Specific Effects of Trabectedin and Lurbinectedin on Human Macrophage Function and Fate—Novel Insights

 , , , ,
, , , ,  , and
, and
Abstract
:Simple Summary
Abstract
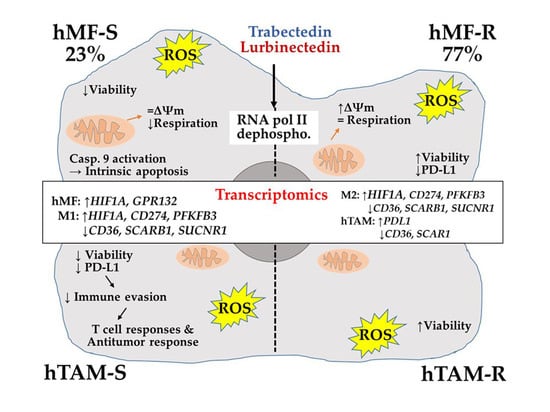
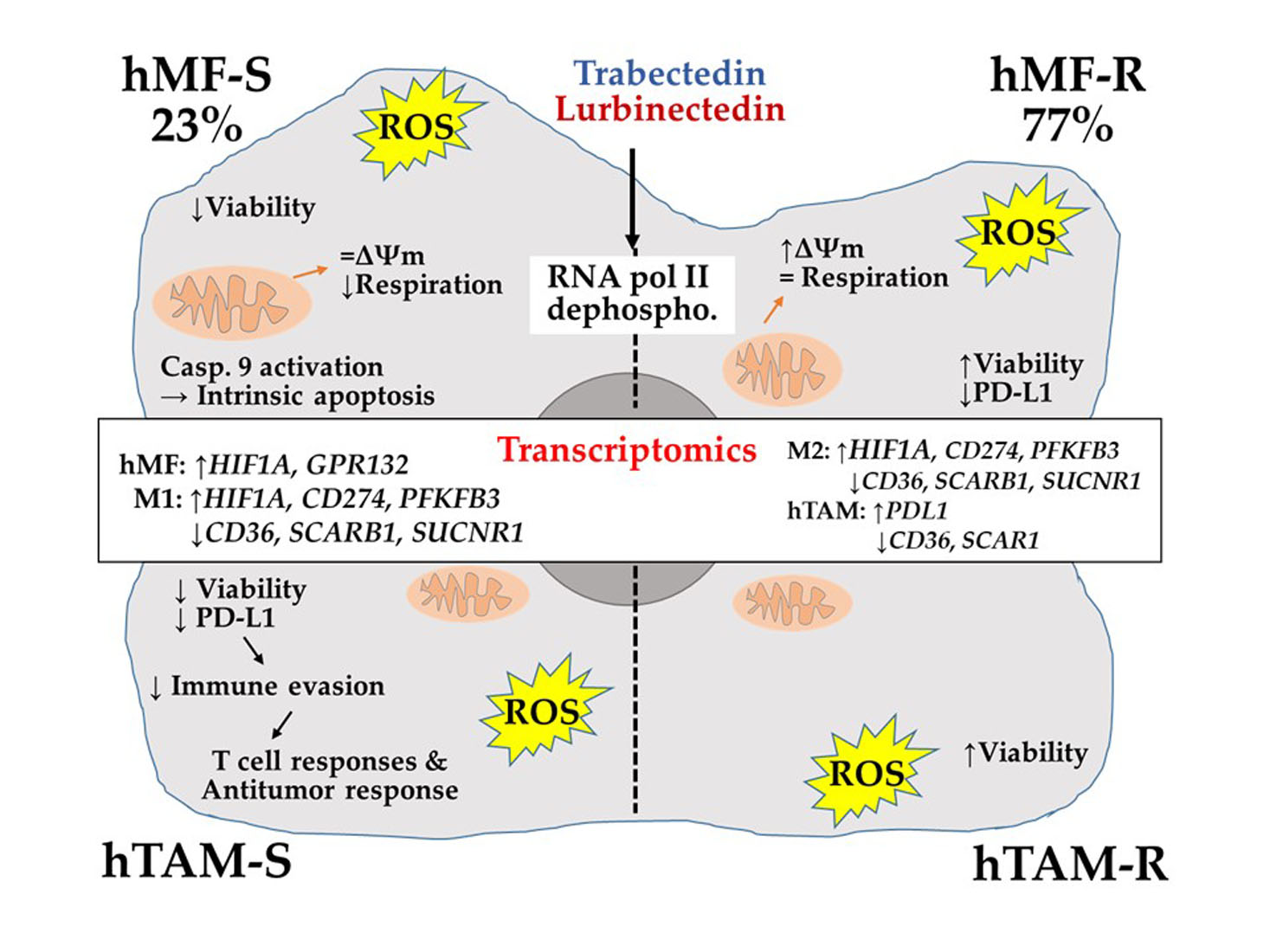
1. Introduction
2. Results
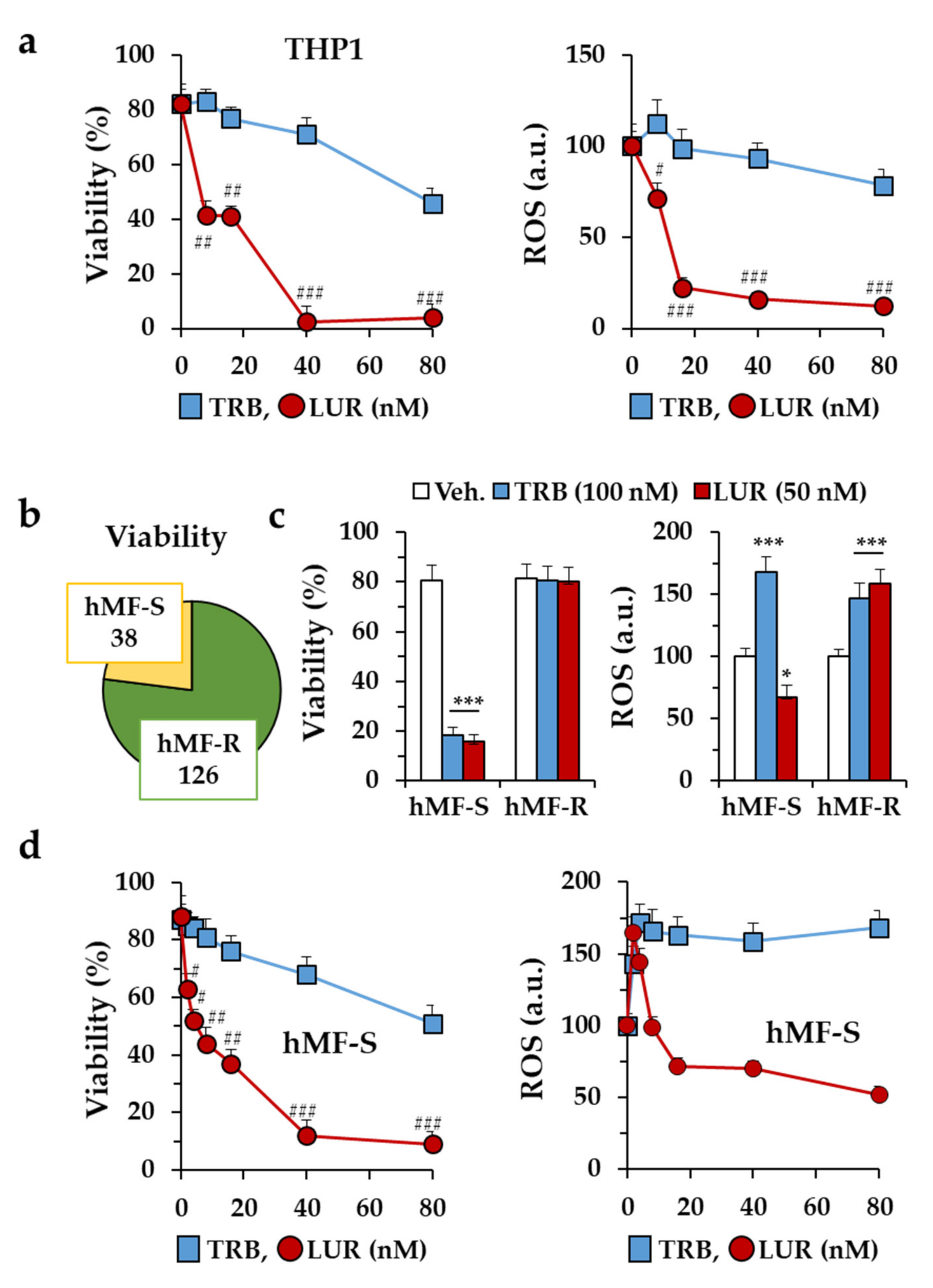
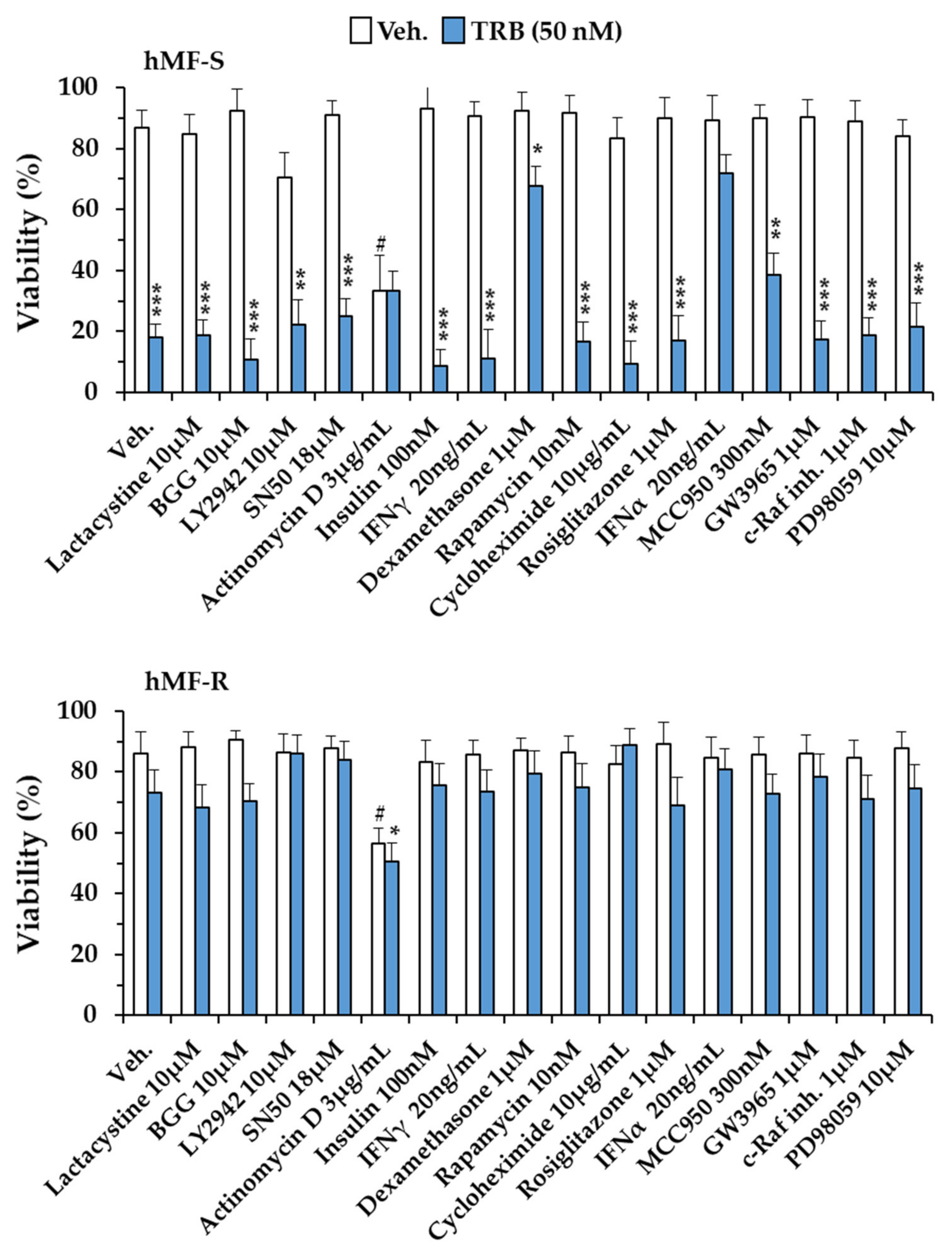
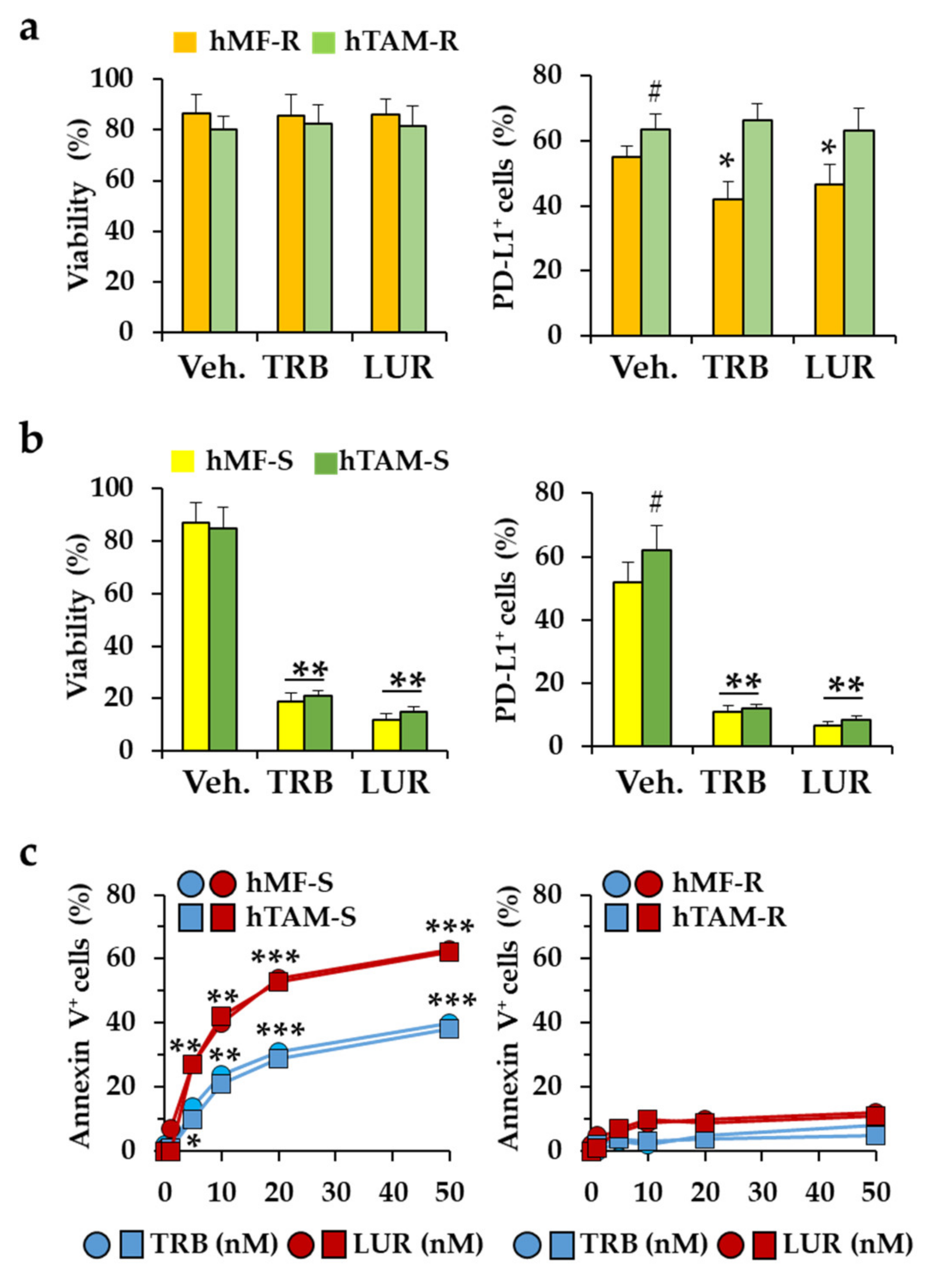
2.1. Differential Responses of Human Monocyte-Derived Macrophages to Trabectedin and Lurbinectedin
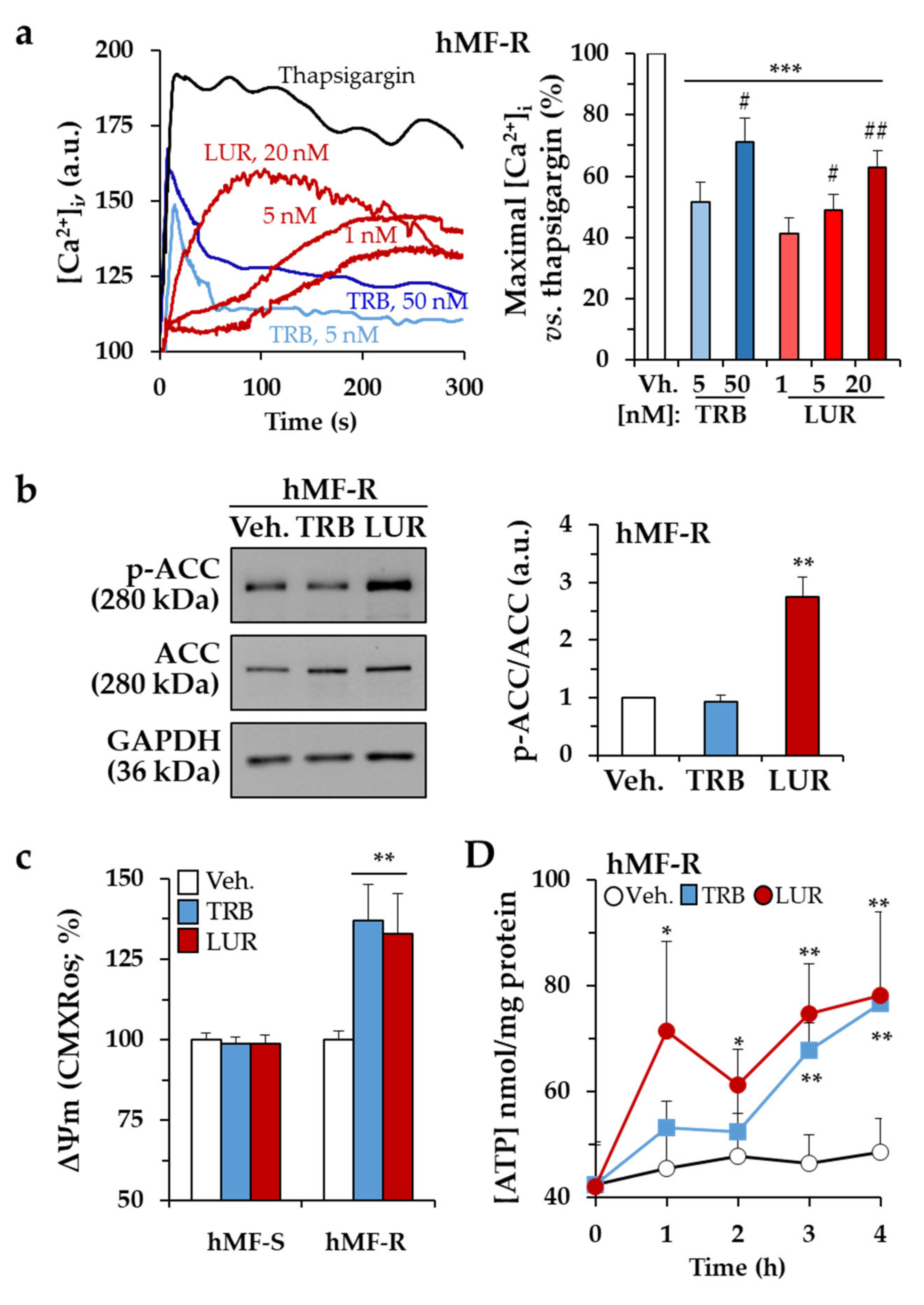
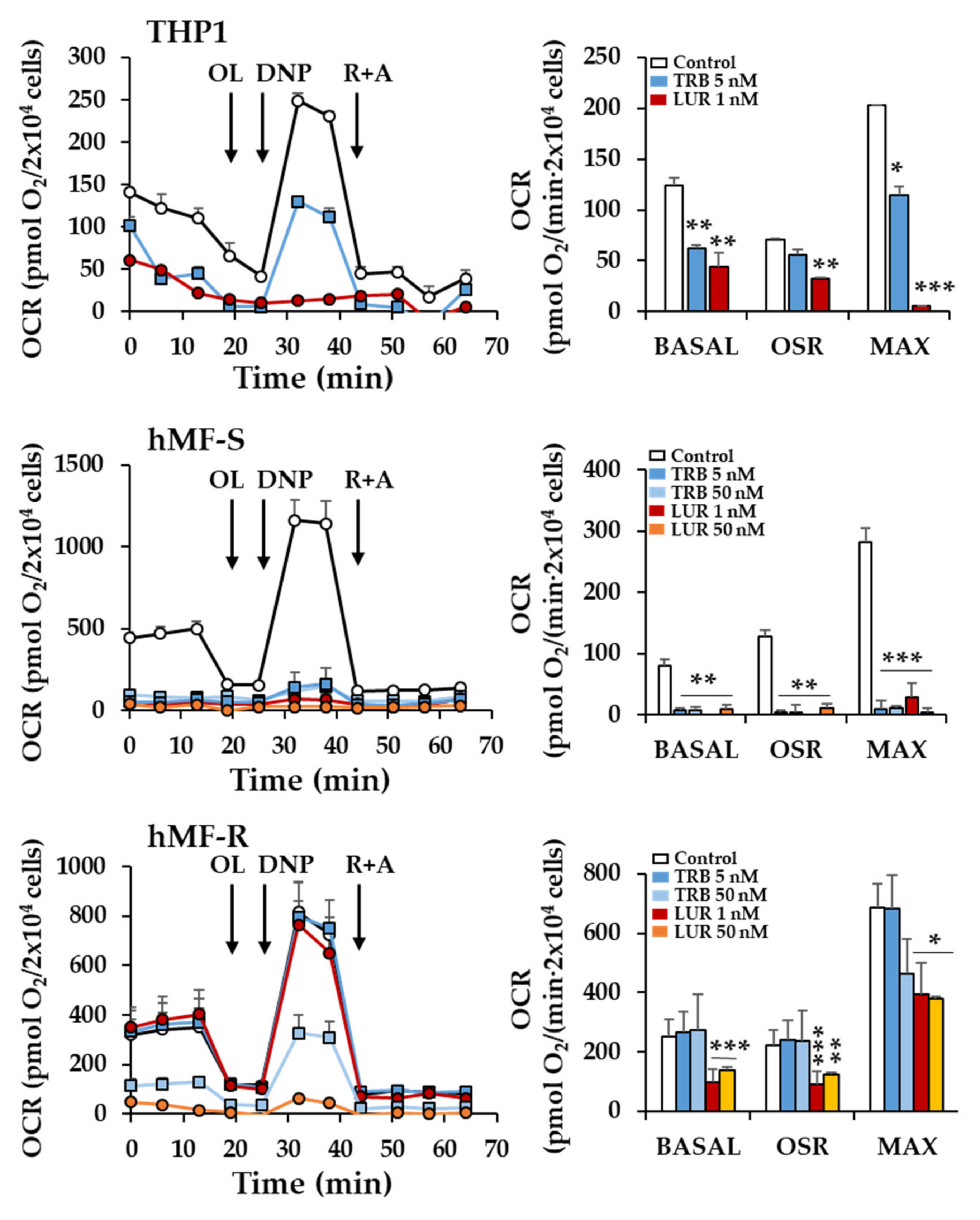
2.2. Treatment of hMF with Trabectedin and Lurbinectedin Promotes Intracellular Ca2+ Rise and Changes in Mitochondrial Respiration
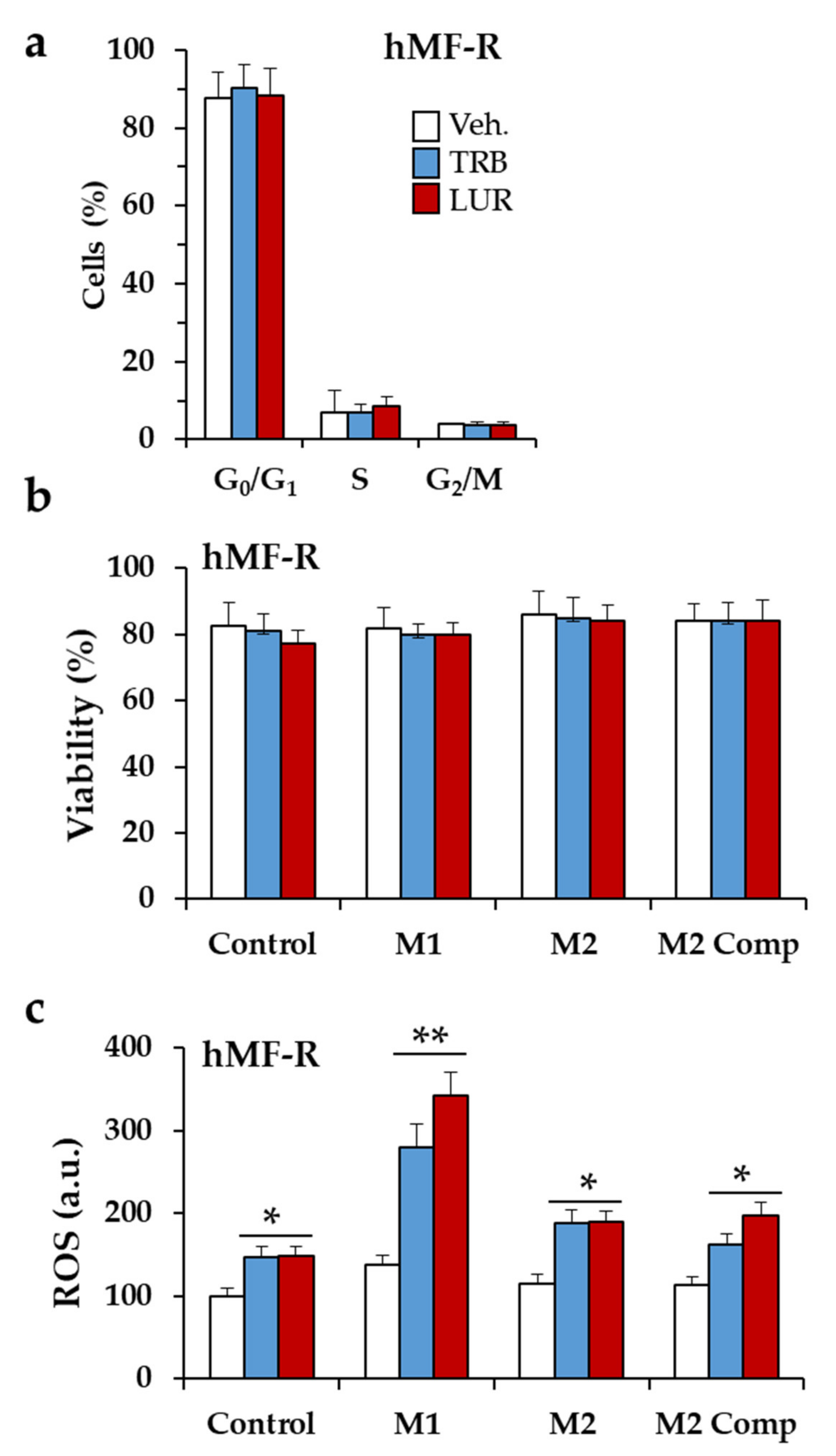
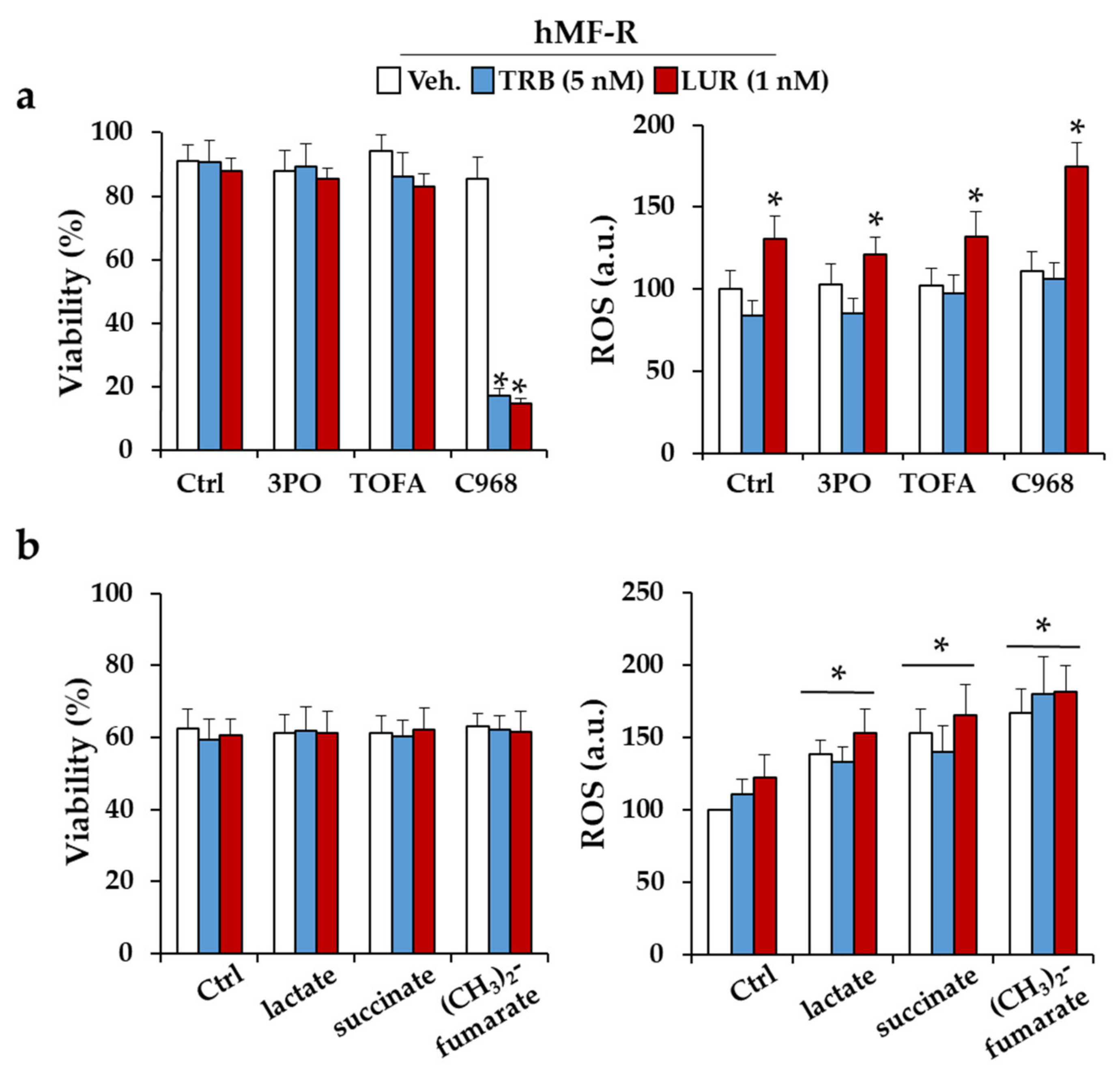
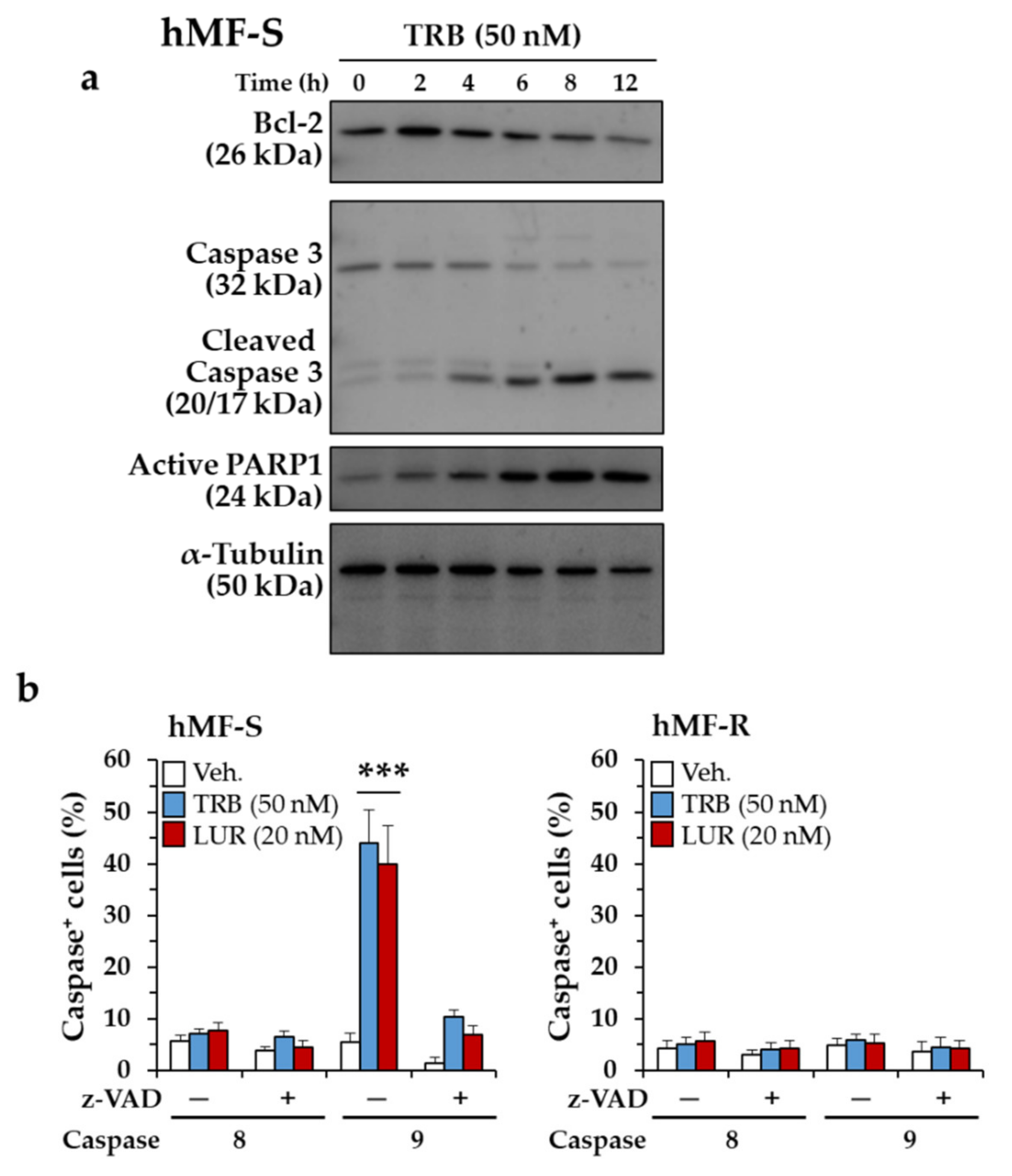
2.3. Glutamine Metabolism Is Required for Survival of hMF-R Treated with Trabectedin and Lurbinectedin
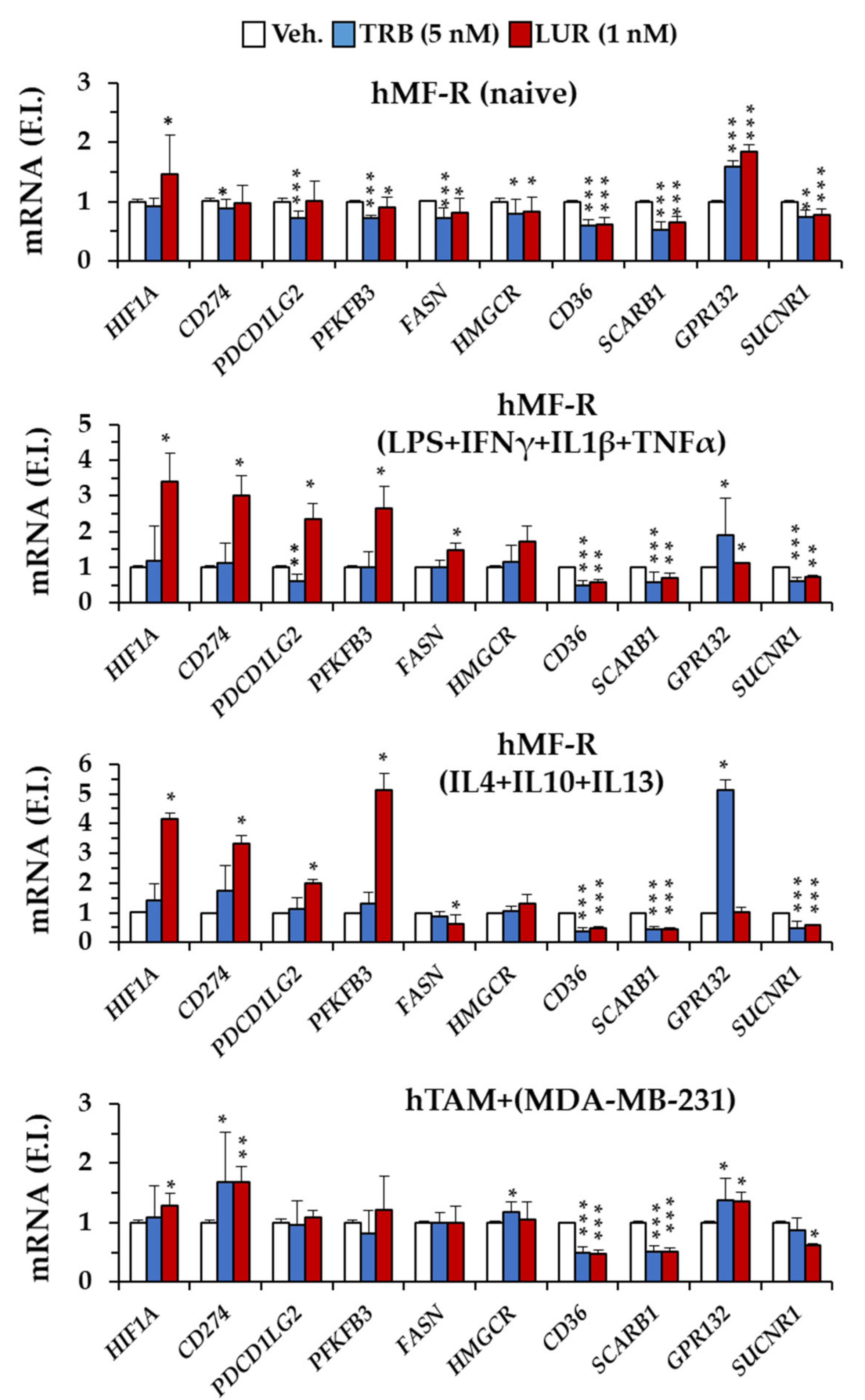
2.4. Specific Gene Profiling of hMF-R Treated with Trabectedin and Lurbinectedin
3. Discussion
4. Materials and Methods
4.1. PharmaMar Antitumoral Drugs
4.2. Human Samples
4.3. Human PMBC Isolation
4.4. hMF Culture and Differentiation
4.5. THP-1 Culture and Differentiation
4.6. hMF Polarization Assays
4.7. MDA-MB-231 Culture and hTAM Generation
4.8. Flow Cytometry Assays
4.9. Immuno-Metabolic Assays
4.10. Apoptosis Assays
4.11. hMF Phagocytosis
4.12. Cell Observer Measurements (Calcium Time-Lapse Assays)
4.13. ATP Measurements in hMFs
4.14. Protein Analysis and Immunoblotting
4.15. Seahorse (Agilent Technologies XF24)
4.16. RNA Isolation and Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cuevas, C.; Francesch, A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009, 26, 322–337. [Google Scholar] [CrossRef]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, L.H.; Zewail-Foote, M. The antitumor agent ecteinascidin 743: Characterization of its covalent DNA adducts and chemical stability. Adv. Exp. Med. Biol. 2001, 500, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Erba, E.; Cavallaro, E.; Damia, G.; Mantovani, R.; Di Silvio, A.; Di Francesco, A.M.; Riccardi, R.; Cuevas, C.; Faircloth, G.T.; D’Incalci, M. The unique biological features of the marine product Yondelis (ET-743, trabectedin) are shared by its analog ET-637, which lacks the C ring. Oncol. Res. 2004, 14, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.Y.; Movva, S. Systemic Therapy for Advanced Soft Tissue Sarcoma. Surg. Clin. North. Am. 2016, 96, 1141–1156. [Google Scholar] [CrossRef]
- Pignata, S.; Pisano, C.; Di Napoli, M.; Cecere, S.C.; Tambaro, R.; Attademo, L. Treatment of recurrent epithelial ovarian cancer. Cancer 2019, 125, 4609–4615. [Google Scholar] [CrossRef]
- Monk, B.J.; Herzog, T.J.; Wang, G.; Triantos, S.; Maul, S.; Knoblauch, R.; McGowan, T.; Shalaby, W.S.W.; Coleman, R.L. A phase 3 randomized, open-label, multicenter trial for safety and efficacy of combined trabectedin and pegylated liposomal doxorubicin therapy for recurrent ovarian cancer. Gynecol. Oncol. 2020, 156, 535–544. [Google Scholar] [CrossRef]
- D’Incalci, M. Trabectedin mechanism of action: What’s new? Future Oncol. 2013, 9, 5–10. [Google Scholar] [CrossRef]
- Rubio, M.J.; Lecumberri, M.J.; Varela, S.; Alarcón, J.; Ortega, M.E.; Gaba, L.; Espinós, J.; Calzas, J.; Barretina, P.; Ruiz, I.; et al. Efficacy and safety of trabectedin in metastatic uterine leiomyosarcoma: A retrospective multicenter study of the Spanish ovarian cancer research group (GEICO). Gynecol. Oncol. reports 2020, 33, 100594. [Google Scholar] [CrossRef]
- Jones, R.L.; Maki, R.G.; Patel, S.R.; Wang, G.; McGowan, T.A.; Shalaby, W.S.; Knoblauch, R.E.; von Mehren, M.; Demetri, G.D. Safety and efficacy of trabectedin when administered in the inpatient versus outpatient setting: Clinical considerations for outpatient administration of trabectedin. Cancer 2019, 125, 4435–4441. [Google Scholar] [CrossRef]
- Kurebayashi, J. Possible treatment strategies for triple-negative breast cancer on the basis of molecular characteristics. Breast Cancer 2009, 16, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Morioka, H.; Takahashi, S.; Araki, N.; Sugiura, H.; Ueda, T.; Takahashi, M.; Yonemoto, T.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Results of sub-analysis of a phase 2 study on trabectedin treatment for extraskeletal myxoid chondrosarcoma and mesenchymal chondrosarcoma. BMC Cancer 2016, 16, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farago, A.F.; Drapkin, B.J.; Lopez-Vilarino de Ramos, J.A.; Galmarini, C.M.; Núñez, R.; Kahatt, C.; Paz-Ares, L. ATLANTIS: A Phase III study of lurbinectedin/doxorubicin versus topotecan or cyclophosphamide/doxorubicin/vincristine in patients with small-cell lung cancer who have failed one prior platinum-containing line. Future Oncol. 2019, 15, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Poveda, A.; Del Campo, J.M.; Ray-Coquard, I.; Alexandre, J.; Provansal, M.; Guerra Alía, E.M.; Casado, A.; Gonzalez-Martin, A.; Fernández, C.; Rodriguez, I.; et al. Phase II randomized study of PM01183 versus topotecan in patients with platinum-resistant/refractory advanced ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1280–1287. [Google Scholar] [CrossRef]
- Cruz, C.; Llop-Guevara, A.; Garber, J.E.; Arun, B.K.; Pérez Fidalgo, J.A.; Lluch, A.; Telli, M.L.; Fernández, C.; Kahatt, C.; Galmarini, C.M.; et al. Multicenter Phase II Study of Lurbinectedin in BRCA-Mutated and Unselected Metastatic Advanced Breast Cancer and Biomarker Assessment Substudy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 3134–3143. [Google Scholar] [CrossRef]
- Cote, G.M.; Choy, E.; Chen, T.; Marino-Enriquez, A.; Morgan, J.; Merriam, P.; Thornton, K.; Wagner, A.J.; Nathenson, M.J.; Demetri, G.; et al. A phase II multi-strata study of lurbinectedin as a single agent or in combination with conventional chemotherapy in metastatic and/or unresectable sarcomas. Eur. J. Cancer 2020, 126, 21–32. [Google Scholar] [CrossRef]
- Benton, C.B.; Chien, K.S.; Tefferi, A.; Rodriguez, J.; Ravandi, F.; Daver, N.; Jabbour, E.; Jain, N.; Alvarado, Y.; Kwari, M.; et al. Safety and tolerability of lurbinectedin (PM01183) in patients with acute myeloid leukemia and myelodysplastic syndrome. Hematol. Oncol. 2019, 37, 96–102. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Yondelis: EPAR – Product Information; European Medicines Agency: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Hajishengallis, G.; Lambris, J.D. Microbial manipulation of receptor crosstalk in innate immunity. Nat. Rev. Immunol. 2011, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Hume, D.A. Macrophages as APC and the dendritic cell myth. J. Immunol. 2008, 181, 5829–5835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saio, M.; Radoja, S.; Marino, M.; Frey, A.B. Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide. J. Immunol. 2001, 167, 5583–5593. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A. Molecular pathways linking inflammation and cancer. Curr. Mol. Med. 2010, 10, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Pasare, C.; Medzhitov, R. Toll-like receptors: Linking innate and adaptive immunity. Microbes Infect. 2004, 6, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragone, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bae, J.-S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediators Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Belgiovine, C.; D’Incalci, M.; Allavena, P.; Frapolli, R. Tumor-associated macrophages and anti-tumor therapies: Complex links. Cell. Mol. Life Sci. 2016, 73, 2411–2424. [Google Scholar] [CrossRef]
- Través, P.G.; Pimentel-Santillana, M.; Rico, D.; Rodriguez, N.; Miethke, T.; Castrillo, A.; Theodorakis, E.A.; Martín-Sanz, P.; Palladino, M.A.; Boscá, L. Anti-inflammatory actions of acanthoic acid-related diterpenes involve activation of the PI3K p110γ/δ subunits and inhibition of NF-κB. Chem. Biol. 2014, 21, 955–966. [Google Scholar] [CrossRef]
- Pulendran, B.; Artis, D. New paradigms in type 2 immunity. Science 2012, 337, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerhahn, S.; Giraudon, C.; Martínez-Díez, M.; Bueren-Calabuig, J.A.; Galmarini, C.M.; Gago, F.; Egly, J.-M. XPF-dependent DNA breaks and RNA polymerase II arrest induced by antitumor DNA interstrand crosslinking-mimetic alkaloids. Chem. Biol. 2011, 18, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Aune, G.J.; Takagi, K.; Sordet, O.; Guirouilh-Barbat, J.; Antony, S.; Bohr, V.A.; Pommier, Y. Von Hippel-Lindau-coupled and transcription-coupled nucleotide excision repair-dependent degradation of RNA polymerase II in response to trabectedin. Clin. Cancer Res. 2008, 14, 6449–6455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique features of trabectedin mechanism of action. Cancer Chemother. Pharmacol. 2016, 77, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Majety, M.; Runza, V.; Lehmann, C.; Hoves, S.; Ries, C.H. A drug development perspective on targeting tumor-associated myeloid cells. FEBS J. 2018, 285, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allavena, P.; Germano, G.; Belgiovine, C.; D’Incalci, M.; Mantovani, A. Trabectedin: A drug from the sea that strikes tumor-associated macrophages. Oncoimmunology 2013, 2, e24614. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I. Biology of ovarian cancer and trabectedin mechanism of action. Future Oncol. 2013, 9, 11–17. [Google Scholar] [CrossRef]
- Zhang, C.-H.; Xu, G.-L.; Jia, W.-D.; Ge, Y.-S.; Wang, W. Can hepatocellular carcinoma be treated by Yondelis through targeting both tumor cells and tumor-associated macrophages? Hepatogastroenterology 2010, 57, 114–116. [Google Scholar] [PubMed]
- Borgoni, S.; Iannello, A.; Cutrupi, S.; Allavena, P.; D’Incalci, M.; Novelli, F.; Cappello, P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. Oncoimmunology 2018, 7, e1393596. [Google Scholar] [CrossRef]
- Brodowicz, T. Trabectedin in soft tissue sarcomas. Future Oncol. 2014, 10, s1–s5. [Google Scholar] [CrossRef] [PubMed]
- Atmaca, H.; Uzunoglu, S. Anti-angiogenic effects of trabectedin (Yondelis; ET-743) on human breast cancer cells. Eur. Cytokine Netw. 2014, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Céspedes, M.V.; Guillén, M.J.; López-Casas, P.P.; Sarno, F.; Gallardo, A.; Álamo, P.; Cuevas, C.; Hidalgo, M.; Galmarini, C.M.; Allavena, P.; et al. Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis. Model. Mech. 2016, 9, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Guseva, N.V.; Rokhlin, O.W.; Glover, R.A.; Cohen, M.B. TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and kills prostate cancer cells independent of p53 status. Cancer Biol. Ther. 2011, 12, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Qiu, L.; Wu, B.; Shen, H.; Zhu, J.; Zhou, L.; Gu, L.; Di, W. TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Mol. Med. Rep. 2013, 8, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Suzuki, M.; Yamamoto, N.; Matsumoto, A.; Iwase, Y.; Yamasaki, K.; Otagiri, M.; Yumita, N. Glutamine Deprivation Enhances Acetyl-CoA Carboxylase Inhibitor-induced Death of Human Pancreatic Cancer Cells. Anticancer Res. 2018, 38, 6683–6689. [Google Scholar] [CrossRef]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Invest. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 2016, 8, 4265–4277. [Google Scholar] [PubMed]
- Allavena, P.; Signorelli, M.; Chieppa, M.; Erba, E.; Bianchi, G.; Marchesi, F.; Olimpio, C.O.; Bonardi, C.; Garbi, A.; Lissoni, A.; et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005, 65, 2964–2971. [Google Scholar] [CrossRef] [Green Version]
- Abate, A.; Rossini, E.; Bonini, S.A.; Fragni, M.; Cosentini, D.; Tiberio, G.A.M.; Benetti, D.; Hantel, C.; Laganà, M.; Grisanti, S.; et al. Cytotoxic Effect of Trabectedin In Human Adrenocortical Carcinoma Cell Lines and Primary Cells. Cancers 2020, 12, 928. [Google Scholar] [CrossRef] [Green Version]
- Hing, J.; Perez-Ruixo, J.J.; Stuyckens, K.; Soto-Matos, A.; Lopez-Lazaro, L.; Zannikos, P. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of trabectedin (ET-743, Yondelis) induced neutropenia. Clin. Pharmacol. Ther. 2008, 83, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Poggesi, I.; Valenzuela, B.; Ouellet, D.; Gonzalez, M.; Hillewaert, V.; Baruchel, S.; Fox, E.; Perez-Ruixo, J.J. Population pharmacokinetics of trabectedin in adolescent patients with cancer. Cancer Chemother. Pharmacol. 2019, 84, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: A single-arm, open-label, phase 2 basket trial. Lancet. Oncol. 2020, 21, 645–654. [Google Scholar] [CrossRef]
- Hortelano, S.; Alvarez, A.M.; Boscá, L. Nitric oxide induces tyrosine nitration and release of cytochrome c preceding an increase of mitochondrial transmembrane potential in macrophages. FASEB J. 1999, 13, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Merino, R.; Ding, L.; Novack, D.; Korsmeyer, S.; Nunez, G. Developmental regulation of the Bcl-2 protein and susceptibility to cell death in B lymphocytes. EMBO J. 1994, 13, 683–691. [Google Scholar] [CrossRef]
- Banerjee, P.; Zhang, R.; Ivan, C.; Galletti, G.; Clise-Dwyer, K.; Barbaglio, F.; Scarfò, L.; Aracil, M.; Klein, C.; Wierda, W.; et al. Trabectedin Reveals a Strategy of Immunomodulation in Chronic Lymphocytic Leukemia. Cancer Immunol. Res. 2019, 7, 2036–2051. [Google Scholar] [CrossRef]
- del Fresno, C.; García-Rio, F.; Gómez-Piña, V.; Soares-Schanoski, A.; Fernández-Ruíz, I.; Jurado, T.; Kajiji, T.; Shu, C.; Marín, E.; Gutierrez del Arroyo, A.; et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: Demonstration in isolated monocytes from cystic fibrosis patients. J. Immunol. 2009, 182, 6494–6507. [Google Scholar] [CrossRef]
- Traves, P.G.; Pimentel-Santillana, M.; Carrasquero, L.M.; Perez-Sen, R.; Delicado, E.G.; Luque, A.; Izquierdo, M.; Martin-Sanz, P.; Miras-Portugal, M.T.; Bosca, L. Selective impairment of P2Y signaling by prostaglandin E2 in macrophages: Implications for Ca2+-dependent responses. J. Immunol. 2013, 190, 4226–4235. [Google Scholar] [CrossRef] [Green Version]
- Hortelano, S.; Castrillo, A.; Alvarez, A.M.; Bosca, L. Contribution of cyclopentenone prostaglandins to the resolution of inflammation through the potentiation of apoptosis in activated macrophages. J. Immunol. 2000, 165, 6525–6531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuevo-Tapioles, C.; Santacatterina, F.; Stamatakis, K.; Núez de Arenas, C.; Gómez de Cedrón, M.; Formentini, L.; Cuezva, J.M. Coordinate β-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat. Commun. 2020, 11, 3606. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody; Provider; Reference (Dilution) |
|---|
| anti-Bcl-2; Cell Signaling (Rabbit mAb); ref. 2870 (1:1000) |
| anti-caspase 3; Cell Signaling (Rabbit mAb); ref. 9662 (1:1000) |
| anti-cleaved caspase 3; Cell Signaling (Rabbit mAb); ref. 9661 (1:1000) |
| anti-PARP-1; R&D Systems (Goat pAb); ref. AF-600-NA (1:1000) anti-PACC; Cell Signaling (Rabbit mAb); ref. 3661 (1:1000) |
| anti-ACC; Cell Signaling (Rabbit mAb); ref. 3676 (1:1000) |
| anti-RNA polymerase II; Abcam; ref. ab5095 (1:1000) |
| anti-α-tubulin; Cell Signaling (Rabbit mAb); ref. D7F10 (1:500) anti-GAPDH; Invitrogen (Mouse mAb); ref. 39-8600 (1:1000) |
| anti-rabbit IgG; Sigma (pAb); ref. A6154 (1:5000) anti-mouse IgG; Invitrogen (Mouse mAb); ref. A9044 (1:5000) |
| mRNA | Forward Primers Sequences | Reverse Primers Sequences |
|---|---|---|
| CD274 | TGGCATTTGCTGAACGCATTT | TGCAGCCAGGTCTAATTGTTTT |
| PD-CD1LG2 | ATTGCAGCTTCACCAGATAGC | AAAGTTGCATTCCAGGGTCAC |
| TLR-4 | TTTGGACAGTTTCCCACATTGA | AAGCATTCCCACCTTTGTTGG |
| IL10 | CGAGATGCCTTCAGCAGAGT | CGCCTTGATGTCTGGGTCTT |
| SUCNR1 | GGAGACGTGCTCTGCATAAG | AGGTGTTCTCGGAAAGGATACTT |
| HIF1A | GAACGTCGAAAAGAAAAGTCTCG | CCTTATCAAGATGCGAACTCACA |
| FASNCD36 | AAGGACCTGTCTAGGTTTGATGC CTTTGGCTTAATGAGACTGGGAC | TGGCTTCATAGGTGACTTCCA GCAACAAACATCACCACACCA |
| HMGCR | TGATTGACCTTTCCAGAGCAAG | CTAAAATTGCCATTCCACGAGC |
| LDLR | TCTGCAACATGGCTAGAGACT | TCCAAGCATTCGTTGGTCCC |
| PFKFB3 | TTGGCGTCCCCACAAAAGT | AGTTGTAGGAGCTGTACTGCTT |
| SCARB1 | AATAAGCCCATGACCCTGAAGC | GCCCCACATGATCTCACCC |
| TNFA | CCAGAGGGAAGAGTTCCCCAGGG | AGGCTTGTCACTCGGGGTTCGAG |
| GPR132 | TGTTCCAGACGGAAGACAAGG | GCGTAGTAGTACCCGGCAA |
| RPLP0 | CAGGCGTCCTCGTGGAAGTGAC | CCAGGTCGCCCTGTCTTCCCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Povo-Retana, A.; Mojena, M.; Stremtan, A.B.; Fernández-García, V.B.; Gómez-Sáez, A.; Nuevo-Tapioles, C.; Molina-Guijarro, J.M.; Avendaño-Ortiz, J.; Cuezva, J.M.; López-Collazo, E.; et al. Specific Effects of Trabectedin and Lurbinectedin on Human Macrophage Function and Fate—Novel Insights. Cancers 2020, 12, 3060. https://doi.org/10.3390/cancers12103060
Povo-Retana A, Mojena M, Stremtan AB, Fernández-García VB, Gómez-Sáez A, Nuevo-Tapioles C, Molina-Guijarro JM, Avendaño-Ortiz J, Cuezva JM, López-Collazo E, et al. Specific Effects of Trabectedin and Lurbinectedin on Human Macrophage Function and Fate—Novel Insights. Cancers. 2020; 12(10):3060. https://doi.org/10.3390/cancers12103060
Chicago/Turabian StylePovo-Retana, Adrián, Marina Mojena, Adrian B. Stremtan, Victoria B. Fernández-García, Ana Gómez-Sáez, Cristina Nuevo-Tapioles, José M. Molina-Guijarro, José Avendaño-Ortiz, José M. Cuezva, Eduardo López-Collazo, and et al. 2020. "Specific Effects of Trabectedin and Lurbinectedin on Human Macrophage Function and Fate—Novel Insights" Cancers 12, no. 10: 3060. https://doi.org/10.3390/cancers12103060