Synthetic Lethal Targeting of Mitotic Checkpoints in HPV-Negative Head and Neck Cancer

 ,
,
Abstract
:1. Introduction
2. Results
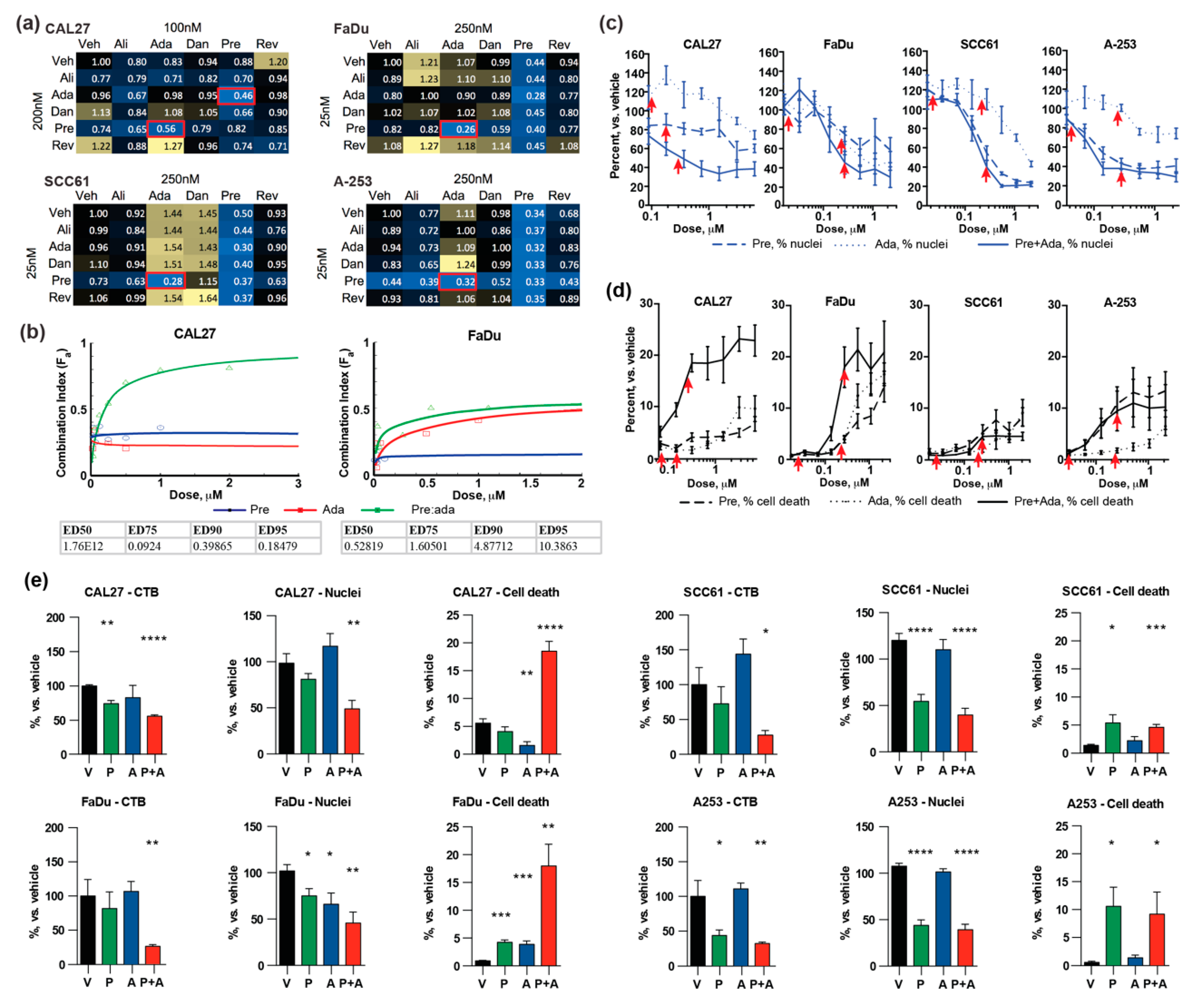
2.1. Profiling Vulnerability to Cell Cycle-Targeted Compounds in Head and Neck Cancer Cell Models
2.2. Potent Combination Activity of Prexasertib and Adavosertib
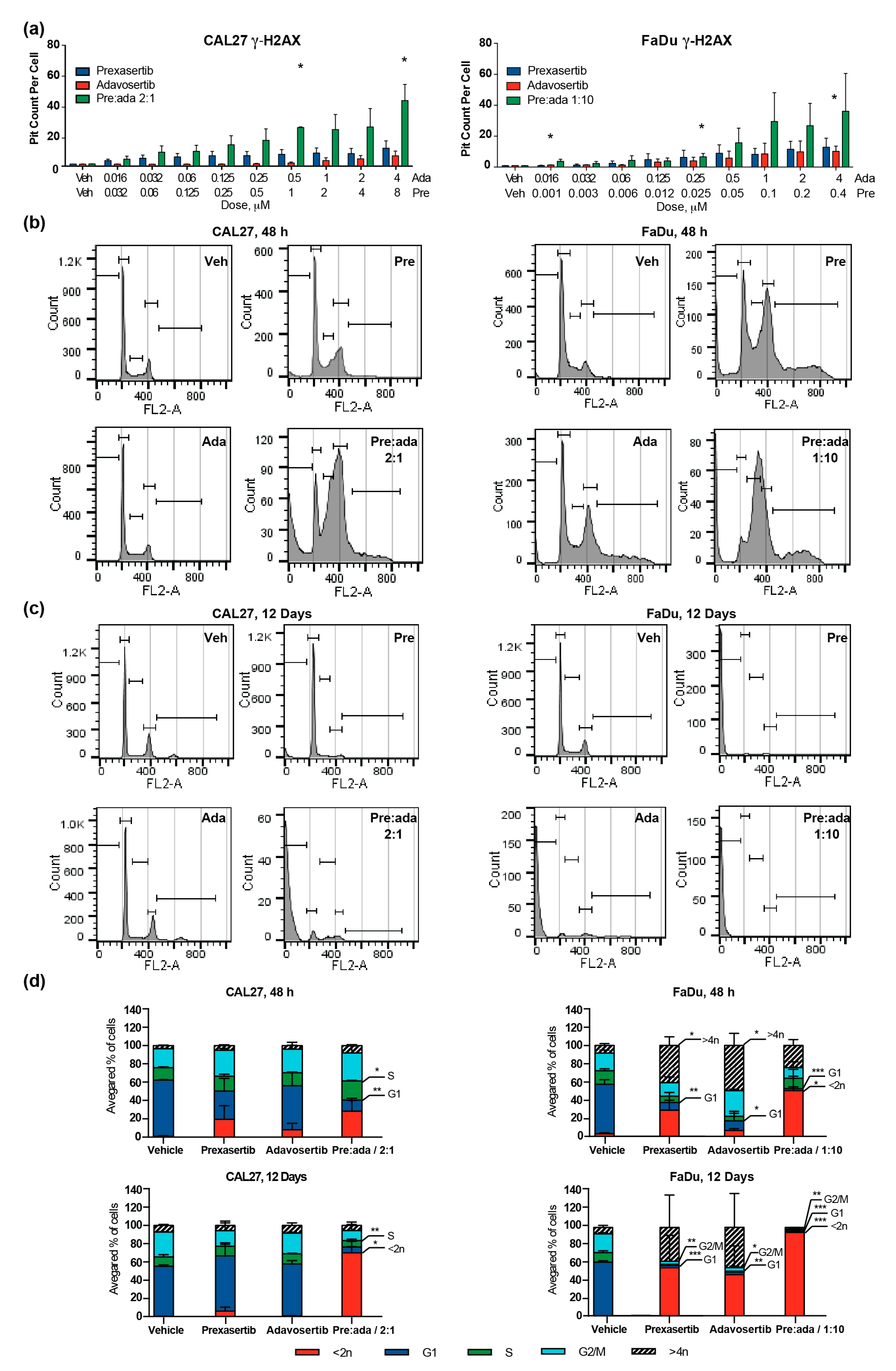
2.3. Combination of Prexasertib and Adavosertib Enhances DNA Breaks and Causes Mitotic Catastrophe
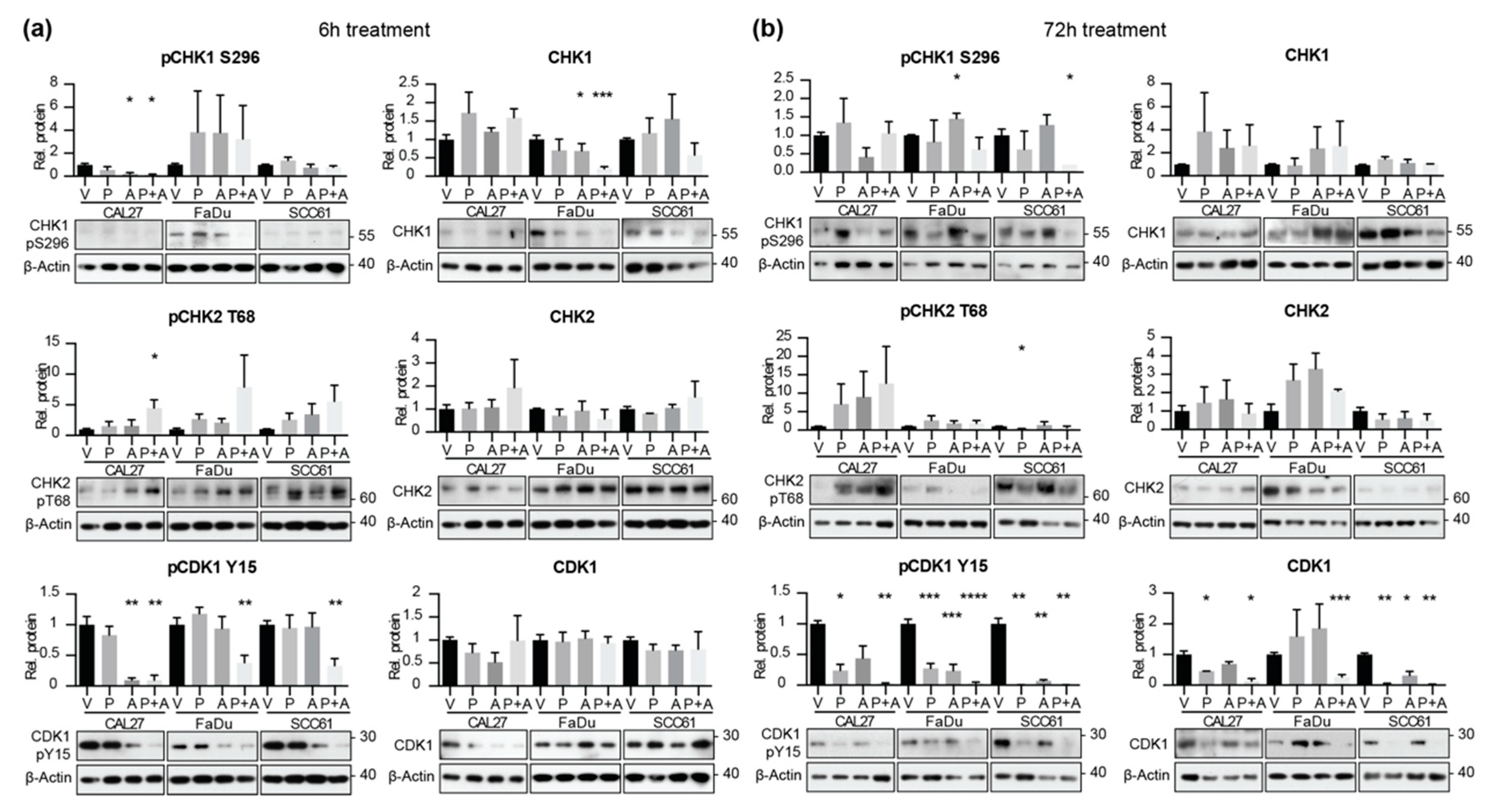
2.4. Combined Application of Prexasertib and Adavosertib Enhances CDK1 Activation
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [CrossRef] [PubMed] [Green Version]
- Cramer, J.D.; Burtness, B.; Le, Q.T.; Ferris, R.L. The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.S.; Karasic, T.B.; DeMichele, A.; Vaughn, D.J.; O’Hara, M.; Perini, R.; Zhang, P.; Lal, P.; Feldman, M.; Gallagher, M.; et al. Palbociclib (PD0332991)-a Selective and Potent Cyclin-Dependent Kinase Inhibitor: A Review of Pharmacodynamics and Clinical Development. JAMA Oncol. 2016, 2, 253–260. [Google Scholar] [CrossRef]
- Beck, T.N.; Georgopoulos, R.; Shagisultanova, E.I.; Sarcu, D.; Handorf, E.A.; Dubyk, C.; Lango, M.N.; Ridge, J.A.; Astsaturov, I.; Serebriiskii, I.G.; et al. EGFR and RB1 as Dual Biomarkers in HPV-Negative Head and Neck Cancer. Molecular Cancer Ther. 2016, 15, 2486–2497. [Google Scholar] [CrossRef] [Green Version]
- Adkins, D.; Ley, J.; Neupane, P.; Worden, F.; Sacco, A.G.; Palka, K.; Grilley-Olson, J.E.; Maggiore, R.; Salama, N.N.; Trinkaus, K.; et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papillomavirus-unrelated head and neck cancer: a multicentre, multigroup, phase 2 trial. Lancet Oncol. 2019. [Google Scholar] [CrossRef]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L., Jr.; Golemis, E.A. Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef]
- Mehra, R.; Serebriiskii, I.G.; Burtness, B.; Astsaturov, I.; Golemis, E.A. Aurora kinases in head and neck cancer. Lancet Oncol. 2013, 14, e425–e435. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Parameswaran, J.; Sandoval-Schaefer, T.; Eoh, K.J.; Yang, D.H.; Zhu, F.; Mehra, R.; Sharma, R.; Gaffney, S.G.; Perry, E.B.; et al. Combined Aurora Kinase A (AURKA) and WEE1 Inhibition Demonstrates Synergistic Antitumor Effect in Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, S.; Cazales, M.; Quaranta, M.; Froment, C.; Trabut, V.; Dozier, C.; Mirey, G.; Bouche, J.P.; Theis-Febvre, N.; Schmitt, E.; et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J. Cell Sci. 2004, 117, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macurek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef]
- van Vugt, M.A.; Bras, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef]
- Den Haese, G.J.; Walworth, N.; Carr, A.M.; Gould, K.L. The Wee1 protein kinase regulates T14 phosphorylation of fission yeast Cdc2. Mol. Biol. Cell 1995, 6, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Lane, H.A.; Nigg, E.A. Cell-cycle control: POLO-like kinases join the outer circle. Trends Cell Biol. 1997, 7, 63–68. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.C.; Misteli, T. Not All DDRs Are Created Equal: Non-Canonical DNA Damage Responses. Cell 2015, 162, 944–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Leu, J.I.; Della Pietra, A.C., 3rd; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Ciceri, P.; Muller, S.; O’Mahony, A.; Fedorov, O.; Filippakopoulos, P.; Hunt, J.P.; Lasater, E.A.; Pallares, G.; Picaud, S.; Wells, C.; et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. [Google Scholar] [CrossRef]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.; Yan, H.; Cho, I.; Fan, H.Y.; Luo, B.; Gai, X.; Bodian, D.L.; Vockley, J.G.; Zhou, Y.; Handorf, E.; et al. Genetic Variants That Presdispose to DNA Double-strand Breaks in Lymphocytes from a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 2015. [Google Scholar] [CrossRef] [Green Version]
- Michelena, J.; Gatti, M.; Teloni, F.; Imhof, R.; Altmeyer, M. Basal CHK1 activity safeguards its stability to maintain intrinsic S-phase checkpoint functions. J. Cell Biol. 2019, 218, 2865–2875. [Google Scholar] [CrossRef] [Green Version]
- Diab, A.; Kao, M.; Kehrli, K.; Kim, H.Y.; Sidorova, J.; Mendez, E. Multiple Defects Sensitize p53-Deficient Head and Neck Cancer Cells to the WEE1 Kinase Inhibition. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, K.; Fang, B.; Koomen, J.M.; Mahajan, N.P. H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat. Struct. Mol. Biol. 2012, 19, 930–937. [Google Scholar] [CrossRef]
- Mahajan, K.; Mahajan, N.P. WEE1 tyrosine kinase, a novel epigenetic modifier. Trends Genet. 2013, 29, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Li, H.; Spier, C.; Mahadevan, D.; LeBlanc, M.; Ul Haq, M.; Huber, B.D.; Flowers, C.R.; Wagner-Johnston, N.D.; Horwitz, S.M.; et al. Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J. Clin. Oncol. 2015, 33, 2399–2404. [Google Scholar] [CrossRef] [PubMed]
- Mendez, E.; Rodriguez, C.P.; Kao, M.C.; Raju, S.; Diab, A.; Harbison, R.A.; Konnick, E.Q.; Mugundu, G.M.; Santana-Davila, R.; Martins, R.; et al. A Phase I Clinical Trial of AZD1775 in Combination With Neoadjuvant Weekly Docetaxel and Cisplatin Before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2740–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neskey, D.M.; Osman, A.A.; Ow, T.J.; Katsonis, P.; McDonald, T.; Hicks, S.C.; Hsu, T.K.; Pickering, C.R.; Ward, A.; Patel, A.; et al. Evolutionary Action score of TP53 (EAp53) identifies high risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef] [Green Version]
- Moser, R.; Xu, C.; Kao, M.; Annis, J.; Lerma, L.A.; Schaupp, C.M.; Gurley, K.E.; Jang, I.S.; Biktasova, A.; Yarbrough, W.G.; et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin. Cancer Res. 2014, 20, 4274–4288. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef] [Green Version]
- Lowery, C.D.; Dowless, M.; Renschler, M.; Blosser, W.; VanWye, A.B.; Stephens, J.R.; Iversen, P.W.; Lin, A.B.; Beckmann, R.P.; Krytska, K.; et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator Across a Range of Preclinical Pediatric Tumor Models. Clin. Cancer Res. 2019, 25, 2278–2289. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Moore, K.; Patel, M.; Grant, S.C.; Burris, H.A., 3rd; William, W.N., Jr.; Jones, S.; Meric-Bernstam, F.; Infante, J.; Golden, L.; et al. Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3263–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, C.; Jonnalagadda, S.; Lingareddy, J.; Awasthi, S.; Gmeiner, W.H.; Palle, K. Prexasertib treatment induces homologous recombination deficiency and synergizes with olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2019, 21, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Bischoff, H.G.; Hwang, J.; Reinhardt, H.C.; Zander, T.; Wang, X.; Hynes, S.; Pitou, C.; Campbell, R.; Iversen, P.; et al. A phase 1 dose-escalation study of checkpoint kinase 1 (CHK1) inhibitor prexasertib in combination with p38 mitogen-activated protein kinase (p38 MAPK) inhibitor ralimetinib in patients with advanced or metastatic cancer. Invest. New Drugs 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-M.; Zhao, B.; Bai, Y.; Iamarino, A.; Gaffney, S.G.; Schlessinger, J.; Lifton, R.P.; Rimm, D.L.; Townsend, J.P. Early and multiple origins of metastatic lineages within primary tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Gaponova, A.V.; Deneka, A.Y.; Beck, T.N.; Liu, H.; Andrianov, G.; Nikonova, A.S.; Nicolas, E.; Einarson, M.B.; Golemis, E.A.; Serebriiskii, I.G. Identification of evolutionarily conserved DNA damage response genes that alter sensitivity to cisplatin. Oncotarget 2017, 8, 19156–19171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abazeed, M.E.; Adams, D.J.; Hurov, K.E.; Tamayo, P.; Creighton, C.J.; Sonkin, D.; Giacomelli, A.O.; Du, C.; Fries, D.F.; Wong, K.K.; et al. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 2013, 73, 6289–6298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Chattopadhyay, N.; Liu, W.J.; Chan, C.; Pignol, J.P.; Reilly, R.M. Optimized digital counting colonies of clonogenic assays using ImageJ software and customized macros: comparison with manual counting. Int. J. Radiat. Biol. 2011, 87, 1135–1146. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | TCGA Provisional | CAL27 | FaDu | SCC61 | A-253 | ||||
|---|---|---|---|---|---|---|---|---|---|
| % Samples | AA Change | Type | AA Change | Type | AA Change | Type | AA Change | Type | |
| TP53 | 72 | p.H193L | Missense | p.R116L | Missense | p.R110L | Missense | p.E48Gfs*66 | Frameshift del |
| NA | Splice site | p.P72R | Polymorphism | ||||||
| CDKN2A | 22 | p.E18* | Stopgain | NA | Splice site | p.A17Pfs*4 | Frameshift del | ||
| CASP8 | 11 | p.II174fs | Frameshift del | ||||||
| p.V460V | Silent | ||||||||
| FAT1 | 23 | p.K3277Nfs*3 | Frameshift del | p.R2567H | Missense | ||||
| HUWE1 | 9 | p.D1193N | Missense | ||||||
| HLA-A | 6 | NA | Splice site | p.D251H | Missense | ||||
| p.E87K | Missense | ||||||||
| p.E87D | Missense | ||||||||
| EP300 | 8 | p.I997V | Missense | ||||||
| p.M2015I | Missense | ||||||||
| CREBBP | 7 | p.T2390N | Missense | p.A2008A | Silent | ||||
| HRAS | 6 | p.C154C | Silent | ||||||
| AJUBA | 6 | p.E310Q | Missense | ||||||
| TGFBR2 | 5 | p.P129Afs*2 | Frameshift ins | p.R537P | Missense | ||||
| HLA-B | 5 | p.I90M | Missense | ||||||
| p.D54G | Missense | ||||||||
| p.Q94H | Missense | ||||||||
| DYSF | 4 | p.A171E | Missense | ||||||
| KDM6A | 4 | p.E1023K | Missense | p.A15_A17del | In-frame del | ||||
| RAC1 | 2.7 | p.L192L | Silent | ||||||
| PTEN | 2.7 | p.L152P | Missense | ||||||
| ZNF233 | 1.8 | p.C389R | Missense | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deneka, A.Y.; Einarson, M.B.; Bennett, J.; Nikonova, A.S.; Elmekawy, M.; Zhou, Y.; Lee, J.W.; Burtness, B.A.; Golemis, E.A. Synthetic Lethal Targeting of Mitotic Checkpoints in HPV-Negative Head and Neck Cancer. Cancers 2020, 12, 306. https://doi.org/10.3390/cancers12020306
Deneka AY, Einarson MB, Bennett J, Nikonova AS, Elmekawy M, Zhou Y, Lee JW, Burtness BA, Golemis EA. Synthetic Lethal Targeting of Mitotic Checkpoints in HPV-Negative Head and Neck Cancer. Cancers. 2020; 12(2):306. https://doi.org/10.3390/cancers12020306
Chicago/Turabian StyleDeneka, Alexander Y., Margret B. Einarson, John Bennett, Anna S. Nikonova, Mohamed Elmekawy, Yan Zhou, Jong Woo Lee, Barbara A. Burtness, and Erica A. Golemis. 2020. "Synthetic Lethal Targeting of Mitotic Checkpoints in HPV-Negative Head and Neck Cancer" Cancers 12, no. 2: 306. https://doi.org/10.3390/cancers12020306