Extracellular Vesicles Orchestrate Immune and Tumor Interaction Networks


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Multiple Pathways for EV Biogenesis
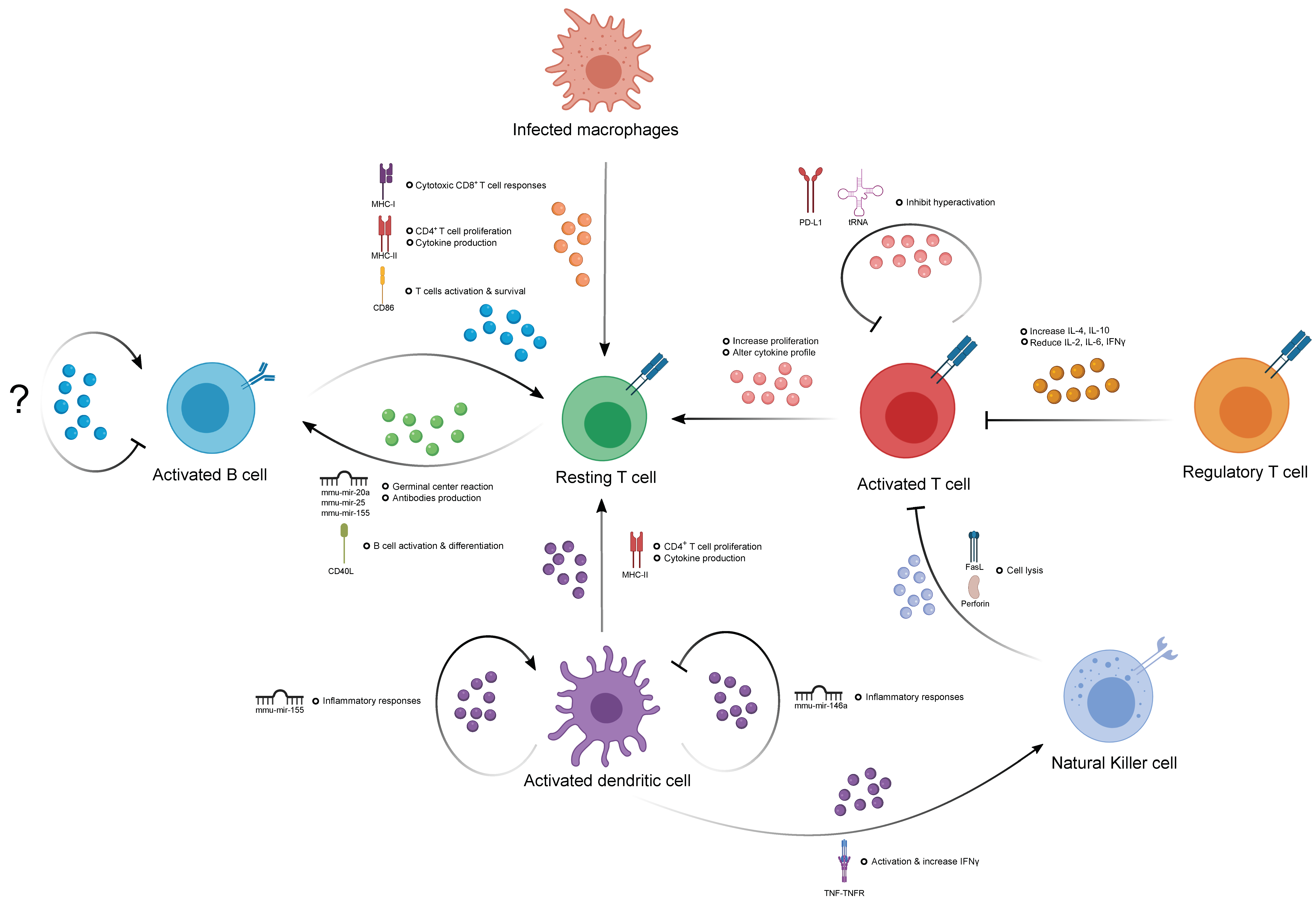
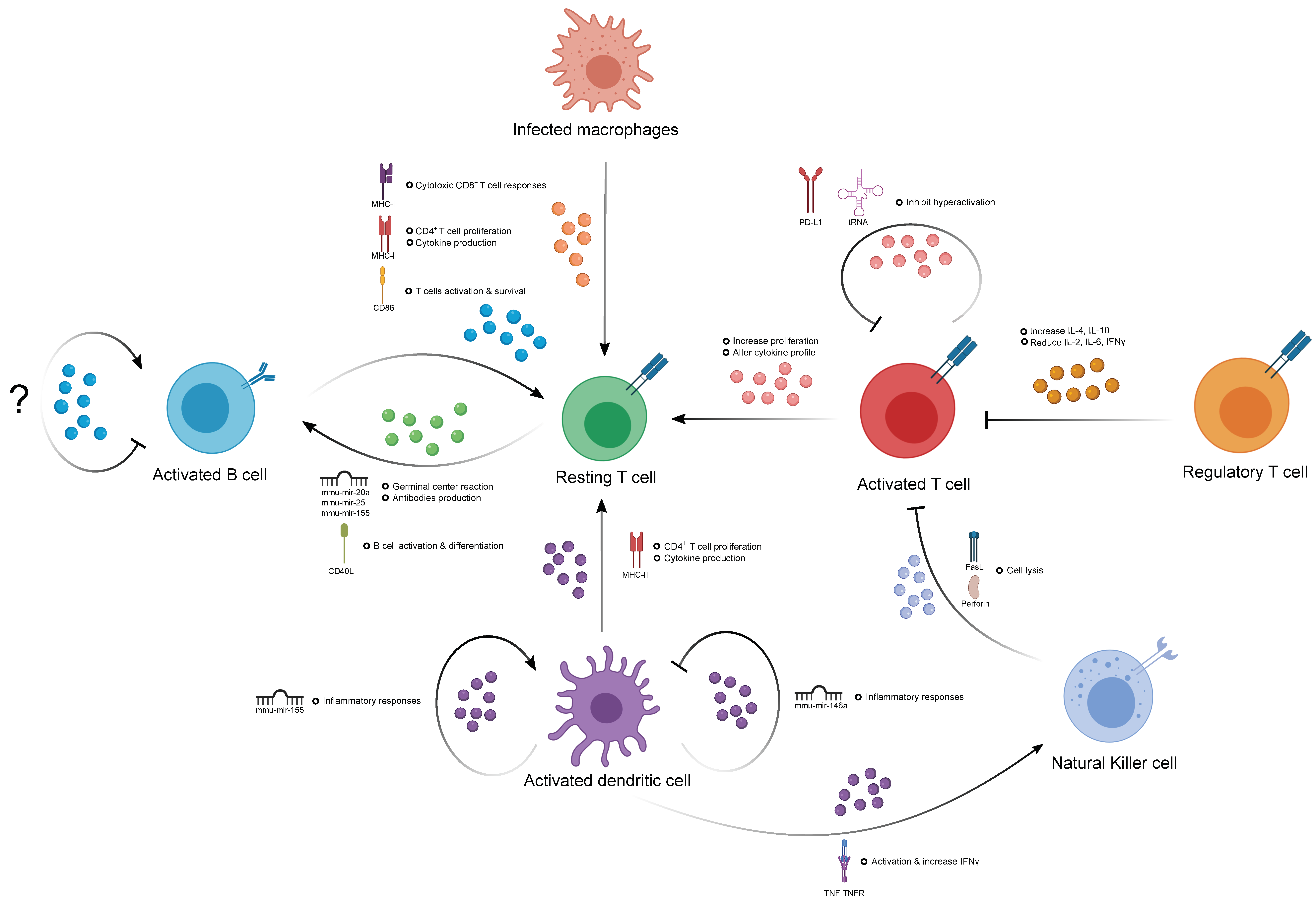
3. The Role of EVs in Immune Homeostasis
3.1. The Contribution of EVs in Humoral Immunity
3.2. The Contribution of EVs in T Cell-Mediated Immunity
3.3. The Contribution of Natural Killer Cell-Derived EVs
3.4. The Contribution of EVs in the Immune Response of Antigen-Presenting Cells
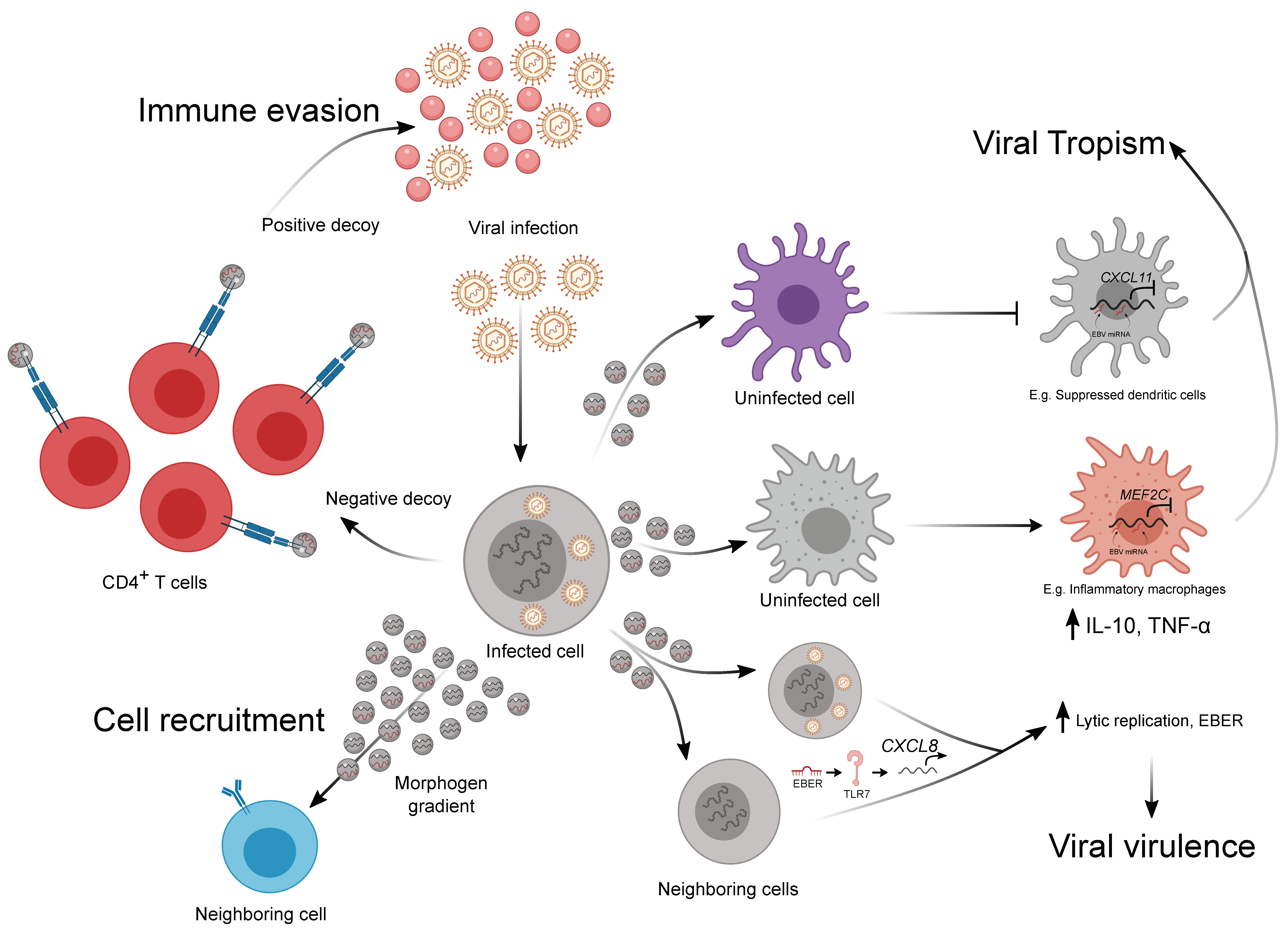
4. The Emerging Role of EVs in Viral–Immune Interactions
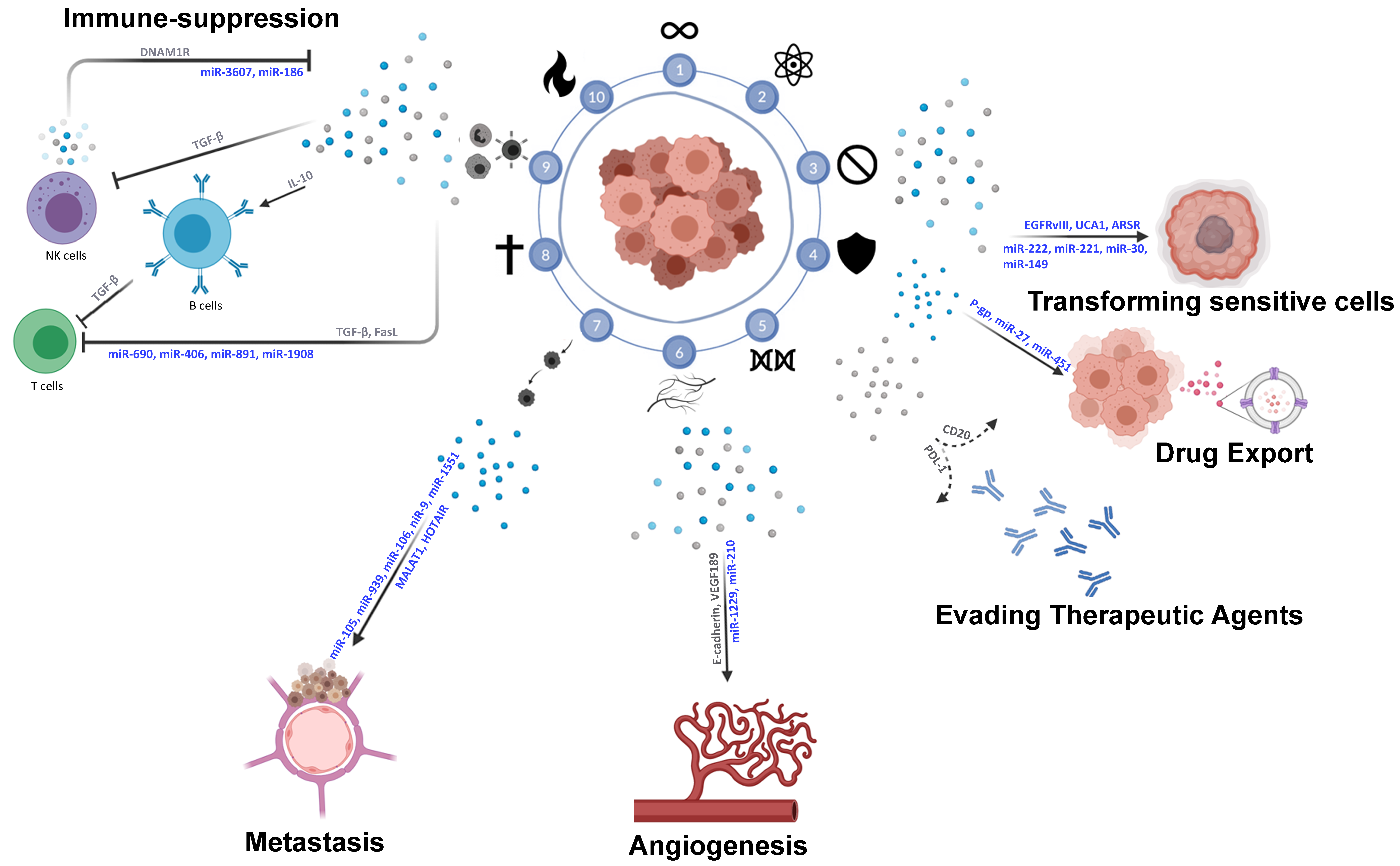
5. EV-Mediated Crosstalk in Immune–Tumor Cell Interactions
5.1. Tumor–Tumor Interactions via EVs
5.2. Tumor–Immune TME Interactions via EVs
5.3. Tumor–Stroma Interactions via EVs
5.4. Cancer Stem Cell Signaling via EVs
6. Mechanisms of Immune–Tumor Cell Communications
6.1. Uptake-Dependent Communication via sncRNA
6.2. Uptake-Dependent Communication via lncRNA
6.3. Uptake-Independent Communication
6.4. EV-Mediated Tumor Resistance to Immunotherapy
7. Future Therapeutic Perspectives for EVs
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kilchert, C.; Wittmann, S.; Vasiljeva, L. The regulation and functions of the nuclear RNA exosome complex. Nat. Rev. Mol. Cell Biol. 2016, 17, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Edgar, J.R. Q&A: What are exosomes, exactly? BMC Biol. 2016, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Chaput, N.; Théry, C. Exosomes: Immune properties and potential clinical implementations. Semin. Immunopathol. 2011, 33, 419–440. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Sigismund, S.; Confalonieri, S.; Ciliberto, A.; Polo, S.; Scita, G.; Di Fiore, P.P. Endocytosis and signaling: Cell logistics shape the eukaryotic cell plan. Physiol. Rev. 2012, 92, 273–366. [Google Scholar] [CrossRef] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Vischer, U.M.; Rosnoblet, C.; Lebrand, C.; Lindsay, M.; Parton, R.G.; Kruithof, E.K.O.; Gruenberg, J. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol. Biol. Cell 2000, 11, 1829–1843. [Google Scholar] [CrossRef] [Green Version]
- Theos, A.C.; Truschel, S.T.; Tenza, D.; Hurbain, I.; Harper, D.C.; Berson, J.F.; Thomas, P.C.; Raposo, G.; Marks, M.S. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 2006, 10, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Stuffers, S.; Wegner, C.S.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Edgar, J.R.; Eden, E.R.; Futter, C.E. Hrs- and CD63-dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic 2014, 15, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of MicroRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Saunderson, S.C.; Schuberth, P.C.; Dunn, A.C.; Miller, L.; Hock, B.D.; Mackay, P.A.; Koch, N.; Jack, R.W.; McLellan, A.D. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J. Immunol. 2008, 180, 8146–8152. [Google Scholar] [CrossRef] [Green Version]
- Admyre, C.; Bohle, B.; Johansson, S.M.; Focke-Tejkl, M.; Valenta, R.; Scheynius, A.; Gabrielsson, S. B cell–derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J. Allergy Clin. Immunol. 2007, 120, 1418–1424. [Google Scholar] [CrossRef]
- Saunderson, S.C.; McLellan, A.D. Role of lymphocyte subsets in the immune response to primary B cell–derived exosomes. J. Immunol. 2017, 199, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Muntasell, A.; Berger, A.C.; Roche, P.A. T cell-induced secretion of MHC class II–peptide complexes on B cell exosomes. EMBO J. 2007, 26, 4263–4272. [Google Scholar] [CrossRef] [Green Version]
- Denzer, K.; Van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; De Groot, C.; Geuze, H.J. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.; Turkes, A.; DeWitt, S.; Steadman, R.; Mason, M.D.; Hallett, M.B. Adhesion and signaling by B cell-derived exosomes: The role of integrins. FASEB J. 2004, 18, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Gutzeit, C.; Nagy, N.; Gentile, M.; Lyberg, K.; Gumz, J.; Vallhov, H.; Puga, I.; Klein, E.; Gabrielsson, S.; Cerutti, A.; et al. Exosomes derived from burkitt’s lymphoma cell lines induce proliferation, differentiation, and class-switch recombination in B cells. J. Immunol. 2014, 192, 5852–5862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, E.C.; Morrish, R.B.; Dillon, M.J.; Leyland, R.; Chahwan, R. Epigenomic Modifications Mediating Antibody Maturation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, R.B.; Hermes, M.; Metz, J.; Stone, N.; Pagliara, S.; Chahwan, R.; Palombo, F. Single Cell Imaging of Nuclear Architecture Changes. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Bala, S.; Bukong, T.; Szabo, G. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Messina, L.; Gutiérrez-Vázquez, C.; Rivas-García, E.; Sánchez-Madrid, F.; De La Fuente, H. Immunomodulatory role of microRNAs transferred by extracellular vesicles. Biol. Cell 2015, 107, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, L.N.; Alonso, M.A. Biogenesis and function of T cell-derived exosomes. Front. Cell Dev. Biol. 2016, 4, 84. [Google Scholar] [CrossRef] [Green Version]
- Wahlgren, J.; Karlson, T.D.L.; Glader, P.; Telemo, E.; Valadi, H. Activated human T cells secrete exosomes that participate in IL-2 mediated immune response signaling. PLoS ONE 2012, 7, e49723. [Google Scholar] [CrossRef] [Green Version]
- Tung, S.L.; Fanelli, G.; Matthews, R.I.; Bazoer, J.; Letizia, M.; Vizcay-Barrena, G.; Faruqu, F.N.; Philippeos, C.; Hannen, R.; Al-Jamal, K.T.; et al. Regulatory T cell extracellular vesicles modify T-effector cell cytokine production and protect against human skin allograft damage. Front. Cell Dev. Biol. 2020, 8, 317. [Google Scholar] [CrossRef]
- Chiou, N.-T.; Kageyama, R.; Ansel, K.M. Selective export into extracellular vesicles and function of tRNA fragments during T cell activation. Cell Rep. 2018, 25, 3356–3370.e4. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Messina, L.; Rodríguez-Galán, A.; De Yébenes, V.G.; Gutiérrez-Vázquez, C.; Tenreiro, S.; Seabra, M.C.; Ramiro, A.R.; Sánchez-Madrid, F. Transfer of extracellular vesicle-micro RNA controls germinal center reaction and antibody production. EMBO Rep. 2020, 21, e48925. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Wang, H.; Peng, H.; Huyan, T.; Cacalano, N.A. Exosomes: Versatile nano mediators of immune regulation. Cancers 2019, 11, 1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broeke, T.T.; Wubbolts, R.; Stoorvogel, W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb. Perspect. Biol. 2013, 5, a016873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, M.; Hu, R.; Runtsch, M.C.; Kagele, D.A.; Mosbruger, T.L.; Tolmachova, T.; Seabra, M.C.; Round, J.L.; Ward, D.M.; O’Connell, R.M. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 2015, 6, 7321. [Google Scholar] [CrossRef] [Green Version]
- Bhatnagar, S.; Shinagawa, K.; Castellino, F.J.; Schorey, J.S. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 2007, 110, 3234–3244. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, J.V.; De Castro, R.O.; Da Silva, E.Z.M.; Silveira, P.P.; Da Silva-Januário, M.E.; Arruda, E.; Jamur, M.C.; Oliver, C.; Aguiar, R.S.; Da Silva, L.L.P. Nef neutralizes the ability of exosomes from CD4+ T cells to act as decoys during HIV-1 infection. PLoS ONE 2014, 9, e113691. [Google Scholar] [CrossRef] [Green Version]
- Bello-Morales, R.; Ripa, I.; López-Guerrero, J.A. Extracellular vesicles in viral spread and antiviral response. Viruses 2020, 12, 623. [Google Scholar] [CrossRef]
- Bello-Morales, R.; López-Guerrero, J.A. Extracellular vesicles in herpes viral spread and immune evasion. Front. Microbiol. 2018, 9, 2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Tsai, M.-H.; Shumilov, A.; Baccianti, F.; Tsao, S.W.; Poirey, R.; Delecluse, H.-J. Epstein–Barr virus ncRNA from a nasopharyngeal carcinoma induces an inflammatory response that promotes virus production. Nat. Microbiol. 2019, 4, 2475–2486. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, H.; Yamakawa, N.; Imadome, K.-I.; Yahata, T.; Kotaki, R.; Ogata, J.; Kakizaki, M.; Fujita, K.; Lu, J.; Yokoyama, K.; et al. Role of exosomes as a proinflammatory mediator in the development of EBV-associated lymphoma. Blood 2018, 131, 2552–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inal, J.M.; Jorfi, S. Coxsackievirus B transmission and possible new roles for extracellular vesicles. Biochem. Soc. Trans. 2013, 41, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.-C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.-H.; Walker, C.M.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Woodson, M.; Neupane, B.; Bai, F.; Sherman, M.B.; Choi, K.H.; Neelakanta, G.; Sultana, H. Exosomes serve as novel modes of tick-borne flavivirus transmission from arthropod to human cells and facilitates dissemination of viral RNA and proteins to the vertebrate neuronal cells. PLoS Pathog. 2018, 14, e1006764. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Feng, Z.; Yue, H.; Bazdar, D.; Mbonye, U.; Zender, C.; Harding, C.V.; Bruggeman, L.; Karn, J.; Sieg, S.F.; et al. Exosomes derived from HIV-1-infected cells promote growth and progression of cancer via HIV TAR RNA. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, T.; Kalamvoki, M. Extracellular vesicles released by herpes simplex virus 1-infected cells block virus replication in recipient cells in a STING-dependent manner. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Tumne, A.; Prasad, V.S.; Chen, Y.; Stolz, D.B.; Saha, K.; Ratner, D.M.; Ding, M.; Watkins, S.C.; Gupta, P. Noncytotoxic suppression of human immunodeficiency virus type 1 transcription by exosomes secreted from CD8+ T cells. J. Virol. 2009, 83, 4354–4364. [Google Scholar] [CrossRef] [Green Version]
- Mack, M.; Kleinschmidt, A.; Brühl, H.; Klier, C.; Nelson, P.J.; Cihak, J.; Plachý, J.; Stangassinger, M.; Erfle, V.; Schlöndorff, D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 2000, 6, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Arakelyan, A.; Fitzgerald, W.; Zicari, S.; Vanpouille, C.; Margolis, L. Extracellular vesicles carry HIV Env and facilitate Hiv infection of human lymphoid tissue. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Schierer, S.; Blume, K.; Dindorf, J.; Wittki, S.; Xiang, W.; Ostalecki, C.; Koliha, N.; Wild, S.; Schuler, G.; et al. HIV-Nef and ADAM17-containing plasma extracellular vesicles induce and correlate with immune pathogenesis in chronic HIV infection. EBioMedicine 2016, 6, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Hubert, A.; Subra, C.; Jenabian, M.-A.; Labrecque, P.-F.T.; Tremblay, C.; Laffont, B.; Provost, P.; Routy, J.-P.; Gilbert, C. Elevated abundance, size, and MicroRNA content of plasma extracellular vesicles in viremic HIV-1+, size, and MicroRNA content of plasma extracellular vesicles in viremic HIV-1+ patients: Correlations with known markers of disease progression. J. Acquir. Immune Defic. Syndr. 2015, 70, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallhov, H.; Gutzeit, C.; Johansson, S.M.; Nagy, N.; Paul, M.; Li, Q.; Friend, S.; George, T.C.; Klein, E.; Scheynius, A.; et al. Exosomes containing glycoprotein 350 released by EBV-transformed B cells selectively target B cells through CD21 and block EBV infection in vitro. J. Immunol. 2011, 186, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; Van Eijndhoven, M.A.J.; Hopmans, E.S.; Lindenberg, J.L.; De Gruijl, T.D.; Würdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keryer-Bibens, C.; Pioche-Durieu, C.; Villemant, C.; Souquère, S.; Nishi, N.; Hirashima, M.; Middeldorp, J.M.; Busson, P. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer 2006, 6, 283. [Google Scholar] [CrossRef] [Green Version]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Raimondo, S.; Saieva, L.; Corrado, C.; Fontana, S.; Flugy, A.; Rizzo, A.; De Leo, G.; Alessandro, R. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun. Signal. 2015, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Wurdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Setti, M.; Osti, D.; Richichi, C.; Ortensi, B.; Del Bene, M.; Fornasari, L.; Beznoussenko, G.; Mironov, A.; Rappa, G.; Cuomo, A.; et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015, 6, 31413–31427. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.-L.; Qu, X.-J.; Zhao, M.-F.; Teng, Y.-E.; Zhang, Y.; Hou, K.-Z.; Jiang, Y.-H.; Yang, X.-H.; Liu, Y. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig. Liver Dis. 2009, 41, 875–880. [Google Scholar] [CrossRef]
- Li, C.; Liu, D.-R.; Li, G.-G.; Wang, H.-H.; Li, X.-W.; Zhang, W.; Wu, Y.-L.; Chen, L. CD97 promotes gastric cancer cell proliferation and invasion through exosome-mediated MAPK signaling pathway. World J. Gastroenterol. 2015, 21, 6215–6228. [Google Scholar] [CrossRef]
- Yang, L.; Wu, X.-H.; Wang, D.; Luo, C.-L.; Chen, L.-X. Bladder cancer cell-derived exosomes inhibit tumor cell apoptosis and induce cell proliferation in vitro. Mol. Med. Rep. 2013, 8, 1272–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotič, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitaš, A.; Peterlin, B. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. OncoImmunology 2016, 5, e1062968. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, Y.; Wang, W.; Zhang, Y.; Chen, Z.; Hao, C.; Zhang, J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4+ T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Ye, S.-B.; Li, Z.-L.; Luo, D.-H.; Huang, B.-J.; Chen, Y.-S.; Zhang, X.-S.; Cui, J.; Zeng, Y.-X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef] [Green Version]
- Bland, C.L.; Byrne-Hoffman, C.N.; Fernandez, A.; Rellick, S.L.; Deng, W.; Ii, D.J.K. Exosomes derived from B16F0 melanoma cells alter the transcriptome of cytotoxic T cells that impacts mitochondrial respiration. FEBS J. 2018, 285, 1033–1050. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Teitz-Tennenbaum, S.; Donald, E.J.; Li, M.; Chang, A.E. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J. Immunol. 2009, 183, 3195–3203. [Google Scholar] [CrossRef]
- Martin, F.; Chan, A.C. B cell immunobiology in disease: Evolving concepts from the clinic. Annu. Rev. Immunol. 2006, 24, 467–496. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.H. CD20+ B cells: The other tumor-infiltrating lymphocytes. J. Immunol. 2010, 185, 4977–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fremd, C.; Schuetz, F.; Sohn, C.; Beckhove, P.; Domschke, C. B cell-regulated immune responses in tumor models and cancer patients. OncoImmunology 2013, 2, e25443. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chalasani, G.; Ng, Y.-H.; Robbins, P.D. Exosomes released from mycoplasma infected tumor cells activate inhibitory B cells. PLoS ONE 2012, 7, e36138. [Google Scholar] [CrossRef] [Green Version]
- Pucci, F.; Garris, C.; Lai, C.P.; Newton, A.; Pfirschke, C.; Engblom, C.; Alvarez, D.; Sprachman, M.; Evavold, C.; Magnuson, A.; et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science 2016, 352, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; An, J.; Huang, S.; He, J.; Zhang, J. Esophageal cancer-derived microvesicles induce regulatory B cells. Cell Biochem. Funct. 2015, 33, 308–313. [Google Scholar] [CrossRef]
- Federici, C.; Shahaj, E.; Cecchetti, S.; Camerini, S.; Casella, M.; Iessi, E.; Camisaschi, C.; Paolino, G.; Calvieri, S.; Ferro, S.; et al. Natural-killer-derived extracellular vesicles: Immune sensors and interactors. Front. Immunol. 2020, 11, 262. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.-H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural killer–derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef]
- Wu, C.-H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell. Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef] [Green Version]
- Di Pace, A.L.; Tumino, N.; Besi, F.; Alicata, C.; Conti, L.A.; Munari, E.; Maggi, E.; Vacca, P.; Moretta, L. Characterization of human NK cell-derived exosomes: Role of DNAM1 receptor in exosome-mediated cytotoxicity against tumor. Cancers 2020, 12, 661. [Google Scholar] [CrossRef] [Green Version]
- Moretta, L.; Pietra, G.; Montaldo, E.; Vacca, P.; Pende, D.; Falco, M.; Del Zotto, G.; Locatelli, F.; Moretta, A.; Mingari, M.C. Human NK cells: From surface receptors to the therapy of leukemias and solid tumors. Front. Immunol. 2014, 5, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; De Almeida, P.; Manieri, N.; Nagata, D.D.A.; Wu, T.D.; Bowles, K.H.; Arumugam, V.; Schartner, J.; Cubas, R.; Mittman, S.; et al. CD226 regulates natural killer cell antitumor responses via phosphorylation-mediated inactivation of transcription factor FOXO1. Proc. Natl. Acad. Sci. USA 2018, 115, E11731–E11740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castriconi, R.; Griffin, K.J.; Kirschner, L.S.; Matyakhina, L.; Stergiopoulos, S.; Robinson-White, A.; Lenherr, S.; Weinberg, F.D.; Claflin, E.; Meoli, E.; et al. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: Critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004, 64, 9180–9184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pende, D.; Spaggiari, G.M.; Marcenaro, S.; Martini, S.; Rivera, P.; Capobianco, A.; Falco, M.; Lanino, E.; Pierri, I.; Zambello, R.; et al. Analysis of the receptor-ligand interactions in the natural killer–mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: Evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112). Blood 2005, 105, 2066–2073. [Google Scholar] [CrossRef]
- Croxford, J.L.; Tang, M.L.F.; Pan, M.F.; Huang, C.W.; Kamran, N.; Phua, C.M.L.; Chng, W.J.; Ng, S.-B.; Raulet, D.H.; Gasser, S. ATM-dependent spontaneous regression of early Eμ-myc–induced murine B-cell leukemia depends on natural killer and T cells. Blood 2013, 121, 2512–2521. [Google Scholar] [CrossRef]
- Guillerey, C.; De Andrade, L.F.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.; Teng, M.W.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J. Clin. Investig. 2015, 125, 2077–2089. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 Interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef]
- Fabbri, M. Natural killer cell–derived vesicular miRNAs: A new anticancer approach? Cancer Res. 2020, 80, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Shi, K.; Qi, K.; Kong, H.; Zhang, J.; Dai, S.; Ye, W.; Deng, T.; He, Q.; Zhou, M. Natural killer cell-derived exosomal miR-3607-3p inhibits pancreatic cancer progression by targeting IL-26. Front. Immunol. 2019, 10, 2819. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Wang, H.; Yu, J.; Zhang, J.; Yang, Z.; Liu, M.; Niu, Y.; Wei, X.; Wang, W.; Li, H.; et al. miR-3607-3p suppresses non-small cell lung cancer (NSCLC) by targeting TGFBR1 and CCNE2. PLoS Genet. 2018, 14, e1007790. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sha, H.-H.; Li, H.-J. Functions and mechanisms of miR-186 in human cancer. Biomed. Pharmacother. 2019, 119, 109428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schlößer, H.; Wang, Z.; Qin, J.; Li, J.; Popp, F.C.; Popp, M.C.; Alakus, H.; Chon, S.-H.; Hansen, H.P.; et al. Tumor-derived extracellular vesicles inhibit natural killer cell function in pancreatic cancer. Cancers 2019, 11, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor- 1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, S.; Zinn, K.; Wang, J.; Zhang, L.; Jia, Y.; Kappes, J.C.; Barnes, S.; Kimberly, R.P.; Grizzle, W.E.; et al. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J. Immunol. 2006, 176, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Huang, L.; Li, Y.; Zhang, X.; Gu, J.; Yan, Y.; Xu, X.; Wang, M.; Qian, H.; Xu, W. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012, 315, 28–37. [Google Scholar] [CrossRef]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal stem cell–derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Bhome, R.; Goh, R.W.; Bullock, M.D.; Pillar, N.; Thirdborough, S.M.; Mellone, M.; Mirnezami, R.; Galea, D.; Veselkov, K.; Gu, Q.; et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: Role in driving cancer progression. Aging 2017, 9, 2666–2694. [Google Scholar] [CrossRef] [Green Version]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [Green Version]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Sergé, A.; Lavaut, M.-N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [Green Version]
- Yeung, C.L.A.; Co, N.-N.; Tsuruga, T.; Yeung, T.-L.; Kwan, S.-Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ha, C.; Dong, H.; Yang, Z.; Ma, Y.; Ding, Y. Cancer-associated fibroblast-derived exosomal microRNA-98-5p promotes cisplatin resistance in ovarian cancer by targeting CDKN1A. Cancer Cell Int. 2019, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Yashiro, M.; Okuno, T.; Kitayama, K.; Masuda, G.; Hirakawa, K.; Ohira, M. CD9-positive exosomes from cancer-associated fibroblasts stimulate the migration ability of scirrhous-type gastric cancer cells. Br. J. Cancer 2018, 118, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Li, H.; Guo, Q.; Zhou, A.; Wang, X.; Li, P.; Zhang, S. Exosomal sonic hedgehog derived from cancer-associated fibroblasts promotes proliferation and migration of esophageal squamous cell carcinoma. Cancer Med. 2020, 9, 2500–2513. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Li, M.; Cao, L.M.; Gu, Q.H.; Deng, P.B.; Tan, Y.; Hu, C.P. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. QJM Int. J. Med. 2019, 112, 581–590. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wang, J.; Li, M.; Cao, C.; Tan, J.; Ma, D.; Gao, Q. TGFβ1 in fibroblasts-derived exosomes promotes epithelial-mesenchymal transition of ovarian cancer cells. Oncotarget 2017, 8, 96035–96047. [Google Scholar] [CrossRef] [Green Version]
- Koch, R.; Demant, M.; Aung, T.; Diering, N.; Cicholas, A.; Chapuy, B.; Wenzel, D.; Lahmann, M.; Güntsch, A.; Kiecke, C.; et al. Populational equilibrium through exosome-mediated Wnt signaling in tumor progression of diffuse large B-cell lymphoma. Blood 2014, 123, 2189–2198. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-B.; Yan, C.; Mu, L.; Mi, Y.; Zhao, H.; Hu, H.; Li, X.-L.; Tao, D.-D.; Wu, Y.-Q.; Gong, J.; et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019, 38, 1951–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.F.D.; Serrano, E.; Patrício, M.I.; Val, M.M.; Albuquerque, P.; Fonseca, J.; Gomes, C.M.F.; Abrunhosa, A.; Paiva, A.; Carvalho, L.; et al. Stroma-derived IL-6, G-CSF and Activin-A mediated dedifferentiation of lung carcinoma cells into cancer stem cells. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liao, T.; Lin, C.; Yuan, L.E.; Lan, H.; Lin, H.; Teng, H.; Chang, H.; Lin, C.; Yang, C.; et al. RAB27B-activated secretion of stem-like tumor exosomes delivers the biomarker microRNA-146a-5p, which promotes tumorigenesis and associates with an immunosuppressive tumor microenvironment in colorectal cancer. Int. J. Cancer 2019, 145, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Cao, H.; Shen, B.; Feng, J. Tumor-derived exosomes in cancer progression and treatment failure. Oncotarget 2015, 6, 37151–37168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.-Y.; Yu, C.-H.; Zhang, H.-H.; Zhang, S.-Z.; Yu, W.-Y.; Yang, Y.; Chen, Q. Exosomal miR-1229 derived from colorectal cancer cells promotes angiogenesis by targeting HIPK2. Int. J. Biol. Macromol. 2019, 132, 470–477. [Google Scholar] [CrossRef]
- Tadokoro, H.; Umezu, T.; Ohyashiki, K.; Hirano, T.; Ohyashiki, J.H. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J. Biol. Chem. 2013, 288, 34343–34351. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Seubert, B.; A Stahl, E.; Dietz, H.C.; Reuning, U.; Moreno-Leon, L.; Ilie, M.; Hofman, P.; Nagase, H.; Mari, B.; et al. Tissue inhibitor of metalloproteinases-1 induces a pro-tumourigenic increase of miR-210 in lung adenocarcinoma cells and their exosomes. Oncogene 2014, 34, 3640–3650. [Google Scholar] [CrossRef]
- Lin, X.-J.; Fang, J.-H.; Yang, X.-J.; Zhang, C.; Yuan, Y.; Zheng, L.; Zhuang, S. Hepatocellular carcinoma cell-secreted exosomal microRNA-210 promotes angiogenesis in vitro and in vivo. Mol. Ther. Nucleic Acids 2018, 11, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood–brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Modica, M.; Regondi, V.; Sandri, M.; Iorio, M.V.; Zanetti, A.; Tagliabue, E.; Casalini, P.; Triulzi, T. Breast cancer-secreted miR-939 downregulates VE-cadherin and destroys the barrier function of endothelial monolayers. Cancer Lett. 2017, 384, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.H.; Zhang, Z.J.; Shang, L.R.; Luo, Y.W.; Lin, Y.F.; Yuan, Y.; Zhuang, S.M. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 2018, 68, 1459–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedy, P.; Medarova, Z. The fundamental role of miR-10b in metastatic cancer. Am. J. Cancer Res. 2018, 8, 1674–1688. [Google Scholar] [PubMed]
- Li, Q.-J.; Zhou, L.; Yang, F.; Wang, G.-X.; Zheng, H.; Wang, D.-S.; He, Y.; Dou, K.-F. MicroRNA-10b promotes migration and invasion through CADM1 in human hepatocellular carcinoma cells. Tumor Biol. 2012, 33, 1455–1465. [Google Scholar] [CrossRef]
- Liao, C.-G.; Kong, L.-M.; Zhou, P.; Yang, X.-L.; Huang, J.-G.; Zhang, H.; Lu, N. miR-10b is overexpressed in hepatocellular carcinoma and promotes cell proliferation, migration and invasion through RhoC, uPAR and MMPs. J. Transl. Med. 2014, 12, 234. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Eun, J.W.; Baek, G.O.; Seo, C.W.; Ahn, H.R.; Kim, S.S.; Cho, S.W.; Cheong, J.Y. Serum exosomal microRNA, miR-10b-5p, as a potential diagnostic biomarker for early-stage hepatocellular carcinoma. J. Clin. Med. 2020, 9, 281. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.-P.; Wang, C.-Y.; Jin, X.-H.; Li, M.; Wang, F.-W.; Huang, W.-J.; Yun, J.-P.; Xu, R.-H.; Cai, Q.-Q.; Xie, D. Acidic microenvironment up-regulates exosomal miR-21 and miR-10b in early-stage hepatocellular carcinoma to promote cancer cell proliferation and metastasis. Theranostics 2019, 9, 1965–1979. [Google Scholar] [CrossRef]
- Dai, G.; Yao, X.; Zhang, Y.; Gu, J.; Geng, Y.; Xue, F.; Zhang, J. Colorectal cancer cell–derived exosomes containing miR-10b regulate fibroblast cells via the PI3K/Akt pathway. Bull Cancer 2018, 105, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef] [PubMed]
- La Shu, S.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Xia, Y.; Wang, Z.; Zheng, J.; Chen, Y.; Li, X.; Wang, Y.; Ming, H. Serum long non coding RNA MALAT-1 protected by exosomes is up-regulated and promotes cell proliferation and migration in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2017, 490, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, Y.; Lu, J.; Sun, Y.; Xiao, H.; Liu, M.; Tian, L. Combined detection of serum exosomal miR-21 and HOTAIR as diagnostic and prognostic biomarkers for laryngeal squamous cell carcinoma. Med. Oncol. 2014, 31, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Berrondo, C.; Flax, J.; Kucherov, V.; Siebert, A.; Osinski, T.; Rosenberg, A.; Fucile, C.; Richheimer, S.; Beckham, C.J. Expression of the long non-coding RNA HOTAIR correlates with disease progression in bladder cancer and is contained in bladder cancer patient urinary exosomes. PLoS ONE 2016, 11, e0147236. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Li, Z.; Li, T.; Zhu, L.; Li, Z.; Tian, N. Long non-coding RNA HOTAIR enhances angiogenesis by induction of VEGFA expression in glioma cells and transmission to endothelial cells via glioma cell derived-extracellular vesicles. Am. J. Transl. Res. 2017, 9, 5012–5021. [Google Scholar]
- Xu, C.-G.; Yang, M.-F.; Ren, Y.-Q.; Wu, C.-H.; Wang, L.-Q. Exosomes mediated transfer of lncRNA UCA1 results in increased tamoxifen resistance in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4362–4368. [Google Scholar]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.-F.; et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef]
- Tang, M.K.S.; Yue, P.Y.K.; Ip, P.P.; Huang, R.-L.; Lai, H.-C.; Cheung, A.N.Y.; Tse, K.Y.; Ngan, H.Y.S.; Wong, A.S.T. Soluble E-cadherin promotes tumor angiogenesis and localizes to exosome surface. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Ko, S.Y.; Lee, W.; Kenny, H.A.; Dang, L.H.; Ellis, L.M.; Jonasch, E.; Lengyel, E.; Naora, H. Cancer-derived small extracellular vesicles promote angiogenesis by heparin-bound, bevacizumab-insensitive VEGF, independent of vesicle uptake. Commun. Biol. 2019, 2, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shedden, K.; Xie, X.T.; Chandaroy, P.; Chang, Y.-T.; Rosania, G.R. Expulsion of small molecules in vesicles shed by cancer cells: Association with gene expression and chemosensitivity profiles. Cancer Res. 2003, 63, 4331–4337. [Google Scholar] [PubMed]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Yu, S.; Li, S.; Wu, J.; Ma, R.; Cao, H.; Zhu, Y.; Feng, J. Exosomes: Decreased sensitivity of lung cancer A549 cells to cisplatin. PLoS ONE 2014, 9, e89534. [Google Scholar] [CrossRef] [Green Version]
- Dusoswa, S.A.; Horrevorts, S.K.; Ambrosini, M.; Kalay, H.; Paauw, N.J.; Nieuwland, R.; Pegtel, M.D.; Würdinger, T.; Van Kooyk, Y.; García-Vallejo, J.-J. Glycan modification of glioblastoma-derived extracellular vesicles enhances receptor-mediated targeting of dendritic cells. J. Extracell. Vesicles 2019, 8, 1648995. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.-C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 2019, 177, 414.e13–427.e13. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Xu, M.; Lu, J.; Mao, L.; Wang, S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer 2019, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torreggiani, E.; Roncuzzi, L.; Perut, F.; Zini, N.; Baldini, N. Multimodal transfer of MDR by exosomes in human osteosarcoma. Int. J. Oncol. 2016, 49, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebawy, M.; Combes, V.; Lee, E.; Jaiswal, R.; Gong, J.; Bonhoure, A.; Grau, G.E.R. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia 2009, 23, 1643–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, C.; Rani, S.; O’Driscoll, L.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef]
- Zhang, F.-F.; Zhu, Y.-F.; Zhao, Q.-N.; Yang, D.-T.; Dong, Y.-P.; Jiang, L.; Xing, W.-X.; Li, X.-Y.; Xing, H.; Shi, M.; et al. Microvesicles mediate transfer of P-glycoprotein to paclitaxel-sensitive A2780 human ovarian cancer cells, conferring paclitaxel-resistance. Eur. J. Pharmacol. 2014, 738, 83–90. [Google Scholar] [CrossRef]
- Levchenko, A.; Mehta, B.M.; Niu, X.; Kang, G.; Villafania, L.; Way, D.; Polycarpe, D.; Sadelain, M.; Larson, S.M. Intercellular transfer of P-glycoprotein mediates acquired multidrug resistance in tumor cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1933–1938. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hu, S.; Wang, J.; Cai, J.; Xiao, L.; Yu, L.; Wang, Z. MiR-27a modulates MDR1/P-glycoprotein expression by targeting HIPK2 in human ovarian cancer cells. Gynecol. Oncol. 2010, 119, 125–130. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, H.; Liu, X.; Evans, B.R.; Medina, D.J.; Liu, C.-G.; Yang, J.-M. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem. Pharmacol. 2008, 76, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-X.; Liu, X.-M.; Lv, M.-M.; Chen, L.; Zhao, J.-H.; Zhong, S.-L.; Ji, M.-H.; Hu, Q.; Luo, Z.; Wu, J.-Z.; et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of MicroRNAs. PLoS ONE 2014, 9, e95240. [Google Scholar] [CrossRef]
- Mao, L.; Li, J.; Chen, W.-X.; Cai, Y.-Q.; Yu, D.-D.; Zhong, S.-L.; Zhao, J.-H.; Zhou, J.-W.; Tang, J.-H. Exosomes decrease sensitivity of breast cancer cells to adriamycin by delivering microRNAs. Tumor Biol. 2015, 37, 5247–5256. [Google Scholar] [CrossRef]
- Wei, Y.; Lai, X.; Yu, S.; Chen, S.; Ma, Y.; Zhang, Y.; Li, H.; Zhu, X.; Yao, L.; Zhang, J. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-D.; Wu, Y.; Zhang, X.-H.; Lv, M.-M.; Chen, W.-X.; Chen, X.; Yang, S.-J.; Shen, H.; Zhong, S.-L.; Tang, J.-H.; et al. Exosomes from adriamycin-resistant breast cancer cells transmit drug resistance partly by delivering miR-222. Tumor Biol. 2016, 37, 3227–3235. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Gong, M.; Hu, Y.; Liu, H.; Zhang, W.; Zhang, M.; Hu, X.; Aubert, D.; Zhu, S.; Wu, L.; et al. Quality and efficiency assessment of six extracellular vesicle isolation methods by nano-flow cytometry. J. Extracell. Vesicles 2020, 9, 1697028. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; Lee, J.; Ahn, B.-C. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Zhang, F.; Li, R.; Yang, Y.; Shi, C.; Shen, Y.; Lu, C.; Chen, Y.; Zhou, W.; Lin, A.; Yu, L.; et al. Specific decrease in B-cell-derived extracellular vesicles enhances post-chemotherapeutic CD8+ T cell responses. Immunity 2019, 50, 738–750.e7. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655–664. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yim, K.H.W.; Al Hrout, A.; Borgoni, S.; Chahwan, R. Extracellular Vesicles Orchestrate Immune and Tumor Interaction Networks. Cancers 2020, 12, 3696. https://doi.org/10.3390/cancers12123696
Yim KHW, Al Hrout A, Borgoni S, Chahwan R. Extracellular Vesicles Orchestrate Immune and Tumor Interaction Networks. Cancers. 2020; 12(12):3696. https://doi.org/10.3390/cancers12123696
Chicago/Turabian StyleYim, Kevin Ho Wai, Ala’a Al Hrout, Simone Borgoni, and Richard Chahwan. 2020. "Extracellular Vesicles Orchestrate Immune and Tumor Interaction Networks" Cancers 12, no. 12: 3696. https://doi.org/10.3390/cancers12123696