Selective Inhibition of Histone Deacetylases 1/2/6 in Combination with Gemcitabine: A Promising Combination for Pancreatic Cancer Therapy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Inhibition of Multiple HDACs Is Required for the Effects of HDAC Inhibitors on PDAC Cells
2.2. Inhibition of HDACs 1, 2, and 6 Accounts for the Effects of HDAC Inhibitors in PDAC Cells
2.3. Pharmacological Inhibition of HDACs 1, 2, and 6 Synergizes with Gemcitabine in PDAC Cells
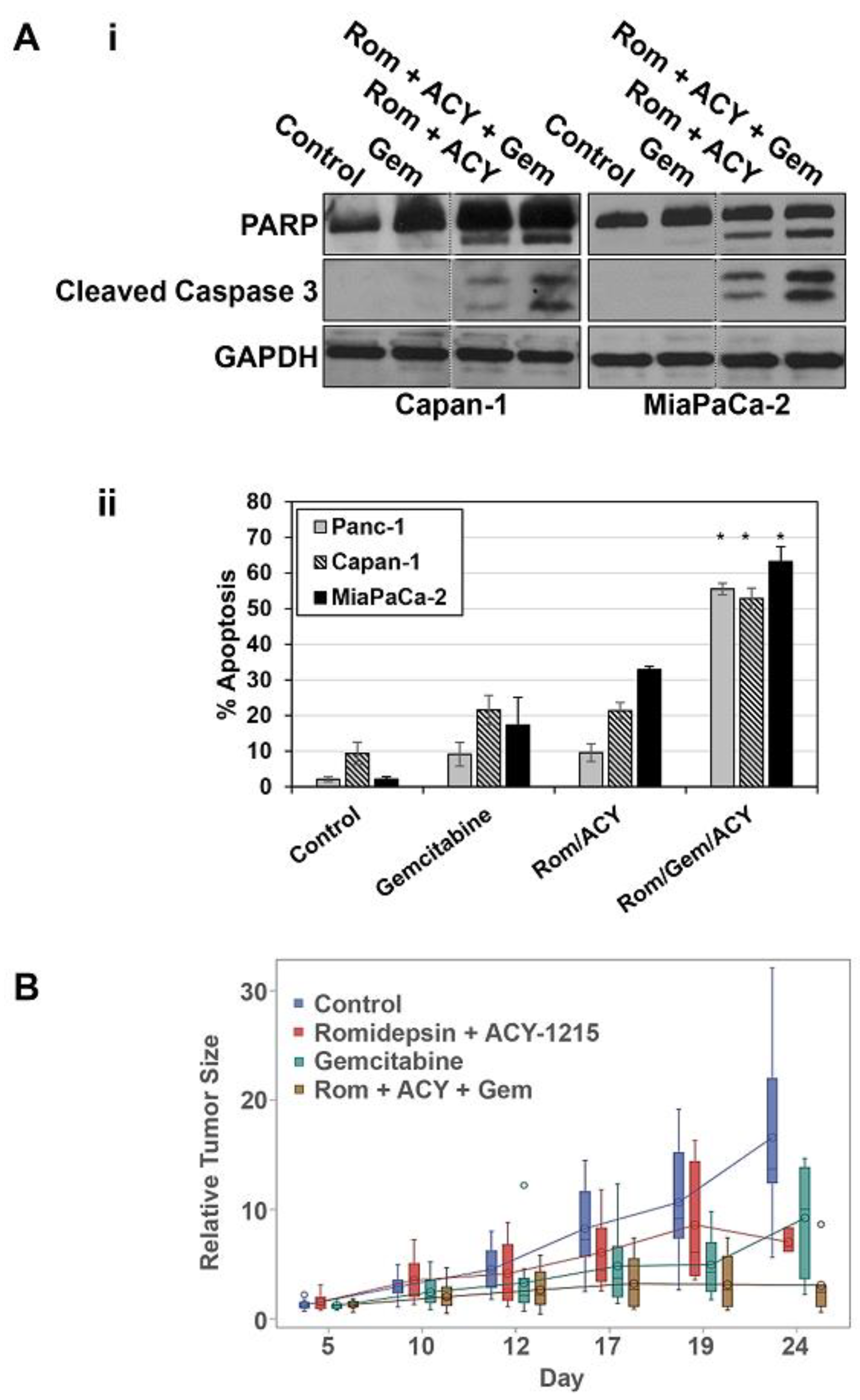
2.4. Selective Inhibition of HDACs 1, 2, and 6 Increases the Apoptosis-Inducing Effects of Gemcitabine In Vitro and Augments the Antitumor Effects of Gemcitabine In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Western Blot Analysis
4.3. MTT Assay
4.4. siRNA Knockdown
4.5. Annexin V-FITC assay
4.6. Mouse Xenograft Studies
4.7. Analysis of Drug Interactions
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Dindyal, S.; Spalding, D. Pancreatic cancer. Medicine 2019, 47, 433–439. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic Cancer. New Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Zhu, H.; Li, T.; Du, Y.; Li, M. Pancreatic cancer: Challenges and opportunities. BMC Med. 2018, 16, 214. [Google Scholar] [CrossRef]
- Picozzi, V.J.; Oh, S.Y.; Edwards, A.; Mandelson, M.T.; Dorer, R.; Rocha, F.G.; Alseidi, A.; Biehl, T.; Traverso, L.W.; Helton, W.S.; et al. Five-Year Actual Overall Survival in Resected Pancreatic Cancer: A Contemporary Single-Institution Experience from a Multidisciplinary Perspective. Ann. Surg. Oncol. 2017, 24, 1722–1730. [Google Scholar] [CrossRef]
- Pusceddu, S.; Ghidini, M.; Torchio, M.; Corti, F.; Tomasello, G.; Niger, M.; Prinzi, N.; Nichetti, F.; Coinu, A.; Di Bartolomeo, M.; et al. Comparative Effectiveness of Gemcitabine plus Nab-Paclitaxel and FOLFIRINOX in the First-Line Setting of Metastatic Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 484. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Nakamura, K.; Akahori, T.; Nagai, M.; Nishiwada, S.; Nakagawa, K.; Ikeda, N.; Sho, M. New Perspective in Pancreatic Cancer. In Alcoholic/Non-Alcoholic Digestive Diseases; Yoshiji, H., Kaji, K., Eds.; Springer: Singapore, 2019; pp. 151–161. [Google Scholar]
- Sohal, D.P.S.; Mangu, P.B.; Khorana, A.A.; Shah, M.A.; Philip, P.A.; O’Reilly, E.M.; Uronis, H.E.; Ramanathan, R.K.; Crane, C.H.; Engebretson, A.; et al. Metastatic Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 2784–2796. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Yao, Y.L.; Sun, J.M.; Davie, J.R.; Seto, E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997, 272, 28001–28007. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Brancolini, C. Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Spratlin, J.L.; Pitts, T.M.; Kulikowski, G.N.; Morelli, M.P.; Tentler, J.J.; Serkova, N.J.; Eckhardt, S.G. Synergistic activity of histone deacetylase and proteasome inhibition against pancreatic and hepatocellular cancer cell lines. Anticancer Res. 2011, 31, 1093–1103. [Google Scholar] [PubMed]
- Moertl, S.; Payer, S.; Kell, R.; Winkler, K.; Anastasov, N.; Atkinson, M.J. Comparison of Radiosensitization by HDAC Inhibitors CUDC-101 and SAHA in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3259. [Google Scholar] [CrossRef]
- Koutsounas, I.; Giaginis, C.; Patsouris, E.; Theocharis, S. Current evidence for histone deacetylase inhibitors in pancreatic cancer. World J. Gastroenterol. 2013, 19, 813–828. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, P.; Donadelli, M.; Costanzo, C.; Moore, P.S.; Palmieri, M.; Scarpa, A. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Archiv. 2006, 448, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Koutsounas, I.; Giaginis, C.; Theocharis, S. Histone deacetylase inhibitors and pancreatic cancer: Are there any promising clinical trials? World J. Gastroenterol. 2013, 19, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Han, Y.; Jiang, Q.; Wang, C.; Chen, X.; Li, X.; Xu, F.; Jiang, Y.; Wang, Q.; Xu, W. Trend of Histone Deacetylase Inhibitors in Cancer Therapy: Isoform Selectivity or Multitargeted Strategy. Med. Res. Rev. 2015, 35, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Fournel, M.; Bonfils, C.; Hou, Y.; Yan, P.T.; Trachy-Bourget, M.-C.; Kalita, A.; Liu, J.; Lu, A.-H.; Zhou, N.Z.; Robert, M.-F.; et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [Google Scholar] [CrossRef]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone Deacetylase (HDAC) Inhibitors with a Novel Connecting Unit Linker Region Reveal a Selectivity Profile for HDAC4 and HDAC5 with Improved Activity against Chemoresistant Cancer Cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Sukhai, M.A.; Hurren, R.; Anyiwe, K.; Mao, X.; Suarez Saiz, F.; Gronda, M.; Eberhard, Y.; et al. Selective Inhibition of Histone Deacetylases Sensitizes Malignant Cells to Death Receptor Ligands. Mol. Cancer Ther. 2010, 9, 246–256. [Google Scholar] [CrossRef]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Budman, D.R.; Tai, J.; Calabro, A.; John, V. The histone deacetylase inhibitor panobinostat demonstrates marked synergy with conventional chemotherapeutic agents in human ovarian cancer cell lines. Investig. New Drugs 2011, 29, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.-H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (Depsipeptide) as a Natural Prodrug That Inhibits Class I Histone Deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Gaisina, I.N.; Tueckmantel, W.; Ugolkov, A.; Shen, S.; Hoffen, J.; Dubrovskyi, O.; Mazar, A.; Schoon, R.A.; Billadeau, D.; Kozikowski, A.P. Identification of HDAC6-Selective Inhibitors of Low Cancer Cell Cytotoxicity. Chem. Med. Chem. 2016, 11, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; He, J.; Zhao, J.; Yun, W.; Xie, C.; Taub, J.W.; Azmi, A.; Mohammad, R.M.; Dong, Y.; Kong, W.; et al. Class I and Class II Histone Deacetylases Are Potential Therapeutic Targets for Treating Pancreatic Cancer. PLoS ONE 2012, 7, e52095. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Ma, J.; Golonzhka, O.; Laumet, G.O.; Gutti, T.; van Duzer, J.H.; Mazitschek, R.; Jarpe, M.B.; Heijnen, C.J.; Kavelaars, A. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 2017, 158, 1126–1137. [Google Scholar] [CrossRef]
- Putcha, P.; Yu, J.; Rodriguez-Barrueco, R.; Saucedo-Cuevas, L.; Villagrasa, P.; Murga-Penas, E.; Quayle, S.N.; Yang, M.; Castro, V.; Llobet-Navas, D.; et al. HDAC6 activity is a non-oncogene addiction hub for inflammatory breast cancers. Breast Cancer Res. 2015, 17, 149. [Google Scholar] [CrossRef]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707. [Google Scholar] [CrossRef]
- Dowdell, K.C.; Pesnicak, L.; Hoffmann, V.; Steadman, K.; Remaley, A.T.; Cohen, J.I.; Straus, S.E.; Rao, V.K. Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, diminishes lymphoproliferation in the Fas -deficient MRL/lpr(-/-) murine model of autoimmune lymphoproliferative syndrome (ALPS). Exp. Hematol. 2009, 37, 487–494. [Google Scholar] [CrossRef][Green Version]
- Zhang, W.; Peyton, M.; Xie, Y.; Soh, J.; Minna, J.D.; Gazdar, A.F.; Frenkel, E.P. Histone deacetylase inhibitor romidepsin enhances anti-tumor effect of erlotinib in non-small cell lung cancer (NSCLC) cell lines. J. Thorac. Oncol. 2009, 4, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Boehm, K.A.; Waterhouse, D.M.; Wagener, D.J.; Krishnamurthi, S.S.; Rosemurgy, A.; Grove, W.; Macdonald, K.; Gulyas, S.; Clark, M.; et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: Results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann. Oncol. 2006, 17, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R.; Spigel, D.R.; Peacock, N.W.; Thompson, D.S.; Greco, F.A.; McCulloch, W.; Burris Iii, H.A. Phase 1 Results From a Study of Romidepsin in Combination With Gemcitabine in Patients With Advanced Solid Tumors. Cancer Investig. 2012, 30, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Yoshizumi, T.; Imura, S.; Sugimoto, K.; Batmunkh, E.; Kanemura, H.; Morine, Y.; Shimada, M. Expression of Hypoxia-Inducible Factor-1α, Histone Deacetylase 1, and Metastasis-Associated Protein 1 in Pancreatic Carcinoma: Correlation With Poor Prognosis With Possible Regulation. Pancreas 2008, 36, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Klieser, E.; Swierczynski, S.; Mayr, C.; Schmidt, J.; Neureiter, D.; Kiesslich, T.; Illig, R. Role of histone deacetylases in pancreas: Implications for pathogenesis and therapy. World J. Gastrointest. Oncol. 2015, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.A.; Hagelkruys, A.; Seiser, C. Transcription and beyond: The role of mammalian class I lysine deacetylases. Chromosoma 2014, 123, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.-O.; Heidecke, C.-D.; Friess, H.; Büchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- Polireddy, K.; Dong, R.; McDonald, P.R.; Wang, T.; Luke, B.; Chen, P.; Broward, M.; Roy, A.; Chen, Q. Targeting Epithelial-Mesenchymal Transition for Identification of Inhibitors for Pancreatic Cancer Cell Invasion and Tumor Spheres Formation. PLoS ONE 2016, 11, e0164811. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nakazono, K.; Tokuda, M.; Mashima, Y.; Dynlacht, B.D.; Itoh, H. HDAC2 promotes loss of primary cilia in pancreatic ductal adenocarcinoma. EMBO Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef]
- Ryu, H.-W.; Shin, D.-H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schäfer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2016, 36, 1804. [Google Scholar] [CrossRef] [PubMed]
- Luchenko, V.L.; Litman, T.; Chakraborty, A.R.; Heffner, A.; Devor, C.; Wilkerson, J.; Stein, W.; Robey, R.W.; Bangiolo, L.; Levens, D.; et al. Histone deacetylase inhibitor-mediated cell death is distinct from its global effect on chromatin. Mol. Oncol. 2014, 8, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Stowe, R.L.; Pinello, C.E.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.Y.; Li, J.-L.; Wang, Y.; Wang, Y.; et al. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef]
- Hsu, A.H.; Lum, M.A.; Shim, K.S.; Frederick, P.J.; Morrison, C.D.; Chen, B.; Lele, S.M.; Sheinin, Y.M.; Daikoku, T.; Dey, S.K.; et al. Crosstalk between PKCalpha and PI3K/AKT Signaling Is Tumor Suppressive in the Endometrium. Cell Rep. 2018, 24, 655–669. [Google Scholar] [CrossRef]
- Hu, Y.P.; Patil, S.B.; Panasiewicz, M.; Li, W.; Hauser, J.; Humphrey, L.E.; Brattain, M.G. Heterogeneity of receptor function in colon carcinoma cells determined by cross-talk between type I insulin-like growth factor receptor and epidermal growth factor receptor. Cancer Res. 2008, 68, 8004–8013. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laschanzky, R.S.; Humphrey, L.E.; Ma, J.; Smith, L.M.; Enke, T.J.; Shukla, S.K.; Dasgupta, A.; Singh, P.K.; Howell, G.M.; Brattain, M.G.; et al. Selective Inhibition of Histone Deacetylases 1/2/6 in Combination with Gemcitabine: A Promising Combination for Pancreatic Cancer Therapy. Cancers 2019, 11, 1327. https://doi.org/10.3390/cancers11091327
Laschanzky RS, Humphrey LE, Ma J, Smith LM, Enke TJ, Shukla SK, Dasgupta A, Singh PK, Howell GM, Brattain MG, et al. Selective Inhibition of Histone Deacetylases 1/2/6 in Combination with Gemcitabine: A Promising Combination for Pancreatic Cancer Therapy. Cancers. 2019; 11(9):1327. https://doi.org/10.3390/cancers11091327
Chicago/Turabian StyleLaschanzky, Richard S., Lisa E. Humphrey, Jihyun Ma, Lynette M. Smith, Thomas J. Enke, Surendra K. Shukla, Aneesha Dasgupta, Pankaj K. Singh, Gillian M. Howell, Michael G. Brattain, and et al. 2019. "Selective Inhibition of Histone Deacetylases 1/2/6 in Combination with Gemcitabine: A Promising Combination for Pancreatic Cancer Therapy" Cancers 11, no. 9: 1327. https://doi.org/10.3390/cancers11091327
APA StyleLaschanzky, R. S., Humphrey, L. E., Ma, J., Smith, L. M., Enke, T. J., Shukla, S. K., Dasgupta, A., Singh, P. K., Howell, G. M., Brattain, M. G., Ly, Q. P., Black, A. R., & Black, J. D. (2019). Selective Inhibition of Histone Deacetylases 1/2/6 in Combination with Gemcitabine: A Promising Combination for Pancreatic Cancer Therapy. Cancers, 11(9), 1327. https://doi.org/10.3390/cancers11091327