Targeting Pyruvate Kinase M2 and Lactate Dehydrogenase A Is an Effective Combination Strategy for the Treatment of Pancreatic Cancer

 ,
, 
Abstract
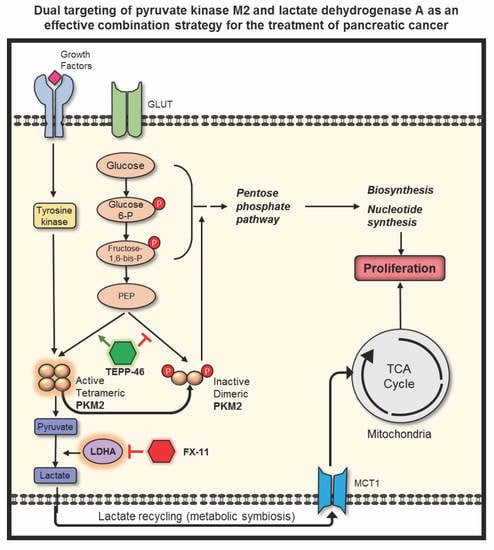
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
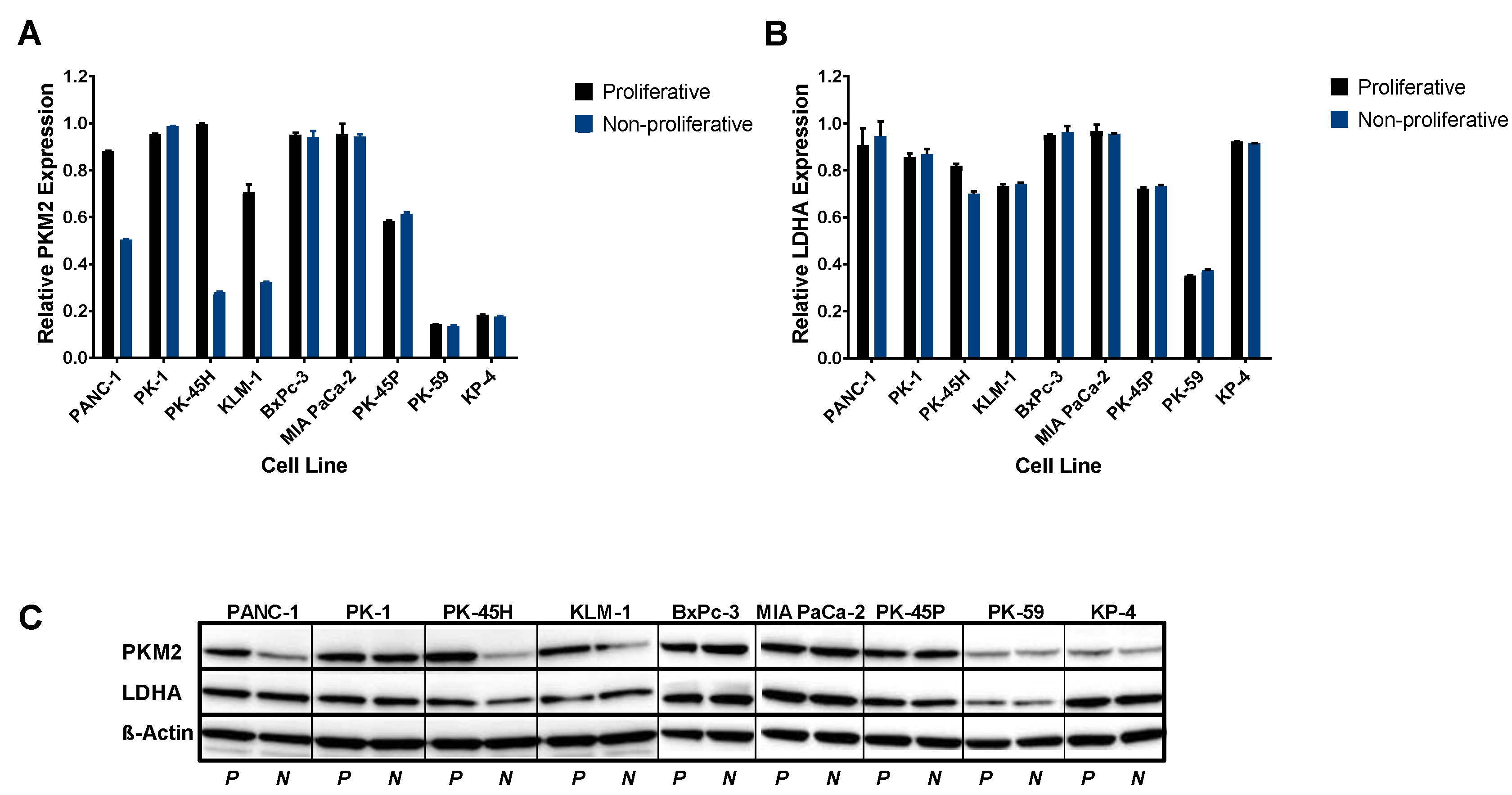
2.1. Both PKM2 and LDHA are Expressed by Pancreatic Cancer Cells
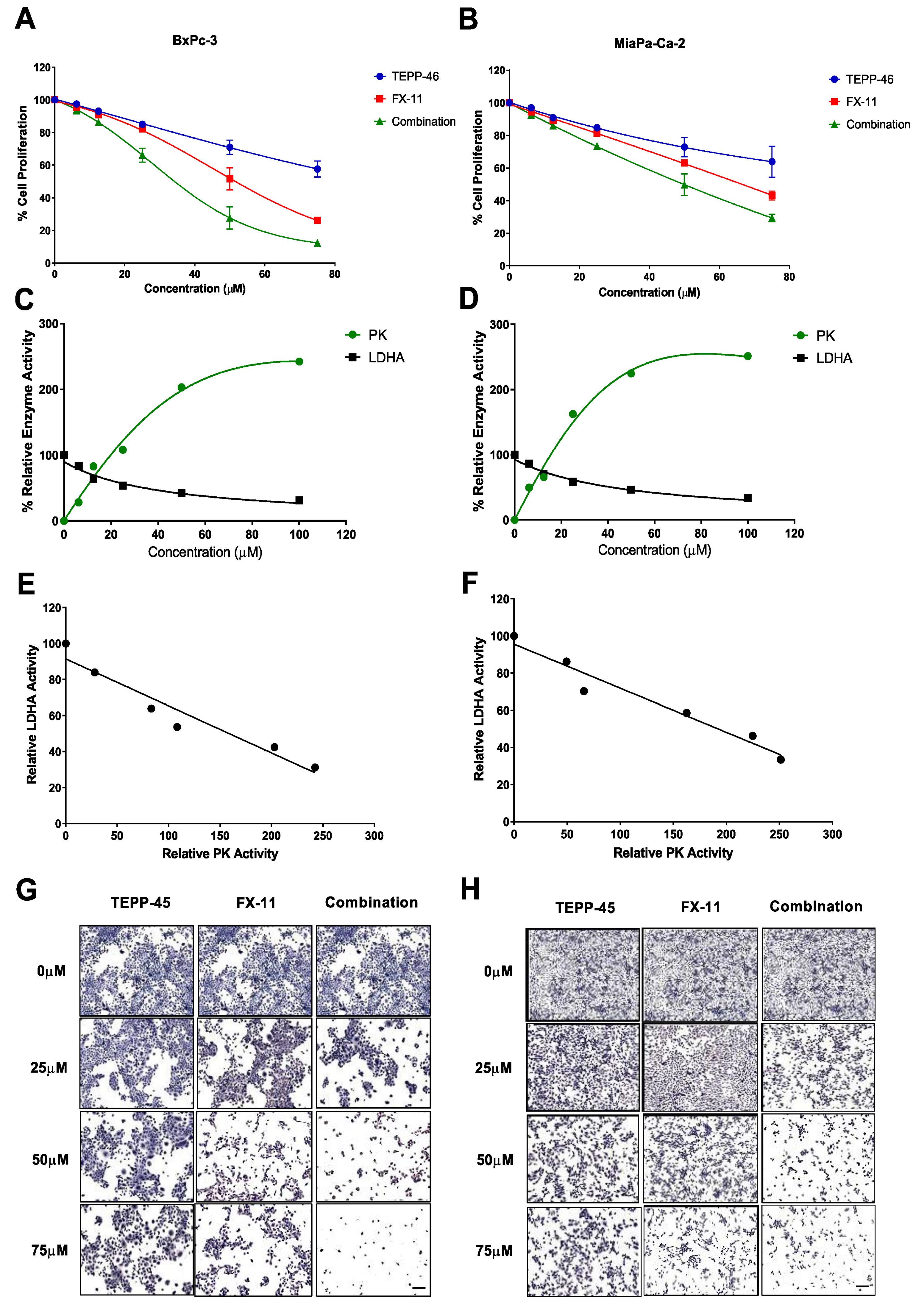
2.2. Synergistic Inhibition of Pancreatic Cancer Cell Proliferation with the Combined Treatment
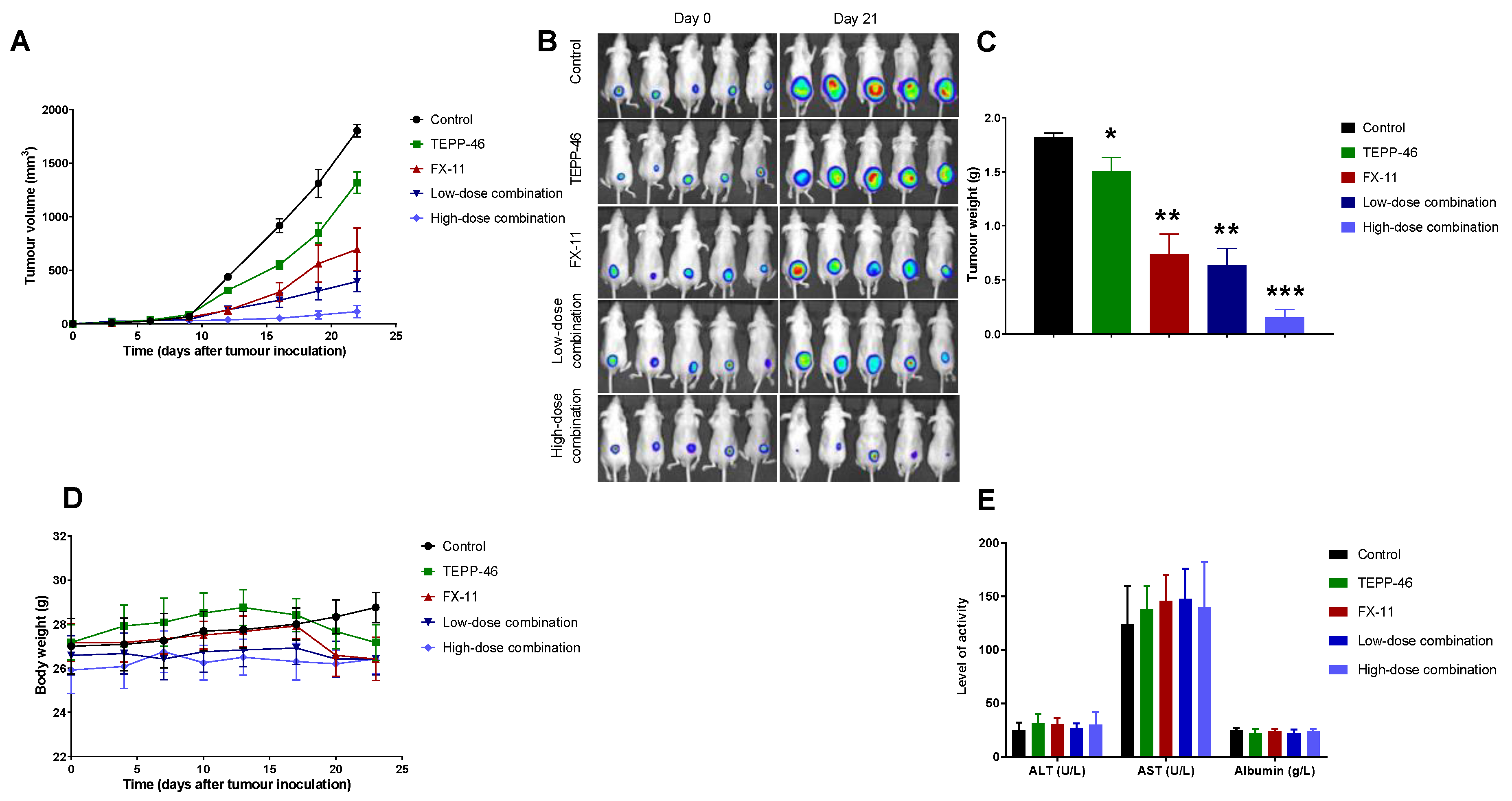
2.3. Combination Therapy Significantly Attenuated Tumor Growth in the Subcutaneous Tumor Model
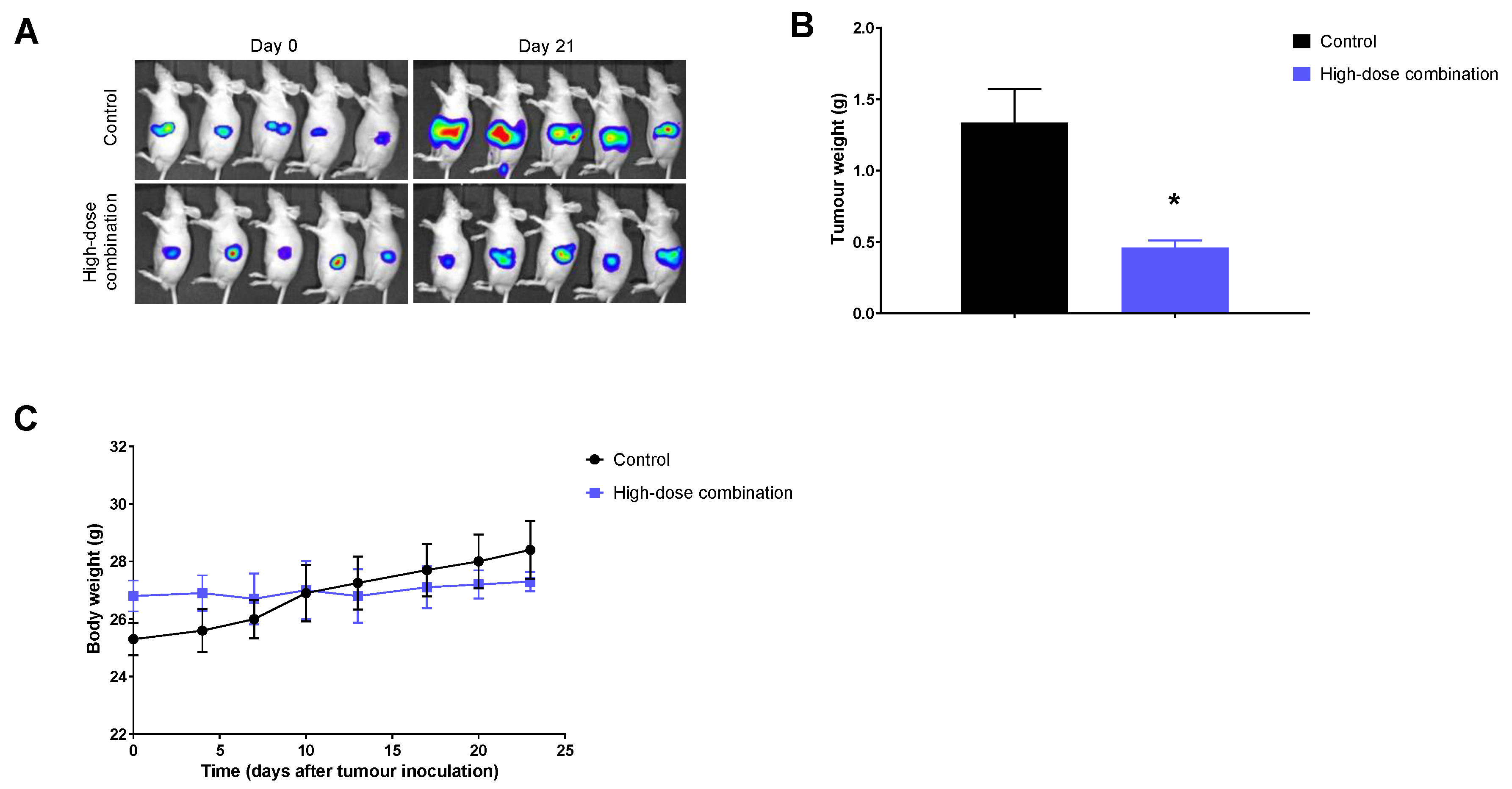
2.4. Significant Therapeutic Efficacy with the Combination Therapy in the Orthotopic Tumor Model
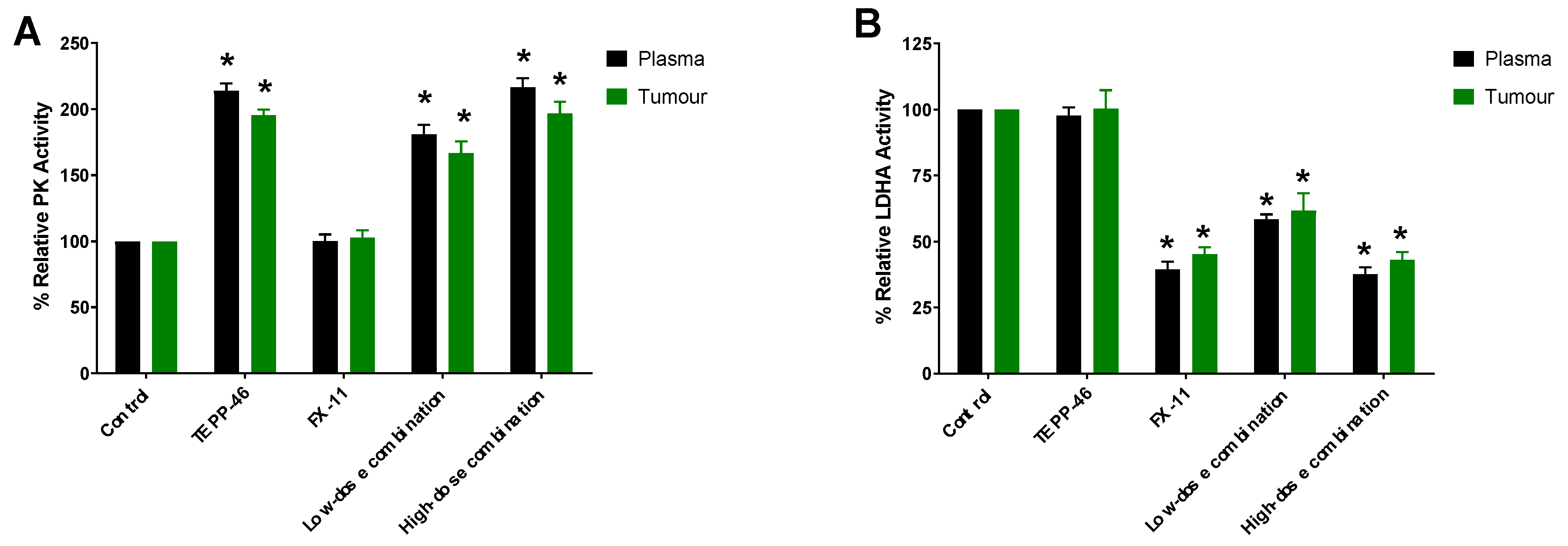
2.5. PK and LDHA Enzyme Activity in Plasma and Tumor Lysates
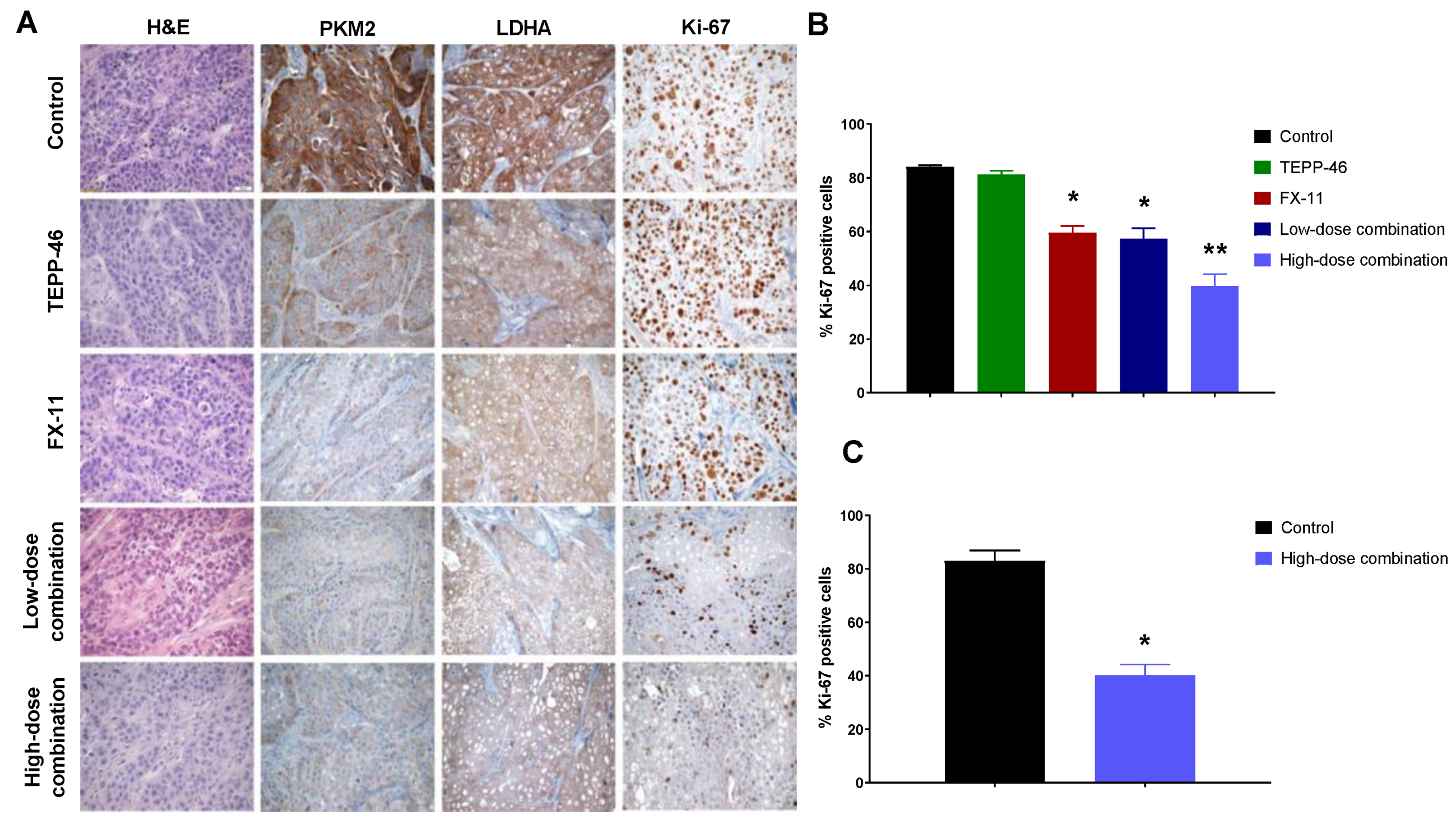
2.6. Reduced PKM2 Detection, LDHA Expression, and Significantly Decreased Proliferation in Tumor Tissues
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Chemicals
4.2. Expression of PKM2 and LDHA in Pancreatic Cancer Cells
4.3. Evaluation of Cell Proliferation Rate
4.4. In Vitro PK and LDHA Enzyme Activity
4.5. Tumor Models and Assessment of In Vivo Efficacy and Toxicity
4.6. Orthotopic Tumor Model
4.7. PK and LDHA Activity in Plasma and Tumor Lysates
4.8. Immunohistochemistry
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.-L.; Myklebust, T.A.; Tervonen, H.; Thursfield, V.; Ransom, D.; Shack, L.; et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 2019. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.; Lee, H.; Park, J.; Kim, S.-H.; Park, J. Targeting Cancer Metabolism—Revisiting the Warburg Effects. Toxicol. Res. 2016, 32, 177–193. [Google Scholar] [CrossRef]
- Yokoyama, M.; Tanuma, N.; Shibuya, R.; Shiroki, T.; Abue, M.; Yamamoto, K.; Miura, K.; Yamaguchi, K.; Sato, I.; Tamai, K.; et al. Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species. Int. J. Oncol. 2018, 52, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Dombrauckas, J.D.; Santarsiero, B.D.; Mesecar, A.D. Structural Basis for Tumor Pyruvate Kinase M2 Allosteric Regulation and Catalysis. Biochemistry 2005, 44, 9417–9429. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Qiao, J.; Yang, J.J.; Liu, Z.-R. Pyruvate Kinase M2 in Blood Circulation Facilitates Tumor Growth by Promoting Angiogenesis. J. Biol. Chem. 2014, 289, 25812–25821. [Google Scholar] [CrossRef]
- Dhar, D.K.; Olde Damink, S.W.M.; Brindley, J.H.; Godfrey, A.; Chapman, M.H.; Sandanayake, N.S.; Andreola, F.; Mazurek, S.; Hasan, T.; Malago, M.; et al. Pyruvate kinase M2 is a novel diagnostic marker and predicts tumour progression in human biliary tract cancer. Cancer 2013, 119, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.-K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-K.; Boxer, M.B.; Vander Heiden, M.G.; Shen, M.; Skoumbourdis, A.P.; Southall, N.; Veith, H.; Leister, W.; Austin, C.P.; Park, H.W.; et al. Evaluation of Thieno[3,2-b]pyrrole[3,2-d]pyridazinones as Activators of the Tumor Cell Specific M2 Isoform of Pyruvate Kinase. Bioorg. Med. Chem. Lett. 2010, 20, 3387–3393. [Google Scholar] [CrossRef] [PubMed]
- Boxer, M.B.; Jiang, J.-K.; Vander Heiden, M.G.; Shen, M.; Skoumbourdis, A.P.; Southall, N.; Veith, H.; Leister, W.; Austin, C.P.; Park, H.W.; et al. Evaluation of Substituted N,N′-Diarylsulfonamides as Activators of the Tumor Cell Specific M2 Isoform of Pyruvate Kinase. J. Med. Chem. 2010, 53, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Tee, S.S.; Park, J.M.; Hurd, R.E.; Brimacombe, K.R.; Boxer, M.B.; Massoud, T.F.; Rutt, B.K.; Spielman, D.M. PKM2 activation sensitizes cancer cells to growth inhibition by 2-deoxy-D-glucose. Oncotarget 2017, 8, 90959–90968. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Dyck, L.; Zasłona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, 1300. [Google Scholar] [CrossRef]
- Xue, J.-J.; Chen, Q.-Y.; Kong, M.-Y.; Zhu, C.-Y.; Gen, Z.-R.; Wang, Z.-L. Synthesis, cytotoxicity for mimics of catalase: Inhibitors of lactate dehydrogenase and hypoxia inducible factor. Eur. J. Med. Chem. 2014, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Rajeshkumar, N.V.; Maitra, A.; Dang, C.V. Conceptual Framework for Cutting the Pancreatic Cancer Fuel Supply. Clin. Cancer Res. 2012, 18, 4285–4290. [Google Scholar] [CrossRef]
- Zhao, D.; Xiong, Y.; Lei, Q.-Y.; Guan, K.-L. LDH-A Acetylation: Implication in Cancer. Oncotarget 2013, 4, 802–803. [Google Scholar] [CrossRef]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med. Chem. 2014, 6, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, W.; Xie, Z.; Shao, Z.; Xie, H.; Qin, G.; Zhao, N. Prognostic relevance of lactate dehydrogenase in advanced pancreatic ductal adenocarcinoma patients. BMC Cancer 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.L.; Liu, J.J.; Dai, Y.H.; Sun, X.G.; Xiong, X.P.; Huang, G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012, 279, 3898–3910. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Deck, L.M.; Royer, R.E.; Chamblee, B.B.; Hernandez, V.M.; Malone, R.R.; Torres, J.E.; Hunsaker, L.A.; Piper, R.C.; Makler, M.T.; Vander Jagt, D.L. Selective Inhibitors of Human Lactate Dehydrogenases and Lactate Dehydrogenase from the Malarial Parasite Plasmodium falciparum. J. Med. Chem. 1998, 41, 3879–3887. [Google Scholar] [CrossRef]
- Yu, Y.; Deck, J.A.; Hunsaker, L.A.; Deck, L.M.; Royer, R.E.; Goldberg, E.; Vander Jagt, D.L. Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem. Pharmacol. 2001, 62, 81–89. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.G.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U.; et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef]
- Yu, S.-L.; Xu, L.-T.; Qi, Q.; Geng, Y.-W.; Chen, H.; Meng, Z.-Q.; Wang, P.; Chen, Z. Serum lactate dehydrogenase predicts prognosis and correlates with systemic inflammatory response in patients with advanced pancreatic cancer after gemcitabine-based chemotherapy. Sci. Rep. 2017, 7, 45194. [Google Scholar] [CrossRef]
- Faloppi, L.; Bianconi, M.; Giampieri, R.; Sobrero, A.; Labianca, R.; Ferrari, D.; Barni, S.; Aitini, E.; Zaniboni, A.; Boni, C.; et al. The value of lactate dehydrogenase serum levels as a prognostic and predictive factor for advanced pancreatic cancer patients receiving sorafenib. Oncotarget 2015, 6, 35087–35094. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Wu, W.; Ni, X.; Kuang, T.; Jin, D.; Wang, D.; Lou, W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumor Biol. 2013, 34, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Li, Z.; Wang, Y.; Qi, B.; Zhang, W.; Ye, J.; Wu, H.; Jiang, H.; Song, L.-N.; Yang, J.; et al. Overexpression of metabolic markers PKM2 and LDH5 correlates with aggressive clinicopathological features and adverse patient prognosis in tongue cancer. Histopathology 2014, 65, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liao, M.; Liu, R.; Chen, J.; Feng, H.; Fu, Z. Overexpression of lactate dehydrogenase-A in human intrahepatic cholangiocarcinoma: Its implication for treatment. World J. Surg. Oncol. 2014, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.H.; Olde Damink, S.W.M.; Malago, M.; Dhar, D.K.; Pereira, S.P. Pyruvate Kinase M2 and Lactate Dehydrogenase A Are Overexpressed in Pancreatic Cancer and Correlate with Poor Outcome. PLoS ONE 2016, 11, e0151635. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979. [Google Scholar] [CrossRef]
- Kolev, Y.; Uetake, H.; Takagi, Y.; Sugihara, K. Lactate Dehydrogenase-5 (LDH-5) Expression in Human Gastric Cancer: Association with Hypoxia-Inducible Factor (HIF-1α) Pathway, Angiogenic Factors Production and Poor Prognosis. Ann. Surg. Oncol. 2008, 15, 2336–2344. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Bougioukas, G.; Didilis, V.; Gatter, K.C.; Harris, A.L. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br. J. Cancer 2003, 89, 877–885. [Google Scholar] [CrossRef]
- Bandara, I.A.; Baltatzis, M.; Sanyal, S.; Siriwardena, A.K. Evaluation of tumor M2-pyruvate kinase (Tumor M2-PK) as a biomarker for pancreatic cancer. World J. Surg. Oncol. 2018, 16, 56. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; Wei, C.; Guo, F.; Chen, Y.; et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol. Cell 2014, 53, 75–87. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Y.; Wang, T.; Hawke, D.H.; Zheng, Y.; Li, X.; Zhou, Q.; Majumder, S.; Bi, E.; Liu, D.X.; et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumor cells. Nat. Commun. 2014, 5, 5566. [Google Scholar] [CrossRef] [PubMed]
- Alves-Filho, J.C.; Pålsson-McDermott, E.M. Pyruvate Kinase M2: A Potential Target for Regulating Inflammation. Front. Immunol. 2016, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 is a PHD3-stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, H.; Jenny, J.Y.; Liu, X.; Liu, Z.-R. Pyruvate Kinase M2 Regulates Gene Transcription by Acting as A Protein Kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, H.K.; Han, Y.-M.; Kim, J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int. J. Biochem. Cell Biol. 2008, 40, 1043–1054. [Google Scholar] [CrossRef]
- Liang, J.; Cao, R.; Zhang, Y.; Xia, Y.; Zheng, Y.; Li, X.; Wang, L.; Yang, W.; Lu, Z. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat. Commun. 2016, 7, 12431. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.H.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.V.; Huang, J.S.; Yin, W.; Albeituni, S.; Rush, J.; Khanal, A.; Yan, J.; Ceresa, B.P.; Frieboes, H.B.; McNally, L.R. Targeted Non-invasive Imaging of EGFR-expressing Orthotopic Pancreatic Cancer using Multispectral Optoacoustic Tomography (MSOT). Cancer Res. 2014, 74, 6271–6279. [Google Scholar] [CrossRef]
- Vassileva, V.; Moriyama, E.H.; De Souza, R.; Grant, J.; Allen, C.J.; Wilson, B.C.; Piquette-Miller, M. Efficacy assessment of sustained intraperitoneal paclitaxel therapy in a murine model of ovarian cancer using bioluminescent imaging. Br. J. Cancer 2008, 99, 2037–2043. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, G.H.; Vassileva, V.; Acedo, P.; Olde Damink, S.W.M.; Malago, M.; Dhar, D.K.; Pereira, S.P. Targeting Pyruvate Kinase M2 and Lactate Dehydrogenase A Is an Effective Combination Strategy for the Treatment of Pancreatic Cancer. Cancers 2019, 11, 1372. https://doi.org/10.3390/cancers11091372
Mohammad GH, Vassileva V, Acedo P, Olde Damink SWM, Malago M, Dhar DK, Pereira SP. Targeting Pyruvate Kinase M2 and Lactate Dehydrogenase A Is an Effective Combination Strategy for the Treatment of Pancreatic Cancer. Cancers. 2019; 11(9):1372. https://doi.org/10.3390/cancers11091372
Chicago/Turabian StyleMohammad, Goran Hamid, Vessela Vassileva, Pilar Acedo, Steven W. M. Olde Damink, Massimo Malago, Dipok Kumar Dhar, and Stephen P. Pereira. 2019. "Targeting Pyruvate Kinase M2 and Lactate Dehydrogenase A Is an Effective Combination Strategy for the Treatment of Pancreatic Cancer" Cancers 11, no. 9: 1372. https://doi.org/10.3390/cancers11091372
APA StyleMohammad, G. H., Vassileva, V., Acedo, P., Olde Damink, S. W. M., Malago, M., Dhar, D. K., & Pereira, S. P. (2019). Targeting Pyruvate Kinase M2 and Lactate Dehydrogenase A Is an Effective Combination Strategy for the Treatment of Pancreatic Cancer. Cancers, 11(9), 1372. https://doi.org/10.3390/cancers11091372