, A.; Hansen, F.K.; Gertzen, C.G.W.; Senger, J.; Marquardt, V.; Marek, L.; Marek, M.; Romier, C.; Remke, M.; et al. Alkoxyurea-Based Histone Deacetylase Inhibitors Increase Cisplatin Potency in Chemoresistant Cancer Cell Lines. J. Med. Chem. 2017, 60, 5334–5348. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, E.C.; Carmody, L.; Tolliday, N.; Crabtree, G.R.; Palmer, M.A.J. Screening for Inhibitors of an Essential Chromatin Remodeler in Mouse Embryonic Stem Cells by Monitoring Transcriptional Regulation. J. Biomol. Screen. 2012, 17, 1221–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marian, C.A.; Stoszko, M.; Wang, L.; Leighty, M.W.; De Crignis, E.; Maschinot, C.A.; Gatchalian, J.; Carter, B.; Chowdhury, B.; Hargreaves, D.C.; et al. Small Molecule Targeting of Specific BAF (mSWI/SNF) Complexes for HIV Latency Reversal. Cell Chem. Boil. 2018, 25, 1443–1455.e14. [Google Scholar] [CrossRef]
- Smith-Roe, S.L.; Nakamura, J.; Holley, D.; Chastain, P.D.; Rosson, G.B.; Simpson, D.A.; Ridpath, J.R.; Kaufman, D.G.; Kaufmann, W.K.; Bultman, S.J. SWI/SNF complexes are required for full activation of the DNA-damage response. Oncotarget 2015, 6, 732–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.M.; Chastain, P.D.; Rosson, G.B.; Groh, B.S.; Weissman, B.E.; Kaufman, D.G.; Bultman, S.J. BRG1 co-localizes with DNA replication factors and is required for efficient replication fork progression. Nucleic Acids Res. 2010, 38, 6906–6919. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Tate, P.; Hu, P.; Tjian, R.; Skarnes, W.C.; Wang, Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc. Natl. Acad. Sci. USA 2008, 105, 6656–6661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidder, B.L.; Palmer, S.; Knott, J. SWI/SNF-Brg1 Regulates Self-Renewal and Occupies Core Pluripotency-Related Genes in Embryonic Stem Cells. STEM CELLS 2009, 27, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Jothi, R.; Ronan, J.L.; Cui, K.; Zhao, K.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. USA 2009, 106, 5187–5191. [Google Scholar] [CrossRef] [Green Version]
- King, H.W.; Klose, R.J. The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife 2017, 6, e22631. [Google Scholar] [CrossRef]
- Lee, G.E.; Kim, J.H.; Taylor, M.; Muller, M.T. DNA Methyltransferase 1-associated Protein (DMAP1) Is a Co-repressor That Stimulates DNA Methylation Globally and Locally at Sites of Double Strand Break Repair*. J. Boil. Chem. 2010, 285, 37630–37640. [Google Scholar] [CrossRef] [Green Version]
- Penicud, K.; Behrens, A. DMAP1 is an essential regulator of ATM activity and function. Oncogene 2013, 33, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Negishi, M.; Saraya, A.; Miyagi, S.; Nagao, K.; Inagaki, Y.; Nishikawa, M.; Tajima, S.; Koseki, H.; Tsuda, H.; Takasaki, Y.; et al. Bmi1 cooperates with Dnmt1-associated protein 1 in gene silencing. Biochem. Biophys. Res. Commun. 2007, 353, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Rountree, M.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef]
- Nettersheim, D.; Heukamp, L.; Fronhoffs, F.; Grewe, M.J.; Haas, N.; Waha, A.; Honecker, F.; Waha, A.; Kristiansen, G.; Schorle, H. Analysis of TET Expression/Activity and 5mC Oxidation during Normal and Malignant Germ Cell Development. PLoS ONE 2013, 8, e82881. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Heimsoeth, A.; Jostes, S.; Schneider, S.; Fellermeyer, M.; Hofmann, A.; Schorle, H. SOX2 is essential for in vivo reprogramming of seminoma-like TCam-2 cells to an embryonal carcinoma-like fate. Oncotarget 2016, 7, 47095–47110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettersheim, D.; Berger, D.; Jostes, S.; Skowron, M.A.; Schorle, H. Deciphering the molecular effects of romidepsin on germ cell tumours: DHRS2 is involved in cell cycle arrest but not apoptosis or induction of romidepsin effectors. J. Cell. Mol. Med. 2018, 23, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Poschmann, G.; Seyfarth, K.; Agbo, D.B.; Klafki, H.-W.; Rozman, J.; Wurst, W.; Wiltfang, J.; Meyer, H.E.; Klingenspor, M.; Stühler, K. High-Fat Diet Induced Isoform Changes of the Parkinson’s Disease Protein DJ-1. J. Proteome Res. 2014, 13, 2339–2351. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2018, 47, D442–D450. [Google Scholar] [CrossRef]
- Eckert, D.; Nettersheim, D.; Heukamp, L.C.; Kitazawa, S.; Biermann, K.; Schorle, H. TCam-2 but not JKT-1 cells resemble seminoma in cell culture. Cell Tissue Res. 2007, 331, 529–538. [Google Scholar] [CrossRef]
- Biermann, K.; Heukamp, L.C.; Steger, K.; Zhou, H.; Franke, F.E.; Sonnack, V.; Brehm, R.; Berg, J.; Bastian, P.J.; Muller, S.C.; et al. Genome-wide expression profiling reveals new insights into pathogenesis and progression of testicular germ cell tumors. Cancer Genom.-Proteom. 2007, 4, 359–367. [Google Scholar]
- Nettersheim, D.; Jostes, S.; Sharma, R.; Schneider, S.; Hofmann, A.; Ferreira, H.; Hoffmann, P.; Kristiansen, G.; Esteller, M.; Schorle, H. BMP Inhibition in Seminomas Initiates Acquisition of Pluripotency via NODAL Signaling Resulting in Reprogramming to an Embryonal Carcinoma. PLoS Genet. 2015, 11, e1005415. [Google Scholar] [CrossRef] [Green Version]
- Nettersheim, D.; Vadder, S.; Jostes, S.; Heimsoeth, A.; Schorle, H. TCam-2 Cells Deficient for SOX2 and FOXA2 Are Blocked in Differentiation and Maintain a Seminoma-Like Cell Fate In Vivo. Cancers 2019, 11, 728. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, J.C.V. An interactive tool for comparing lists with Venn Diagrams. 2007. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html.bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 1 March 2020).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; Kamath, A.; McDade, F.; Rogers, D.; Brooks, A.N.; Zhu, J.; Haussler, D. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Skanderup, A.J.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward primer | Reverse primer | Tan | Cycles |
|---|---|---|---|---|
| ARID1A-5″ | TCTTGCCCATCTGATCCATT | CCAACAAAGGAGCCACCAC | 60 °C | 40 |
| ARID1A-flank. guideRNA A,B | CGCAGCAAGGACATGGGTA | ATGGAGTCTGGCCCTGTTGA | 60 °C | 40 |
| ARID1A-flank. guideRNA C | TCTCAGCAGTCCCAGCAAAC | AGGCAAGCTGGAGGGTCTT | 60 °C | 40 |
| ARID1B | CAAGGGGATCAGAGCAACCC | CTACCTGGGATACTTGCAGGA | 60 °C | 45 |
| ATF3 | AAGAACGAGAAGCAGCATTTGAT | TTCTGAGCCCGGACAATACAC | 60 °C | 40 |
| ATM | TGGATCCAGCTATTTGGTTTGA | CCAAGTATGTAACCAACAATAGAAGAAGTAG | 60 °C | 40 |
| ATR | CAGCTTTGTGCCATTTACTG | CTACCTCAATTCCAAGCACA | 60 °C | 40 |
| CDKN1A | CCTCATCCCGTGTTCTCCTTT | GTACCACCCAGCGGACAAGT | 60 °C | 40 |
| CHEK1 | ATATGAAGCGTGCCGTAGACT | TGCCTATGTCTGGCTCTATTCTG | 60 °C | 40 |
| DUSP1 | GTACATCAAGTCCATCTGAC | GGTTCTTCTAGGAGTAGACA | 60 °C | 40 |
| ETAA1 | GAGAATTTCCATACATTTCCCCTTT | CTAAACAAGGAAGTAATTTGGTACAATCAA | 60 °C | 40 |
| FGF4 | TTCTTCGGGCCATGAGCAG | CCGAAGAAAGTGCACCAAGG | 60 °C | 40 |
| FOS | GAGAGCTGGTAGTTAGTAGCATGTTGA | AATTCCAATAATGAACCCAATAGATTAGTTA | 60 °C | 45 |
| GADD45B | GTCGGCCAAGTTGATGAAT | CACGATGTTGATGTCGTTGT | 60 °C | 40 |
| GAPDH | TGCCAAATATGATGACATCAAGAA | GGAGTGGGTGTCGCTGTTG | 60 °C | 45 |
| ID2 | TCAGCCTGCATCACCAGAGA | CTGCAAGGACAGGATGCTGATA | 60 °C | 40 |
| MLH1 | CTTGTACCCCCCGGAGAAG | TGCAACATCTCCCGGAGAAC | 60 °C | 40 |
| P53 | TTGCAATAGGTGTGCGTCAGA | AGTGCAGGCCAACTTGTTCAG | 60 °C | 40 |
| SMARCA4 | CAGCATGCCAAGGATTTCAAG | CGATCCGCTCGTTCTCTTTC | 60 °C | 40 |
| SMARCB1 | AACGTCAGCGGGTTCAAAT | GCCTTCACCTGGAACATGAA | 60 °C | 40 |
| TOP2A | AGTCATTCCACGAATAACCA | TTCACACCATCTTCTTGAG | 60 °C | 40 |
| TOPBP1 | TGTGACTGGCTTATGTGGCT | TGGCACACTCATACTTCTGACC | 60 °C | 40 |
| TP53BP1 | ATTGAGGATACGGAACCCATGT | TGCTGGATTCATCAGGATACTATCA | 60 °C | 40 |
| XPC | GGCCAAAGGTCTGCTCATCA | GTCCACCTCCTGCATCTGTG | 60 °C | 40 |
| ZMYND11 | TTGTTAAACGTGCCATGACC | GCATGTGTGGAGACAGAGGA | 60 °C | 40 |
| ARID1A genotyping | GCGGTACCCGATGACCATGC | TACTGGAGGTCATTGAGGGG | 60 °C | 45 |
| ARID1A guideRNA A | GCGGTACCCGATGACCATGC | |||
| ARID1A guideRNA B | ATGGTCATCGGGTACCGCTG | |||
| ARID1A guideRNA C | CCCCTCAATGACCTCCAGTA |
| Antibody | Company | Clone | Order No. | Dilution | Application |
|---|---|---|---|---|---|
| primary antibodies | |||||
| ARID1A | Cell Signaling Technology | D2A8U | 12354 | 1:500 | Western Blot |
| Beta-Actin | Sigma-Aldrich | AC-15 | A5441 | 1:20,000 | Western Blot |
| GAPDH | Abcam | 6C5 | ab8245 | 1:30,000 | Western Blot |
| Vinculin | Merck/Sigma | V284 | 05–386 | 1:2000 | Western Blot |
| Phospho-Histone H2A.X | Cell Signaling Technology | Ser139 | 2577 | 1:50 | IF |
| secondary Abs | |||||
| Polyclonal Rabbit Anti-Mouse HRP | Dako | P026002-2 | 1:1000 | Western Blot | |
| Polyclonal Goat Anti-Rabbit HRP | Dako | P044801-2 | 1:2000 | Western Blot | |
| Goat anti-Mouse IgG (H+L) Alexa Fluor 488 | Thermo Fisher Scientific | A11029 | 1:2000 | IF/FACS | |
| Goat anti-Rabbit IgG (H+L) Alexa Fluor 488 | Thermo Fisher Scientific | A11034 | 1:2000 | IF/FACS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurz, L.; Miklyaeva, A.; Skowron, M.A.; Overbeck, N.; Poschmann, G.; Becker, T.; Eul, K.; Kurz, T.; Schönberger, S.; Calaminus, G.; et al. ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 While ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition. Cancers 2020, 12, 905. https://doi.org/10.3390/cancers12040905
Kurz L, Miklyaeva A, Skowron MA, Overbeck N, Poschmann G, Becker T, Eul K, Kurz T, Schönberger S, Calaminus G, et al. ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 While ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition. Cancers. 2020; 12(4):905. https://doi.org/10.3390/cancers12040905
Chicago/Turabian StyleKurz, Lukas, Alissa Miklyaeva, Margaretha A. Skowron, Nina Overbeck, Gereon Poschmann, Teresa Becker, Katharina Eul, Thomas Kurz, Stefan Schönberger, Gabriele Calaminus, and et al. 2020. "ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 While ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition" Cancers 12, no. 4: 905. https://doi.org/10.3390/cancers12040905