MDA-9/Syntenin (SDCBP) Is a Critical Regulator of Chemoresistance, Survival and Stemness in Prostate Cancer Stem Cells

 and
and
Abstract
:
1. Introduction
2. Results
2.1. MDA-9 Expression Is Elevated in the Unique Self-Renewing PCSC Subpopulation in Prostate Cancer
2.2. mda-9 Is Co-Expressed in PCSCs with Stem Cell Markers and Stemness-Regulating Genes
2.3. MDA-9 Over-Expression Leads to Expression of a Stem-Like Phenotype
2.4. MDA-9 Activates Downstream Signaling, Which Regulates Self-Renewal in PCSCs
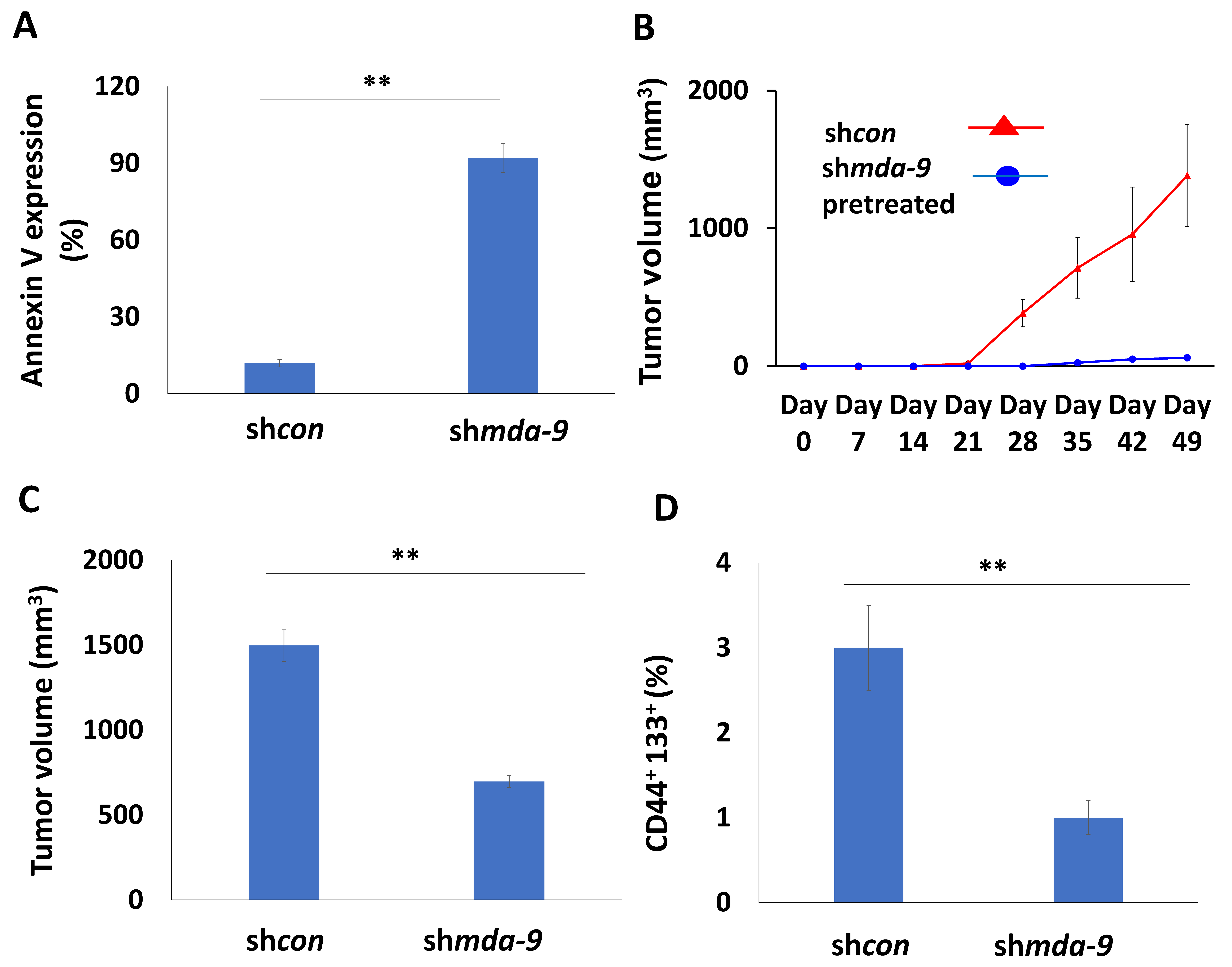
2.5. MDA-9 Maintains PCSC-Mediated Survival and Tumorigenicity
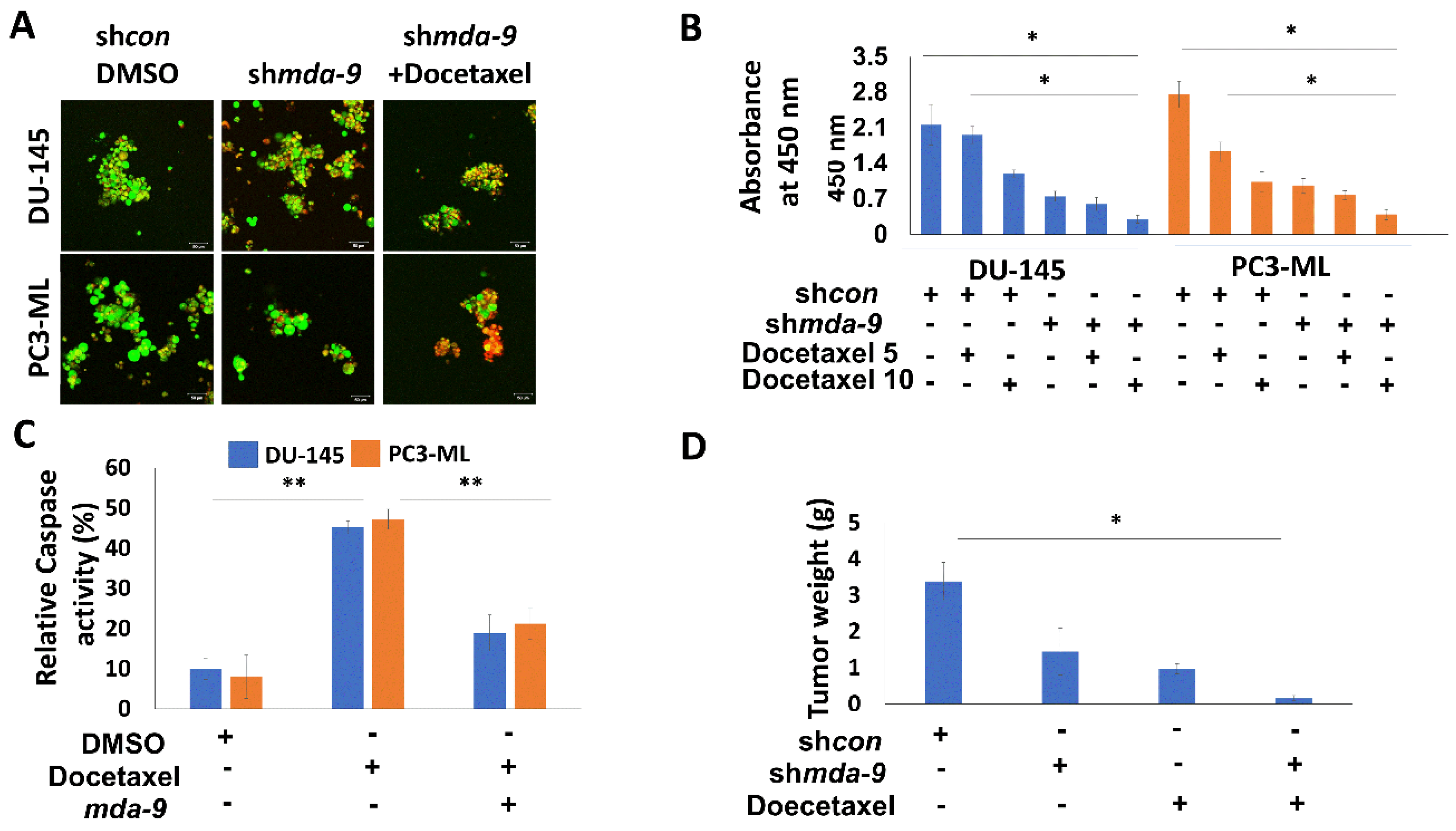
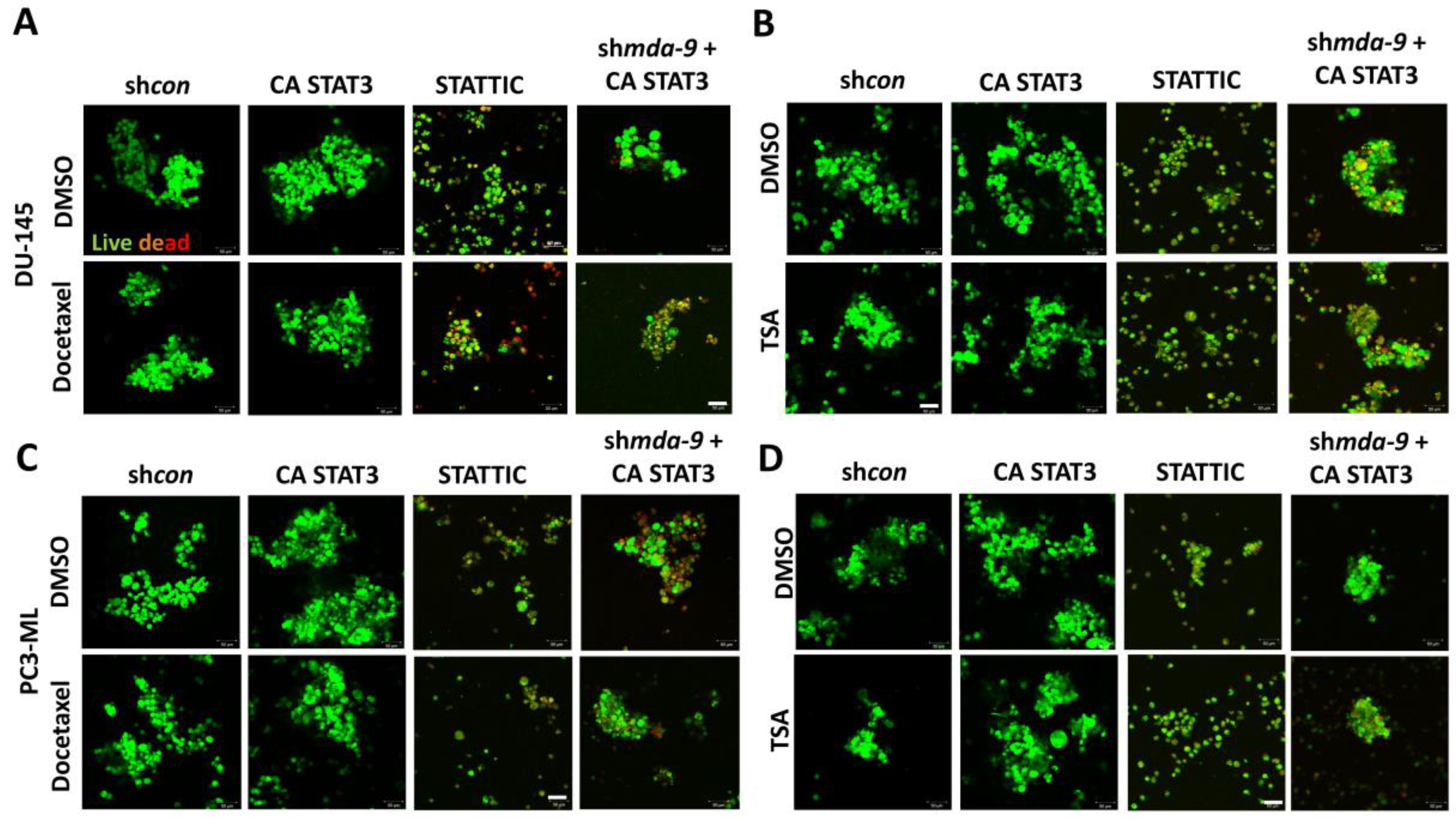
2.6. Suppression of MDA-9 Sensitizes PCSCs to Multiple Chemotherapeutic Drugs via STAT3
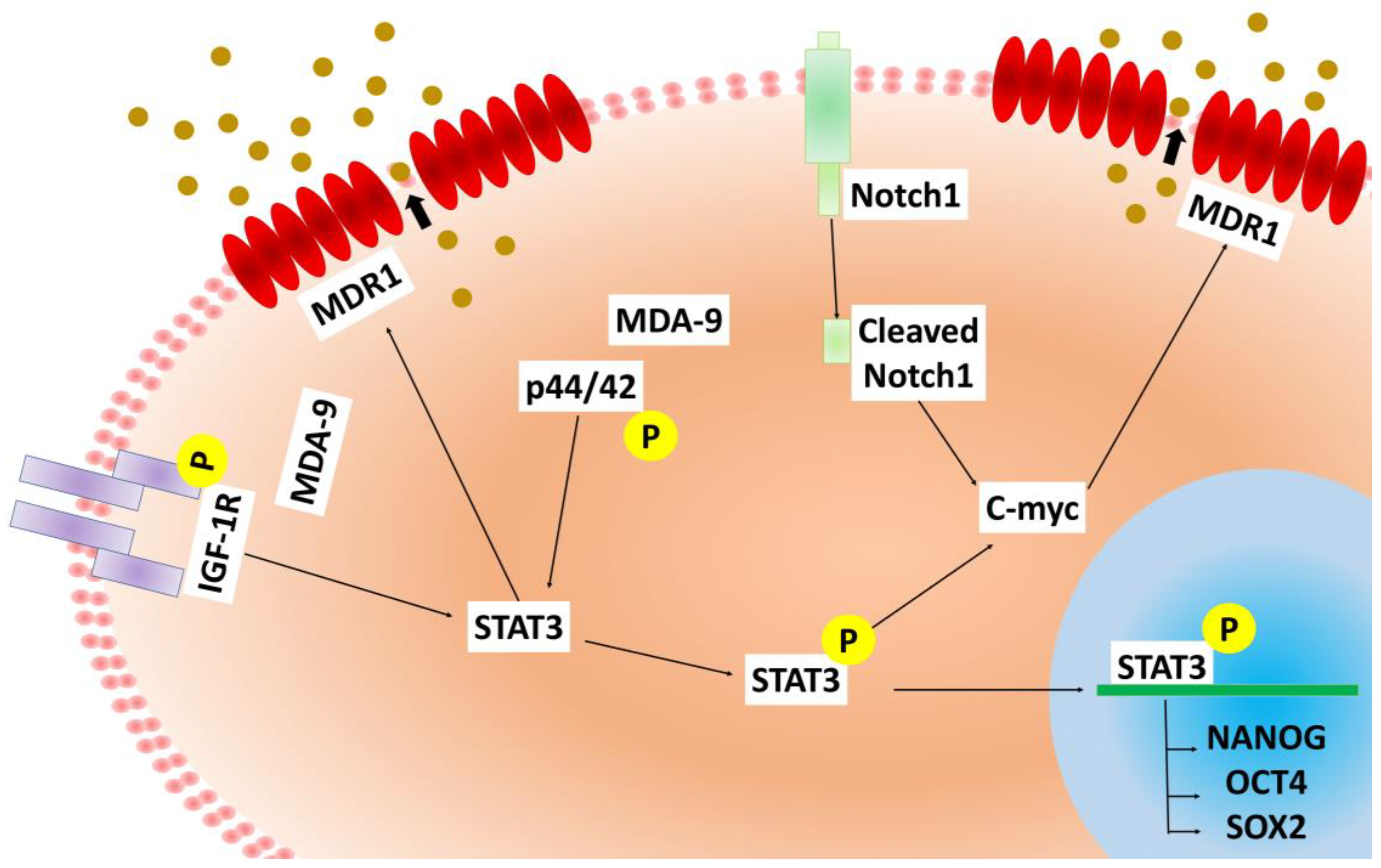
2.7. MDA-9 Mediates PCSC Chemoresistance through the STAT3-MDR1 Axis
2.8. C-myc Regulation by MDA-9 Is Essential for Stem Cell Renewal, Maintenance, Survival and MDR1 Expression
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Tissue Samples
4.2. Isolation and Culture of Putative Human PCSCs and NSCCs
4.3. Isolation and Culture of Primary Prostate Epithelial Stem Cells
4.4. Promoter Reporter Assays
4.5. Reverse Transcription Polymerase Chain Reaction
4.6. Western Blotting
4.7. Immunofluorescent Staining and Confocal Microscopy
4.8. Live Cell Imaging
4.9. Live/Dead Cell Assay
4.10. Cell Proliferation
4.11. Flow Cytometry Sorting and Analysis
4.12. Intracellular Flow Cytometry
4.13. Tumorigenicity Studies
4.14. Peptide Blocking Studies
4.15. shRNA Knockdown
4.16. Chemotherapeutic Studies
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taitt, H.E. Global Trends and Prostate Cancer: A Review of Incidence, Detection, and Mortality as Influenced by Race, Ethnicity, and Geographic Location. Am. J. Mens. Health 2018, 12, 1807–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Lang, S.H.; Frame, F.M.; Collins, A.T. Prostate cancer stem cells. J. Pathol. 2009, 217, 299–306. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 393–401. [Google Scholar] [CrossRef]
- Raff, M. Adult stem cell plasticity: Fact or artifact? Annu. Rev. Cell Dev. Biol. 2003, 19, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.G.; Patrawala, L.; Calhoun, T.; Bhatia, B.; Choy, G.; Schneider-Broussard, R.; Jeter, C. Prostate cancer stem/progenitor cells: Identification, characterization, and implications. Mol. Carcinog. 2007, 46, 1–14. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.T.; Habib, F.K.; Maitland, N.J.; Neal, D.E. Identification and isolation of human prostate epithelial stem cells based on α2β1-integrin expression. J. Cell Sci. 2001, 114, 3865–3872. [Google Scholar]
- Rybak, A.P.; Bristow, R.G.; Kapoor, A. Prostate cancer stem cells: Deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget 2015, 6, 1900–1919. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Gao, W.Q. The Notch pathway in prostate development and cancer. Differentiation 2008, 76, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, E.Y.; Miller, K.; Nelson, J.; Gleave, M.; Fizazi, K.; Moul, J.W.; Nathan, F.E.; Higano, C.S. Detection of previously unidentified metastatic disease as a leading cause of screening failure in a phase III trial of zibotentan versus placebo in patients with nonmetastatic, castration resistant prostate cancer. J. Urol. 2012, 188, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellawell, G.O.; Turner, G.D.; Davies, D.R.; Poulsom, R.; Brewster, S.F.; Macaulay, V.M. Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res. 2002, 62, 2942–2950. [Google Scholar]
- Zhao, Y.; Bao, Q.; Renner, A.; Camaj, P.; Eichhorn, M.; Ischenko, I.; Angele, M.; Kleespies, A.; Jauch, K.W.; Bruns, C. Cancer stem cells and angiogenesis. Int. J. Dev. Biol. 2011, 55, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Birnie, R.; Bryce, S.D.; Roome, C.; Dussupt, V.; Droop, A.; Lang, S.H.; Berry, P.A.; Hyde, C.F.; Lewis, J.L.; Stower, M.J.; et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol. 2008, 9, R83. [Google Scholar] [CrossRef] [Green Version]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef]
- Gu, G.; Yuan, J.; Wills, M.; Kasper, S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007, 67, 4807–4815. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.; Motiani, K.; Giridhar, P.V.; Kasper, S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp. Biol. Med. 2013, 238, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Moltzahn, F.; Thalmann, G.N. Cancer stem cells in prostate cancer. Transl. Androl. Urol. 2013, 2, 242–253. [Google Scholar] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse-Gehling, P.; Fargeas, C.A.; Dittfeld, C.; Garbe, Y.; Alison, M.R.; Corbeil, D.; Kunz-Schughart, L.A. CD133 as a biomarker for putative cancer stem cells in solid tumours: Limitations, problems and challenges. J. Pathol. 2013, 229, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Dhir, R.; Ni, Z.; Lou, W.; DeMiguel, F.; Grandis, J.R.; Gao, A.C. Stat3 activation in prostatic carcinomas. Prostate 2002, 51, 241–246. [Google Scholar] [CrossRef]
- Ni, Z.; Lou, W.; Leman, E.S.; Gao, A.C. Inhibition of constitutively activated Stat3 signaling pathway suppresses growth of prostate cancer cells. Cancer Res. 2000, 60, 1225–1228. [Google Scholar]
- Horinaga, M.; Okita, H.; Nakashima, J.; Kanao, K.; Sakamoto, M.; Murai, M. Clinical and pathologic significance of activation of signal transducer and activator of transcription 3 in prostate cancer. Urology 2005, 66, 671–675. [Google Scholar] [CrossRef]
- Liu, X.; He, Z.; Li, C.H.; Huang, G.; Ding, C.; Liu, H. Correlation analysis of JAK-STAT pathway components on prognosis of patients with prostate cancer. Pathol. Oncol. Res. 2012, 18, 17–23. [Google Scholar] [CrossRef]
- Tam, L.; McGlynn, L.M.; Traynor, P.; Mukherjee, R.; Bartlett, J.M.; Edwards, J. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br. J. Cancer 2007, 97, 378–383. [Google Scholar] [CrossRef]
- Kroon, P.; Berry, P.A.; Stower, M.J.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettiar, S.; Li, C.; Li, P.K.; et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013, 73, 5288–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Garcia, A.J.; Wu, M.; Lawson, D.A.; Witte, O.N.; Wu, H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1480–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Oyan, A.M.; Liu, R.; Hua, Y.; Zhang, J.; Hovland, R.; Popa, M.; Liu, X.; Brokstad, K.A.; Simon, R.; et al. Generation of prostate tumor-initiating cells is associated with elevation of reactive oxygen species and IL-6/STAT3 signaling. Cancer Res. 2013, 73, 7090–7100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Frame, F.M.; Pellacani, D.; Collins, A.T.; Simms, M.S.; Mann, V.M.; Jones, G.D.; Meuth, M.; Bristow, R.G.; Maitland, N.J. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br. J. Cancer 2013, 109, 3023–3033. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer cell 2012, 22, 373–388. [Google Scholar] [CrossRef] [Green Version]
- Moitra, K. Overcoming Multidrug Resistance in Cancer Stem Cells. Biomed. Res. Int. 2015, 2015, 635745. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Noonan, K.E.; Beck, C.; Holzmayer, T.A.; Chin, J.E.; Wunder, J.S.; Andrulis, I.L.; Gazdar, A.F.; Willman, C.L.; Griffith, B.; Von Hoff, D.D.; et al. Quantitative analysis of MDR1 (multidrug resistance) gene expression in human tumors by polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1990, 87, 7160–7164. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Castellón, E.A.; Valenzuela, R.; Lillo, J.; Castillo, V.; Contreras, H.R.; Gallegos, I.; Mercado, A.; Huidobro, C. Molecular signature of cancer stem cells isolated from prostate carcinoma and expression of stem markers in different Gleason grades and metastasis. Biol. Res. 2012, 45, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kegelman, T.P.; Wu, B.; Das, S.K.; Talukdar, S.; Beckta, J.M.; Hu, B.; Emdad, L.; Valerie, K.; Sarkar, D.; Furnari, F.B.; et al. Inhibition of radiation-induced glioblastoma invasion by genetic and pharmacological targeting of MDA-9/Syntenin. Proc. Natl. Acad. Sci. USA 2017, 114, 370–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, D.; Boukerche, H.; Su, Z.Z.; Fisher, P.B. MDA-9/Syntenin: More than just a simple adapter protein when it comes to cancer metastasis. Cancer Res. 2008, 68, 3087–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. Evolving Strategies for Therapeutically Targeting Cancer Stem Cells. Adv. Cancer Res. 2016, 131, 159–191. [Google Scholar] [PubMed]
- Talukdar, S.; Das, S.K.; Pradhan, A.K.; Emdad, L.; Shen, X.N.; Windle, J.J.; Sarkar, D.; Fisher, P.B. Novel function of MDA-9/Syntenin (SDCBP) as a regulator of survival and stemness in glioma stem cells. Oncotarget 2016, 7, 54102–54119. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Pradhan, A.K.; Bhoopathi, P.; Shen, X.N.; August, L.A.; Windle, J.J.; Sarkar, D.; Furnari, F.B.; Cavenee, W.K.; Das, S.K.; et al. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc. Natl. Acad. Sci. USA 2018, 11, 5768–5773. [Google Scholar] [CrossRef] [Green Version]
- Das, S.K.; Bhutia, S.; Kegelman, T.P.; Peachy, L.; Oyesanya, R.A.; Dasgupta, S.; Sokhi, U.K.; Azab, B.; Dash, R.; Quinn, B.A.; et al. MDA-9/syntenin: A positive gatekeeper of melanoma metastasis. Front. Biosci. Landmark Ed. 2012, 17, 1–15. [Google Scholar] [CrossRef]
- Qian, X.L.; Li, Y.Q.; Yu, B.; Gu, F.; Liu, F.F.; Li, W.D.; Zhang, X.M.; Fu, L. Syndecan binding protein (SDCBP) is overexpressed in estrogen receptor negative breast cancers, and is a potential promoter for tumor proliferation. PLoS ONE 2013, 8, e60046. [Google Scholar] [CrossRef] [Green Version]
- Abdulghani, J.; Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Zellweger, T.; Alanen, K.; Mirtti, T.; et al. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 2008, 172, 1717–1728. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Menacho, I.; van der Sluis, T.; Hollema, H.; Gimm, O.; Buys, C.H.; Magee, A.I.; Isacke, C.M.; Hofstra, R.M.; Eggen, B.J. Ras/ERK1/2-mediated STAT3 Ser727 phosphorylation by familial medullary thyroid carcinoma-associated RET mutants induces full activation of STAT3 and is required for c-fos promoter activation, cell mitogenicity, and transformation. J. Biol. Chem. 2007, 282, 6415–6424. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zong, C.S.; Hermanto, U.; Lopez-Bergami, P.; Ronai, Z.; Wang, L.H. RACK1 recruits STAT3 specifically to insulin and insulin-like growth factor 1 receptors for activation, which is important for regulating anchorage-independent growth. Mol. Cell. Biol. 2006, 26, 413–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkach, M.; Rosemblit, C.; Rivas, M.A.; Proietti, C.J.; Diaz Flaque, M.C.; Mercogliano, M.F.; Beguelin, W.; Maronna, E.; Guzman, P.; Gercovich, F.G.; et al. p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr. Relat. Cancer 2013, 20, 197–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiuchi, N.; Nakajima, K.; Ichiba, M.; Fukada, T.; Narimatsu, M.; Mizuno, K.; Hibi, M.; Hirano, T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J. Exp. Med. 1999, 189, 63–73. [Google Scholar] [CrossRef]
- Barre, B.; Vigneron, A.; Coqueret, O. The STAT3 transcription factor is a target for the Myc and riboblastoma proteins on the Cdc25A promoter. J. Biol. Chem. 2005, 280, 15673–15681. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P.; et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.J.; Wilson, A.; Trumpp, A. More than just proliferation: Myc function in stem cells. Trends Cell Biol. 2005, 15, 128–137. [Google Scholar] [CrossRef]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef] [Green Version]
- Yun, E.J.; Zhou, J.; Lin, C.J.; Hernandez, E.; Fazli, L.; Gleave, M.; Hsieh, J.T. Targeting Cancer Stem Cells in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 670–679. [Google Scholar] [CrossRef] [Green Version]
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol. Cell. Biol. 2007, 27, 4444–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Jiang, M.; Lu, Y.; Chen, H.; Sun, J.; Wu, S.; Ku, W.Y.; Nakagawa, H.; Kita, Y.; Natsugoe, S.; et al. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 2013, 12, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukerche, H.; Su, Z.Z.; Emdad, L.; Baril, P.; Balme, B.; Thomas, L.; Randolph, A.; Valerie, K.; Sarkar, D.; Fisher, P.B. MDA-9/Syntenin: A positive regulator of melanoma metastasis. Cancer Res. 2005, 65, 10901–10911. [Google Scholar] [CrossRef] [Green Version]
- Kegelman, T.P.; Das, S.K.; Hu, B.; Bacolod, M.D.; Fuller, C.E.; Menezes, M.E.; Emdad, L.; Dasgupta, S.; Baldwin, A.S.; Bruce, J.N.; et al. MDA-9/syntenin is a key regulator of glioma pathogenesis. Neuro Oncol. 2014, 16, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Das, S.K.; Bhutia, S.K.; Sokhi, U.K.; Azab, B.; Su, Z.Z.; Boukerche, H.; Anwar, T.; Moen, E.L.; Chatterjee, D.; Pellecchia, M.; et al. Raf kinase inhibitor RKIP inhibits MDA-9/syntenin-mediated metastasis in melanoma. Cancer Res. 2012, 72, 6217–6226. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tang, D.G. Prostate cancer stem cells and their potential roles in metastasis. J. Surg. Oncol. 2011, 103, 558–562. [Google Scholar] [CrossRef]
- Das, S.K.; Pradhan, A.K.; Bhoopathi, P.; Talukdar, S.; Shen, X.N.; Sarkar, D.; Emdad, L.; Fisher, P.B. The MDA-9/Syntenin/IGF1R/STAT3 Axis Directs Prostate Cancer Invasion. Cancer Res. 2018, 78, 2852–2863. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.W.; Fojo, A.; Chin, J.E.; Roninson, I.B.; Richert, N.; Pastan, I.; Gottesman, M.M. Human multidrug-resistant cell lines: Increased mdr1 expression can precede gene amplification. Science 1986, 232, 643–645. [Google Scholar] [CrossRef]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Parekh, H.; Wiesen, K.; Simpkins, H. Acquisition of taxol resistance via P-glycoprotein- and non-P-glycoprotein-mediated mechanisms in human ovarian carcinoma cells. Biochem. Pharmacol. 1997, 53, 461–470. [Google Scholar] [CrossRef]
- Wang, Y.C.; Juric, D.; Francisco, B.; Yu, R.X.; Duran, G.E.; Chen, K.G.; Chen, X.; Sikic, B.I. Regional activation of chromosomal arm 7q with and without gene amplification in taxane-selected human ovarian cancer cell lines. Genes Chromosomes Cancer 2006, 45, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’Leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Zarrintaj, P.; Mostafapoor, F.; Milan, P.B.; Saeb, M.R. Theranostic Platforms Proposed for Cancerous Stem Cells: A Review. Curr. Stem. Cell Res. Ther. 2019, 14, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Miki, J.; Rhim, J.S. Prostate cell cultures as in vitro models for the study of normal stem cells and cancer stem cells. Prostate Cancer Prostatic Dis. 2008, 11, 32–39. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | DU-145 | ARCaP-M | PC3-ML | |||
|---|---|---|---|---|---|---|
| Genes | Non-Stem Cancer Cell | Cancer Stem Cell | Non-Stem Cancer Cell | Cancer Stem Cell | Non-Stem Cancer Cell | Cancer Stem Cell |
| mda-9 | 1 ± 0.05 | 3.48 ± 0.10 | 1 ± 0.20 | 8.30 ± 0.07 | 1 ± 0.07 | 5.67 ± 0.25 |
| Stemness genes | ||||||
| Nanog | 1 ± 0.03 | 12.50 ± 0.02 | 1 ± 0.02 | 8.64 ± 0.11 | 1 ± 0.14 | 13.88 ± 2.0 |
| Sox2 | 1 ± 0.20 | 2.94 ± 0.04 | 1 ± 0.32 | 2.49 ± 0.06 | 1 ± 0.19 | 4.48 ± 1.93 |
| Oct4 | 1 ± 0.07 | 20.00 ± 0.01 | 1 ± 0.11 | 5.51 ± 0.01 | 1 ± 0.22 | 13.01 ± 0.45 |
| c-myc | 1 ± 0.15 | 2.44 ± 0.06 | 1 ± 0.09 | 2.39 ± 0.05 | 1 ± 0.03 | 3.93 ± 1.7 |
| Cell Line | CSC Population (%) shcon | CSC Population (%) shmda-9 |
|---|---|---|
| DU-145 | 40.3 ± 5.2 | 11 ± 5.5 |
| PC3-ML | 15.4 ± 3.5 | 3.5 ± 1.2 |
| ARCaP-M | 29.8 ± 3.7 | 9.9 ± 2.4 |
| Genes | DU-145 | ARCaP-M | PC3-ML | |||
|---|---|---|---|---|---|---|
| Shcon | Shmda-9 | Shcon | Shmda-9 | Shcon | Shmda-9 | |
| mda-9 | 1 ± 0.07 | 0.2 ± 0.15 | 1 ± 0.05 | 0.3 ± 0.07 | 1 ± 0.06 | 0.1 ± 0.12 |
| Stemness genes | ||||||
| Nanog | 1 ± 0.02 | 0.55 ± 0.04 | 1 ± 0.03 | 0.10 ± 0.04 | 1 ± 0.02 | 0.15 ± 0.02 |
| Sox2 | 1 ± 0.06 | 0.32 ± 0.00 | 1 ± 0.01 | 0.26 ± 0.03 | 1 ± 0.08 | 0.10 ± 0.05 |
| Oct4 | 1 ± 0.11 | 0.14 ± 0.04 | 1 ± 0.25 | 0.10 ± 0.01 | 1 ± 0.05 | 0.03 ± 0.00 |
| c-myc | 1 ± 0.01 | 0.17 ± 0.03 | 1 ± 0.02 | 0.05 ± 0.01 | 1 ± 0.02 | 0.09 ± 0.00 |
| % Expression | DU-145 shcon | DU-145 shmda-9 | ARCaP-M shcon | ARCaP-M shmda-9 | PC3-ML shcon | PC3-ML shmda-9 |
|---|---|---|---|---|---|---|
| Protein | ||||||
| P-STAT3 | 62.3 ± 9.8 | 15.4 ± 4.1 | 6.5 ± 0.6 | 2.2 ± 0.3 | 41.3 ± 7.6 | 29.6 ± 3.1 |
| STAT3 | 85.1 ± 8.6 | 87.4 ± 2.7 | 24.6 ± 1.9 | 25.9 ± 5.2 | 62.7 ± 7.9 | 65.7 ± 4.6 |
| P-SRC | 68.2 ± 6.5 | 11.5 ± 5.3 | 37.9 ± 5.6 | 12.6 ± 3.8 | 53.9 ± 4.2 | 37.1 ± 2.7 |
| SRC | 77.4 ± 5.1 | 75.2 ± 4.5 | 39.4 ± 5.8 | 34.1 ± 3.5 | 69.9 ± 6.7 | 63.3 ± 5.1 |
| P-p44/42 | 18.8 ± 4.4 | 1.4 ± 0.07 | 21.2 ± 4.7 | 6.4 ± 0.6 | 6.4 ± 1.5 | 1.7 ± 0.2 |
| p44/42 | 21.4 ± 2.9 | 4.8 ± 2.9 | 21.6 ± 9.5 | 11.9 ± 2.9 | 16.2 ± 2.4 | 6.5 ± 1.2 |
| P-IGF1R | 20.2 ± 5.3 | 9.7 ± 2.5 | 18.5 ± 2.9 | 10.5 ± 3.1 | 7.2 ± 1.1 | 1.8 ± 0.4 |
| IGF1R | 35.7 ± 4.9 | 36.9 ± 7.2 | 34.8 ± 7.4 | 36.2 ± 4.4 | 25.6 ± 5.7 | 27.4 ± 3.2 |
| NOTCH1 | 94.3 ± 4.7 | 31.5 ± 8.5 | 49.7 ± 9.1 | 36.2 ± 2.6 | 74.7 ± 8.8 | 36.4 ± 4.6 |
| DLL1 | 41.8 ± 8.2 | 13.7 ± 2.6 | 44.5 ± 12.6 | 32.9 ± 3.3 | 49.4 ± 6.9 | 27.6 ± 2.6 |
| Numb | 22.3 ± 4.8 | 44.3 ± 5.9 | 0.5 ± 0.1 | 7.3 ± 1.7 | 9.81 ± 2.4 | 19.5 ± 3.8 |
| C-Myc | 53.8 ± 2.2 | 25.4 ± 3.5 | 60.6 ± 9.6 | 39.6 ± 7.6 | 53.0 ± 3.1 | 37.8 ± 1.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talukdar, S.; Das, S.K.; Pradhan, A.K.; Emdad, L.; Windle, J.J.; Sarkar, D.; Fisher, P.B. MDA-9/Syntenin (SDCBP) Is a Critical Regulator of Chemoresistance, Survival and Stemness in Prostate Cancer Stem Cells. Cancers 2020, 12, 53. https://doi.org/10.3390/cancers12010053
Talukdar S, Das SK, Pradhan AK, Emdad L, Windle JJ, Sarkar D, Fisher PB. MDA-9/Syntenin (SDCBP) Is a Critical Regulator of Chemoresistance, Survival and Stemness in Prostate Cancer Stem Cells. Cancers. 2020; 12(1):53. https://doi.org/10.3390/cancers12010053
Chicago/Turabian StyleTalukdar, Sarmistha, Swadesh K. Das, Anjan K. Pradhan, Luni Emdad, Jolene J. Windle, Devanand Sarkar, and Paul B. Fisher. 2020. "MDA-9/Syntenin (SDCBP) Is a Critical Regulator of Chemoresistance, Survival and Stemness in Prostate Cancer Stem Cells" Cancers 12, no. 1: 53. https://doi.org/10.3390/cancers12010053