1. Introduction
The endoplasmic reticulum (ER) orchestrates protein folding and export. This function is sensitive to cellular alterations in energy levels, redox status, or calcium homeostasis [
1], resulting in accumulation of misfolded proteins in the ER lumen, a condition known as ER stress [
2]. The detection and resolution of ER stress relies on three major ER-spanning transmembrane proteins, PERK, inositol requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6), which trigger the unfolded protein response (UPR). The UPR induces global translational and transcriptional changes to improve the ER protein-folding capacity. However, if ER stress cannot be resolved, the UPR shifts from pro-survival to pro-apoptotic signaling pathways [
2]. Several lines of evidence suggest a pivotal role for the PERK branch in the cellular outcome of ER stress [
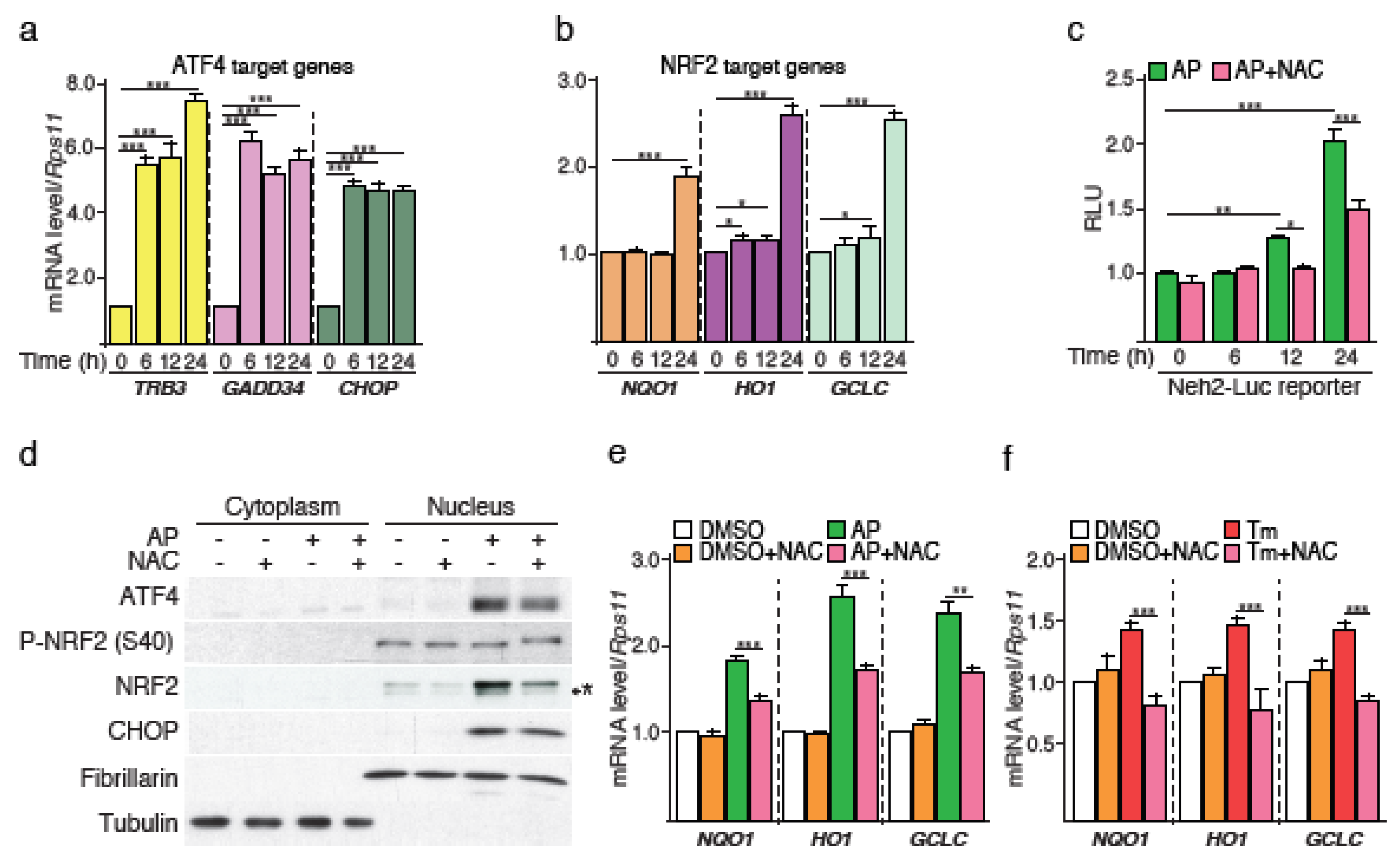
3]. Activated PERK phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α) that reduces protein synthesis but paradoxically increases the translation of mRNAs such as that of ATF4. ATF4 in turn initiates a transcriptional program including the up-regulation of the transcription factor C/EBP homologous protein (CHOP). ATF4 and CHOP cooperatively stimulate the expression of genes encoding functions in protein synthesis, and the reactivated translation rate generates significant amounts of reactive oxygen species (ROS) that induce apoptosis [
4,
5]. As part of the mechanisms engaged to limit oxidative insults, PERK has been shown to directly phosphorylate the master regulator of the cellular antioxidant response nuclear factor erythroid 2-related factor 2 (NRF2), leading to NRF2/Kelch-like ECH-associated protein 1 (KEAP1) complex dissociation and NRF2 stabilization and activation [
6]. In addition, IRE1 can also activate the NRF2 and the subsequent oxidative stress response [
7], indicating that the molecular events governing NRF2 activity upon ER stress are multifaceted. Apart from the classical activation of NRF2 relying on the induced-dissociation of the NRF2/KEAP1 complex, evidence suggests that NRF2 regulation also occurs at the transcriptional level and that increased transcription promotes resistance to ROS [
8,
9,
10]. Notably, chromatin immunoprecipitation (ChIP) sequencing data from human mammary cells have exposed the direct binding of ATF4 to the promoter of the
NRF2 gene (
NFE2L2) upon ER stress [
11], suggesting that PERK-mediated regulation of NRF2 might be more complex than previously anticipated.
Herein, we present an alternative mechanism to the direct phosphorylation by PERK that contributes to the protective NRF2-induced antioxidant response upon PERK activation.
3. Discussion
ER stress-associated PERK activation has previously been reported to induce rapid and direct phosphorylation of NRF2 [
6]. Although this mechanism results in a ROS-independent activation of NRF2 during ER stress, we report that ROS generated during late stages of PERK-dependent protein synthesis recovery, also contribute to triggering the cytoprotective NRF2 antioxidant program. In addition, PERK activation triggers an ATF4-dependent control of
NRF2 mRNA abundance that reinforces the cytoprotective function of NRF2. These results show that the PERK pathway can engage different mechanisms to activate NRF2 during ER stress.
Lu and colleagues notably demonstrated that PERK-dependent changes in gene expression are mainly reliant on transcription factors downstream of eIF2α phosphorylation [
12], which has raised questions about the physiological importance of transcription activation by alternative PERK substrates such as NRF2 [
2]. Our findings may reconcile these apparently discrepant results since the different mechanisms of NRF2 activation are theoretically not mutually exclusive but rather complementary or alternative.
The PERK pathway induced an ATF4-dependent
NRF2 mRNA increase and the binding of ATF4 to the CARE of
Nfe2l2 promoter region supporting a direct regulation of
NRF2 gene expression. Interestingly, no conserved CARE was found in the orthologous region of the murine
Nfe2l2 promoter and no ATF4 binding to this promoter has been described in mouse embryonic fibroblasts [
4], suggesting a species-specific
NRF2 regulation by ATF4. Fv2E-PERK-mediated ATF4-dependent increase in NRF2 protein amounts occurred relatively rapidly but did not readily translate into NRF2 activation. Considering that at steady-state, the abundance of NRF2 is maintained at a level significantly lower than that of KEAP1 [
18], we hypothesize that a proportion of KEAP1 may act as a floodgate trapping the newly produced NRF2. Only when significant amounts of ROS are generated during protein synthesis recovery, is KEAP1 massively released from NRF2 leading to a strong NRF2 activation (
Figure 4).
In the context of tumorigenesis, there has been a renewed interest in NRF2 as a driver of cancer progression and resistance to therapy, over the last years. Many cancer cells hijack ER stress signaling to promote progression [
2], thus our findings suggest that ATF4-dependent NRF2 activation could be aiding in this process, therefore supporting the interest in targeting NRF2 in cancer treatment.
In summary, we show that in addition to the reported rapid and direct phosphorylation of NRF2 by PERK, the PERK-eIF2α-ATF4 pathway controls NRF2 expression in human cells, which is at least in part activated by upcoming ER stress-associated oxidative stimuli. This work broadens our understanding of how cells cope with oxidative stress generated during ER stress.
4. Materials and Methods
4.1. Cell Culture and Treatments
NCI-H358 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% v/v penicillin/streptomycin solution. Puromycine (2 µg/mL) was used for the selection of Fv2E-PERK NCI-H358 cells, generated in-house. A549 cells were cultured in DMEM high glucose supplemented with 10% fetal bovine serum (FBS), glutamax and 1% v/v penicillin/streptomycin and HBEC-3KT were maintained in KSFM supplemented with bovine pituitary extract and recombinant human EGF. The cells were maintained at 37 °C in a 5% CO2 incubator.
To induce ER stress, cells were treated with 0.5 μg/mL tunicamycin (Sigma-Aldrich, ref: T7765, St Louis, MO, USA) or 30 nM thapsigargin (Sigma-Aldrich, ref: T9033, St Louis, MO, USA) added to the culture medium. To induce oxidative stress or NRF2-mediated response, cells were treated with 100 μM H2O2 (Millipore, ref: 107209). To activate the Fv2E-PERK fusion protein cells were treated with 0.2 nM AP20187 (Clonetech, ref: 635060, St Louis, MO, USA). An equivalent volume of DMSO was used for control experiments. Leucine starvation experiments were performed using a DMEM/Nutrient Mixture: F-12 (DMEM/F12) medium devoid of leucine, glutamine, lysine and methionine (Sigma-Aldrich, ref: D9785, St Louis, MO, USA), in which glutamine, lysine, methionine and 10% dialyzed serum were subsequently added. The control medium for these experiments was supplemented with leucine.
4.2. siRNA and Plasmid Transfections
NCI-H358 cells were transfected with ON-TARGETplus SMART pool human NFE2L2 siRNA (L-003755-00-0010), ON-TARGETplus non-targeting siRNA (D-001810-03-50) (Dharmacon, GE Healthcare) or human ATF4 siRNA (sc-35112) (Santa Cruz Biotechnology, Dallas, TX, USA). All siRNA transfections were performed using 50 nM siRNA and HiPerFect reagent (Qiagen, Venlo, Netherland), according to the manufacturer’s traditional transfection protocol, for 72 h followed by treatments. Cells were seeded either onto 6-well plates (25 × 104 cells per well), onto 24-well plates (5 × 104 cells per well), or 96-well plates (2 × 104 cells per well).
For NRF2 overexpression experiments, cells seeded onto 6-well plates were transfected with a pcDNA3 vector encoding the Flag-NRF2 (addgene #36971) or luciferase by using the Attractene reagent (Qiagen), according to the manufacturer’s transfection protocol.
4.3. Cell Extracts and Western Blot Analysis
Whole cell extracts were prepared from cultured cells lysed in the radioimmunoprecipitation assay (RIPA) protein buffer containing proteases and phosphatases inhibitors (Roche, Basel, Switzerland), and obtained by centrifugation at 16,000× g for 15 min at 4 °C.
Subcellular fractionation was performed as follows: cells were detached by trypsin-EDTA and resuspended in 10 mM HEPES, pH 8.0, containing 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT and protease and phosphatase inhibitors, incubated on ice for 10 min, to be then lysed by adding 0.1% NP-40. Cytoplasmic fraction was obtained by centrifugation at 1500× g for 10 min at 4 °C. The pellet, representing the nuclear fraction, was dissolved in the RIPA protein buffer containing protease and phosphatase inhibitors, and incubated on ice for 30 min. The nuclear fraction was obtained by centrifugation at 1500× g for 30 min at 4 °C. Tubulin and Fibrillarin were used as loading control for purity of cytoplasmic and nuclear fractions, respectively.
Protein concentrations of the cellular extracts were determined using the DC Protein Assay (Bio-Rad). Equal amounts of proteins (20 μg) were separated by SDS-PAGE and then transferred onto nitrocellulose membranes (Biorad, Hercules, CA, USA). Membranes were incubated in the blocking buffer, 5% milk or Bovine Serum Albumin (BSA) in Tris-Buffered Saline/Tween 20 (TBST), for 1 h at room temperature, then incubated overnight at 4°C with the appropriate primary antibodies, diluted in TBST containing 5% milk or BSA. Membranes were washed three times with TBST, incubated for 1 h at room temperature with the appropriate secondary antibodies, diluted in TBST containing 5% milk, and again washed three times with TBST. Detection by enhanced chemiluminescence was performed using the Clarity western ECL substrate (Bio-Rad, Hercules, CA, USA).
For NRF2-flag immunoprecipitation, cells were transfected with the vector encoding the Flag-NRF2. 30 h later, cells were treated with PERK activators for 6 h. Then, cells were collected and lysed with the IP buffer (Tris-HCl 20mM pH 7.4, NaCl 300 mM, EDTA 1mM, NP40 0.5%, glycerol 10% complemented with proteases and phosphatases inhibitors). Supernatants were incubated with the Flag M2 beads (Sigma-Aldrich) overnight at 4 °C. Then beads were washed three times with IP buffer and samples elution was performed in Laemmli buffer at 95 °C for 5min.
The primary antibodies used were purchased either from Santa Cruz Biotechnology: ATF4 (sc-200 and sc-390063) and α-Tubulin (sc-23950); from Cell Signaling Technology: PERK (3192), eIF2α (9722), P-eIF2α (3398), CHOP (2895), P-Threonine (9381) and p58IPK (2940); from Abcam: NRF2 (ab62352), P-NRF2 Ser40 (ab76026) and Fibrillarin (ab4566); from BD Biosciences: BiP (610978); from Enzo life sciences: FKBP12 (ALX-210-142); or from Millipore: Puromycin clone 12D10 (MABE343). The HRP-conjugated secondary antibodies were supplied by Jackson Laboratories (anti-rabbit and anti-mouse antibodies). Western blot images are representative of three independent experiments Proteins bands quantification was performed using the ImageJ software (NIH, Bethesda, MD, USA).
4.4. SUnSET Assay
Rates of protein synthesis in NCI-H358 cultured cells were evaluated using the surface sensing of translation (SUnSET) method as previously described [
14]. NCI-H358 cells were seeded onto 6-well plates (2 × 10
5 cells per well) and treated with 0.2 nM AP20187 for the indicated times. Cells were then incubated with 5 μg/mL puromycin (Sigma-Aldrich P9620, St Louis, MO, USA), added directly into the medium for 15 min at 37 °C, 5% CO
2, prior to be harvested and processed to prepare whole cell extracts in the RIPA buffer. The amount of puromycin incorporated into nascent peptides was then evaluated by Western blot analysis by using anti-puromycin antibody.
4.5. Luciferase Assay
Luciferase assays were performed in NCI-H358 cells seeded onto 24 well-plates (12 × 10
4 cells per well), transiently co-transfected with Neh2-luc reporter plasmid [
16] (0.4 μg for each sample) pRL-SV40 internal control vector (used at ratio of 10:1) and Attractene reagent (Qiagen), according to the manufacturer’s transfection protocol. Cells were collected in 100 µL of passive lysis buffer and analyzed for Firefly and Renilla luciferase activities with the Dual-Luciferase Reporter Assay System (Promega, ref: E1910), according to the manufacturer’s instructions. Luciferase activities were measured with a microplate reader (Tecan, Infinite M200 PRO, Life Science). The assays were repeated at least three times in triplicate.
4.6. Cell Viability Assay
Cell viability was determined using the CellTiter-Blue assay (Promega ref: G8081). NCI-H358 cells were plated onto 24-wells plate (5 × 104 cells per well) prior to the NRF2 knock-down and the appropriate treatment (5mM NAC treatment prior to AP). CellTiter-Blue reagent was added to each well (100 µL per well), to the replaced medium (500 µL) and the plates were incubated at for 1 h 37 °C, 5% CO2. Fluorescence intensity (545 nm/600 nm excitation/emission) was measured using a microplate reader (Clariostar, BMG LABTECH). Cell viability is expressed as the mean percentage compared to the control. The assays were repeated at least three times in triplicate.
4.7. ROS Detection
Intracellular ROS measurement was performed using the fluorescent probe 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA; C6827, Invitrogen Molecular Probes). Briefly, NCI-H358 cells were seeded onto 96-well plate (2 × 104 cells per well). Cells were pre-incubated with 10 µM CM-H2DCFDA, diluted in PBS supplemented with 10% FBS, for 30 min at 37 °C, 5% CO2. Following the treatment, fluorescence intensity (490 nm/530 nm excitation/emission) was measured using a microplate reader (Clariostar, BMG LABTECH). A sister plate for each experiment was used to determine the cell number by CellTiter-Blue assay, and a calibration curve was obtained by plating an increasing the number of cells per well (2.5–40 × 103). The assays were repeated at least three times in triplicate. The results were presented as the fold increase in ROS per cell.
4.8. RNA Extraction and RT-qPCR
Total cellular RNA was extracted using the NucleoSpin RNA Kit (Macherey-Nagel), according to the manufacturer’s protocol. For cDNA synthesis, 0.5 µg of RNA were reverse transcribed using Superscript II reverse transcriptase (Invitrogen, ref: 18064014, Carlsbad, CA, USA) with random primers (Invitrogen, ref: S0142, Carlsbad, CA, USA), according to the manufacturer’s instructions. cDNA was then amplified by qPCR using specific primers listed in
Supplementary Table S1 and the SYBR Green Master Mix (Biorad, Hercules, CA, USA). qPCR was performed using the CFX connect real-time PCR system (Biorad, Hercules, CA, USA). Expression of target genes was normalized against endogenous RPS11 mRNA levels, used as an internal control, and assessed using the comparative ΔΔCT method. qPCR experiments were repeated at least three times in triplicate. Primer list:
NQO1 Fw: GAAGAGCACTGATCGTACTGG, Rev: GGATACTGAAAGTTCGCAGGG,
HO1 Fw: TCTTCGCCCCTGTCTACTTC, Rev: CTTCACATAGCGCTGCATGG,
GCLC Fw: CCTGTCTGGGGAGAAAGTTC, Rev: ACTCGGACATTGTTCCTCCG,
NRF2 Fw: ACATCGAGAGCCCAGTCTTC, Rev: AGCTCCTCCCAAACTTGCTC,
TRB3 Fw: TGGTACCCAGCTCCTCTACG, Rev: GACAAAGCGACACAGCTTGA, RPS11 Fw: AGCAGCCGACCATCTTTC, Rev: ATAGCCTCCTTGGGTGTCTTG,
GADD34 Fw: CTGTGATCGCTTCTGGCA Rev: GGAAGAAAGGGTGGGCATC and
CHOP Fw: GGTATGAGGACCTGCAAGAGGT Rev: CTTGTGACCTCTGCTGGTTCTG.
4.9. Chromatin Immunoprecipitation (ChIP) Assay
Chromatin immunoprecipitation (ChIP) was performed using the SimpleChIP Plus Enzymatic Chromatin IP Kit (Magnetic Beads) (9005; Cell Signalling Technology, Danvers, MA, USA, Country). Briefly, NCI-H358 cells were seeded in 150 mm in diameter dishes (4 × 10
6 cells per dish) and 3 dishes per treatment were used. Following the treatment with 0.2 nM AP20187 for 6 h, cells were washed with ice-cold PBS and cross-linked with 1% formaldehyde at room temperature for 10 min, to be then harvested in ice-cold PBS containing protease and phosphatase inhibitors. Chromatin was prepared and fragmented by partial digestion with Micrococcal Nuclease, followed by a mild sonication (Bioruptor Diagenode, Seraing, Belgium), according to the manufacturer’s protocol. Chromatin immunoprecipitations were performed using the ATF4 mouse monoclonal antibody (11815, Cell Signalling Technology), normal rabbit IgG antibody and ChIP-Grade Protein G Magnetic Beads both supplied by the kit. After reversal of protein-DNA cross-links, DNA was purified using spin columns. DNA enrichment was analyzed by qPCR using 1 µL of the template and the primers listed in
Supplementary Table S1. The results were calculated as a signal relative to the total amount of input chromatin (ΔCT adjusted to % input), and fold-enrichment values are presented. Primer list:
Nfe2l2 Fw: GCTGAGCTTCCGAAAATCCC, Rev: GGGAGCTAACGGAGACCTC,
Trib3 Fw: GCGGATGCAGAGGAGAGA, Rev: CACTTCCGCTGCGAGTCT, Negative region Fw: CCCATGTCCCAGGAAGAAG, Rev: AGTCCTGGAAGGGGTAGTGG.
4.10. Statistical Analyses
Statistical analyses were performed using the GraphPad Prism 6 software (GraphPad Software, La Jolla, CA, USA) via one-way ANOVA (with Holm-Sidak’s post-test correction for multiple comparisons). All data are expressed as means ± SEM of at least three independent experiments, * p < 0.05, ** p < 0.01, *** p < 0.001.

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}



