RNA-Based Detection of Gene Fusions in Formalin-Fixed and Paraffin-Embedded Solid Cancer Samples

 , , and
, , and
Abstract
:1. Introduction
2. Results
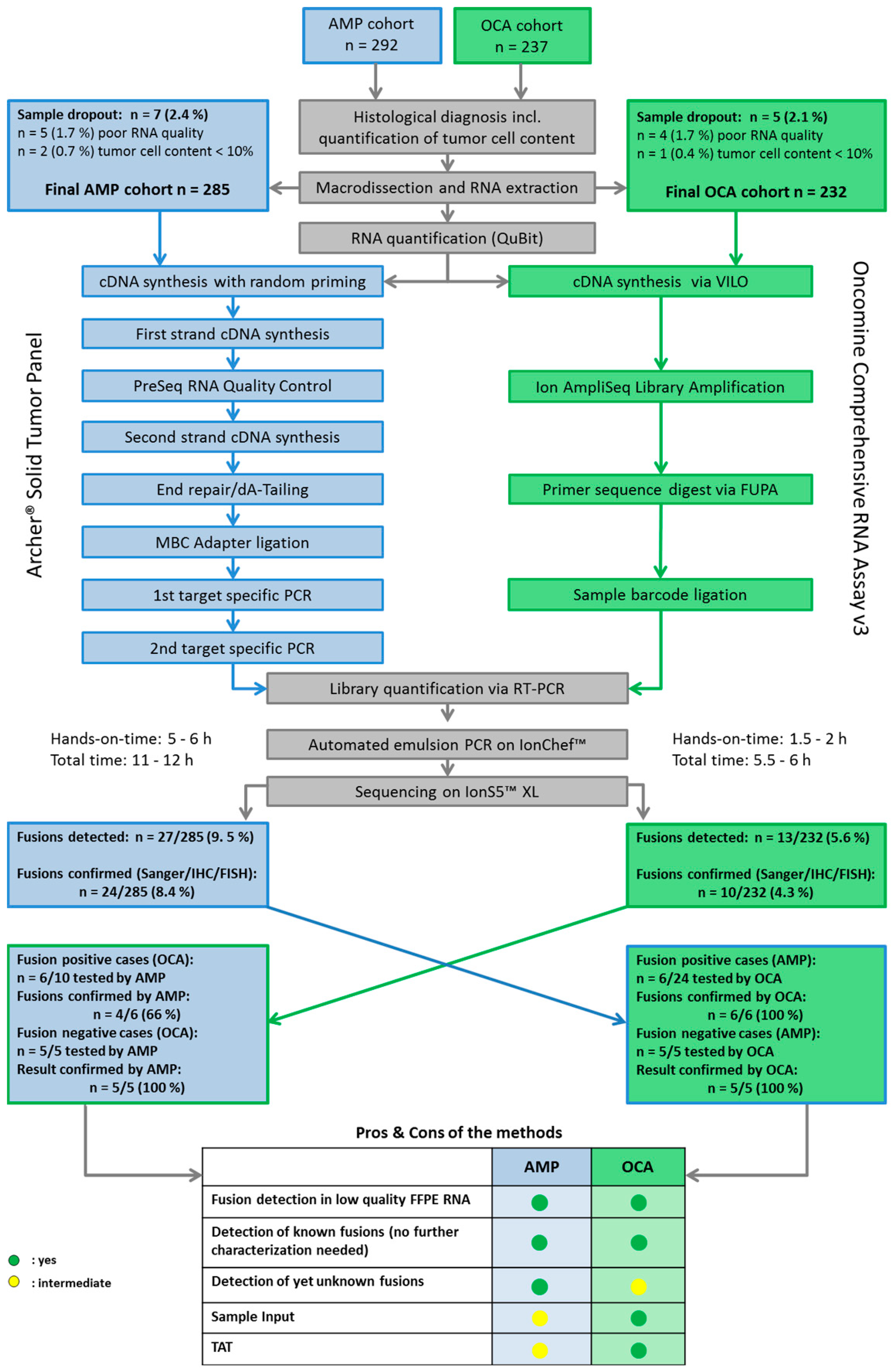
2.1. Cohort Characteristics, Tumor Cell Content and Sample Dropouts
2.2. Turnaround Time and RNA Yields
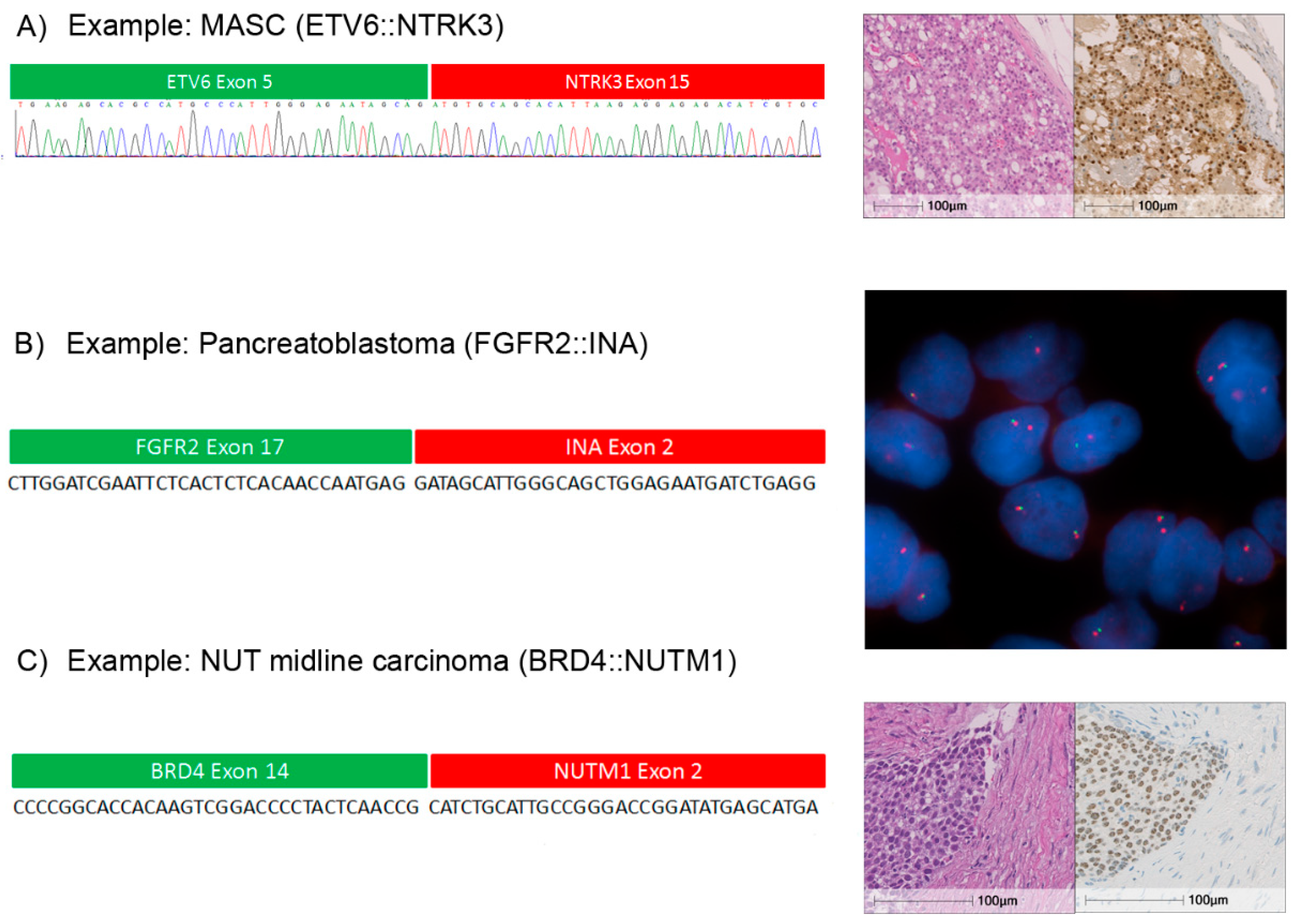
2.3. Detection and Validation of Gene Fusions
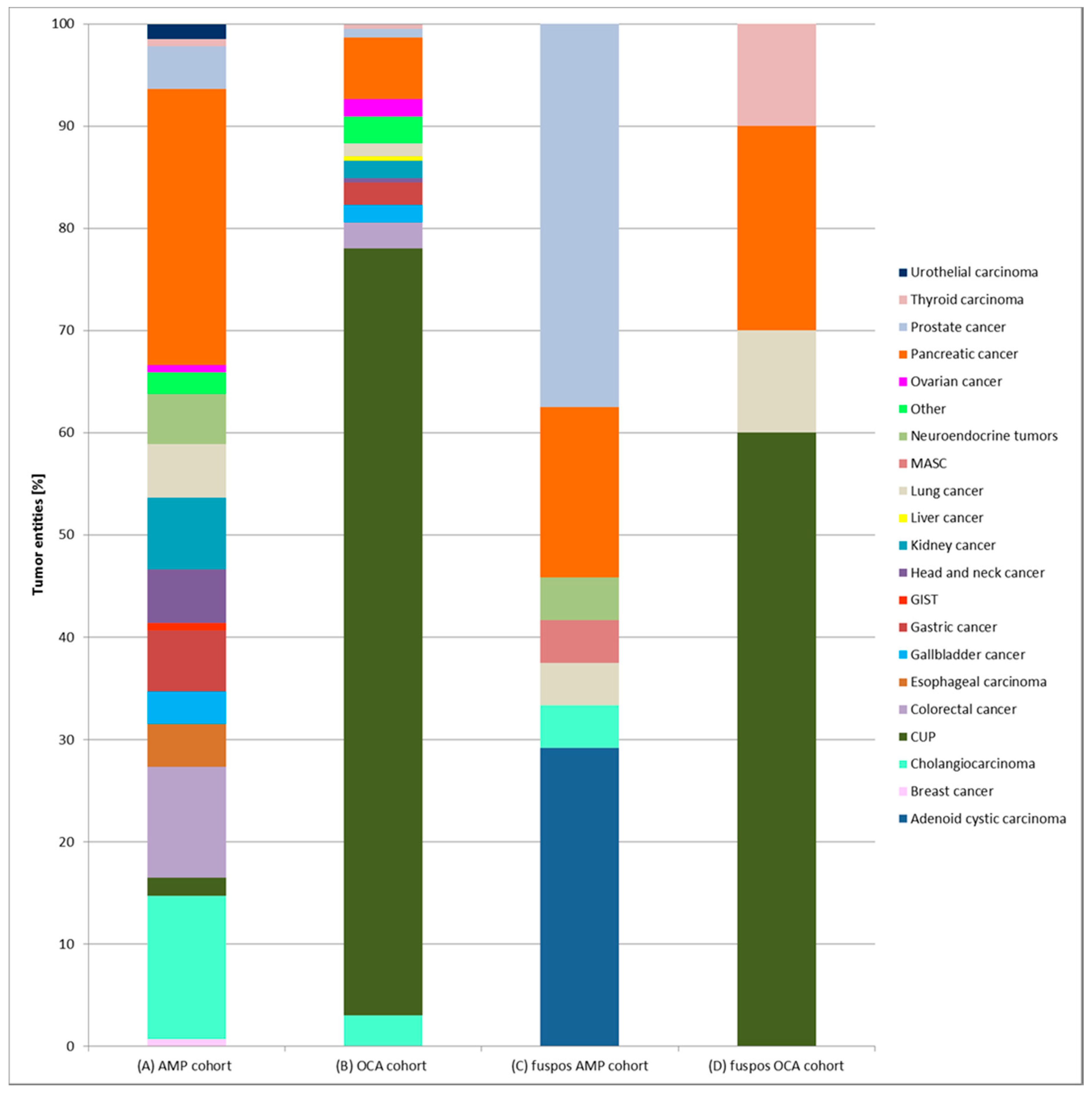
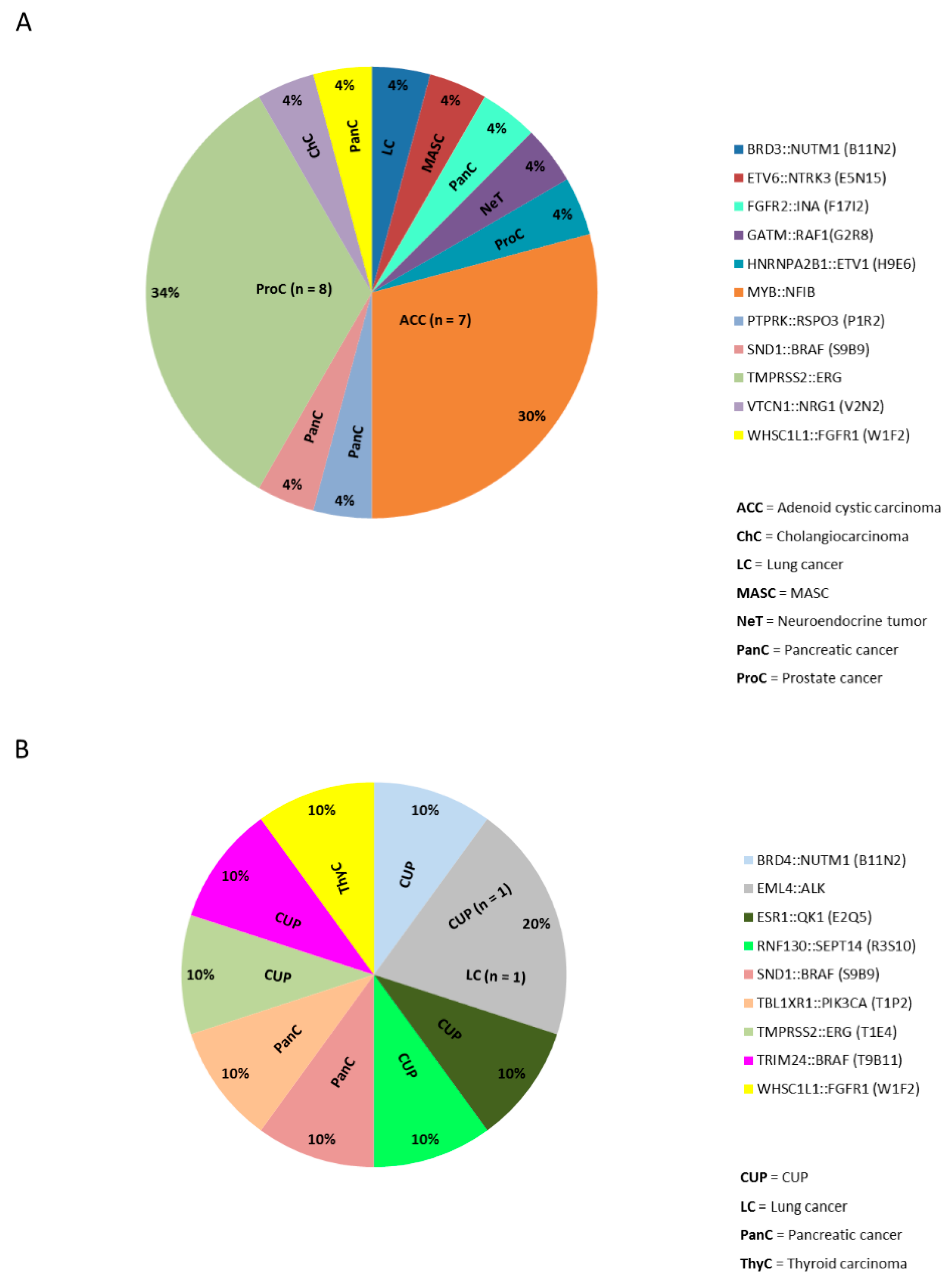
2.4. Gene Fusions in Different Cancer Types
2.5. Clinical and Diagnostic Implications of Gene Fusions
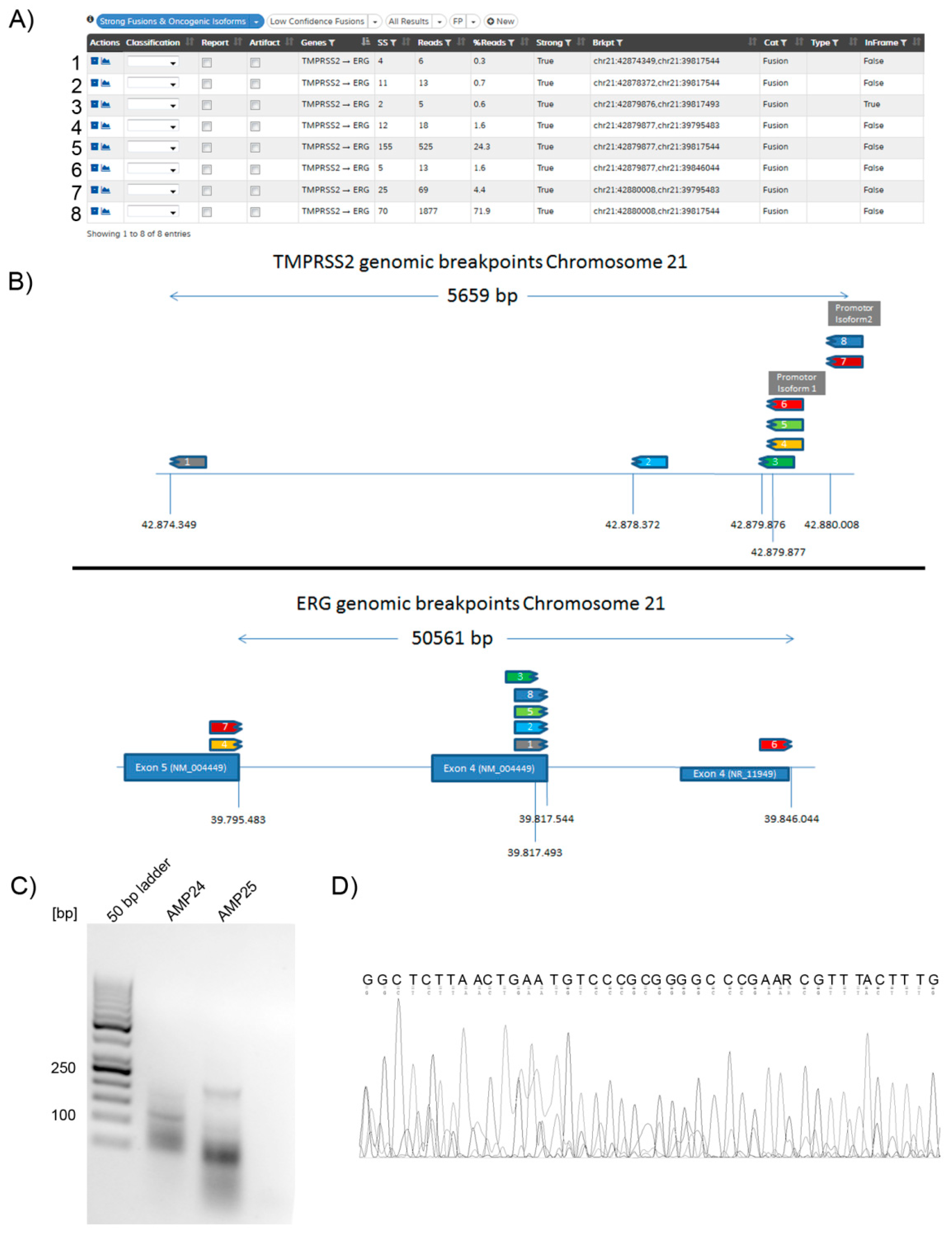
2.6. Gene Fusion Isoforms
2.7. Identification of Previously Unknown Gene Fusions
2.8. Individual Cases
3. Discussion
4. Material and Methods
4.1. RNA Extraction, Library Preparation and Semiconductor Sequencing
4.2. Library Preparation for the Oncomine™ Comprehensive RNA Panel v3
4.3. Library Preparation for AMP Based NGS (Archer Dx) Translocation Detection
4.4. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Sanger Sequencing Validation
4.5. Immunohistochemistry
4.6. FISH Analyses
4.7. Research Ethics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef]
- Melo, J.V. The molecular biology of chronic myeloid leukaemia. Leukemia 1996, 10, 751–756. [Google Scholar]
- Ewing, J. The Classic: Diffuse endothelioma of bone. Proceedings of the New York Pathological Society. Clin. Orthop. Relat. Res. 2006, 450, 25–27. [Google Scholar] [CrossRef]
- Koelsche, C.; Hartmann, W.; Schrimpf, D.; Stichel, D.; Jabar, S.; Ranft, A.; Reuss, D.E.; Sahm, F.; Jones, D.T.W.; Bewerunge-Hudler, M.; et al. Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod. Pathol. 2018, 31, 1246–1256. [Google Scholar] [CrossRef] [Green Version]
- Baldauf, M.C.; Gerke, J.S.; Orth, M.F.; Dallmayer, M.; Baumhoer, D.; de Alava, E.; Hartmann, W.; Kirchner, T.; Grunewald, T.G.P. Are EWSR1-NFATc2-positive sarcomas really Ewing sarcomas? Mod. Pathol. 2018, 31, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular characterization of cancers with NTRK gene fusions. Mod. Pathol. 2019, 32, 147–153. [Google Scholar] [CrossRef]
- Lei, J.T.; Shao, J.; Zhang, J.; Iglesia, M.; Chan, D.W.; Cao, J.; Anurag, M.; Singh, P.; He, X.; Kosaka, Y.; et al. Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell Rep. 2018, 24, 1434–1444 e7. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Ippen, F.M.; Hafeez, U.; Gan, H.K. Emerging Gene Fusion Drivers in Primary and Metastatic Central Nervous System Malignancies: A Review of Available Evidence for Systemic Targeted Therapies. Oncologist 2018, 23, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Scolnick, J.A.; Dimon, M.; Wang, I.C.; Huelga, S.C.; Amorese, D.A. An Efficient Method for Identifying Gene Fusions by Targeted RNA Sequencing from Fresh Frozen and FFPE Samples. PLoS ONE 2015, 10, e0128916. [Google Scholar] [CrossRef] [PubMed]
- Reeser, J.W.; Martin, D.; Miya, J.; Kautto, E.A.; Lyon, E.; Zhu, E.; Wing, M.R.; Smith, A.; Reeder, M.; Samorodnitsky, E.; et al. Validation of a Targeted RNA Sequencing Assay for Kinase Fusion Detection in Solid Tumors. J. Mol. Diagn. 2017, 19, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, K.E.; Karlsson, A.; Leveen, P.; Brunnstrom, H.; Reutersward, C.; Holm, K.; Jonsson, M.; Annersten, K.; Rosengren, F.; Jirstrom, K.; et al. Clinical framework for next generation sequencing based analysis of treatment predictive mutations and multiplexed gene fusion detection in non-small cell lung cancer. Oncotarget 2017, 8, 34796–34810. [Google Scholar] [CrossRef] [Green Version]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. 2019, 25, 4712–4722.18. [Google Scholar] [CrossRef]
- Volckmar, A.L.; Leichsenring, J.; Kirchner, M.; Christopoulos, P.; Neumann, O.; Budczies, J.; de Oliveira, C.M.M.; Rempel, E.; Buchhalter, I.; Brandt, R.; et al. Combined targeted DNA and RNA sequencing of advanced NSCLC in routine molecular diagnostics: Analysis of the first 3,000 Heidelberg cases. Int. J. Cancer 2019, 145, 649–661. [Google Scholar] [CrossRef]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, T.; Endris, V.; Leichsenring, J.; Reiling, A.; Neumann, O.; Volckmar, A.L.; Kirchner, M.; Allgauer, M.; Schirmacher, P.; Kramer, A.; et al. Comparative genetic profiling aids diagnosis and clinical decision making in challenging cases of CUP syndrome. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Leichsenring, J.; Volckmar, A.L.; Kirchner, M.; Kazdal, D.; Kriegsmann, M.; Stogbauer, F.; Bockmayr, T.; Klauschen, F.; Herth, F.J.F.; Penzel, R.; et al. Targeted deep sequencing of effusion cytology samples is feasible, informs spatiotemporal tumor evolution, and has clinical and diagnostic utility. Genes Chromosomes Cancer 2018, 57, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.L.; Walsh, K.; Diamond, A.; Oniscu, A.; Deans, Z.C. Validation of the OncomineTM focus panel for next-generation sequencing of clinical tumour samples. Virchows Arch. 2018, 473, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Hutchinson, K.E.; Frampton, G.M.; Chalmers, Z.R.; Johnson, A.; Shi, C.; Elvin, J.; Ali, S.M.; Ross, J.S.; Basturk, O.; et al. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov. 2014, 4, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Surrey, L.F.; Straka, J.; Luo, M.; Lin, F.; Harding, B.; Resnick, A.C.; Storm, P.B.; Buccoliero, A.M.; Santi, M.; et al. Novel FGFR2-INA fusion identified in two low-grade mixed neuronal-glial tumors drives oncogenesis via MAPK and PI3K/mTOR pathway activation. Acta Neuropathol. 2018, 136, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Pfarr, N.; Stenzinger, A.; Penzel, R.; Warth, A.; Dienemann, H.; Schirmacher, P.; Weichert, W.; Endris, V. High-throughput diagnostic profiling of clinically actionable gene fusions in lung cancer. Genes Chromosomes Cancer 2016, 55, 30–44. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, K.; Buttner, R.; Langer, T.; Ukena, D. Pathological-anatomical diagnosis according to the German lung cancer guideline 2018. Pathologe 2018, 39, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J. Thorac. Oncol. 2018, 13, 323–358. [Google Scholar]
- Lara, P.N., Jr.; Heilmann, A.M.; Elvin, J.A.; Parikh, M.; de Vere White, R.; Gandour-Edwards, R.; Evans, C.P.; Pan, C.X.; Schrock, A.B.; Erlich, R.; et al. TMPRSS2-ERG fusions unexpectedly identified in men initially diagnosed with nonprostatic malignancies. JCO Precis. Oncol. 2017, 1, 1–6. [Google Scholar]
- Wang, J.; Cai, Y.; Ren, C.; Ittmann, M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006, 66, 8347–8351. [Google Scholar] [CrossRef] [PubMed]
- St John, J.; Powell, K.; Conley-Lacomb, M.K.; Chinni, S.R. TMPRSS2-ERG Fusion Gene Expression in Prostate Tumor Cells and Its Clinical and Biological Significance in Prostate Cancer Progression. J. Cancer Sci. Ther. 2012, 4, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Endris, V.; Penzel, R.; Warth, A.; Muckenhuber, A.; Schirmacher, P.; Stenzinger, A.; Weichert, W. Molecular diagnostic profiling of lung cancer specimens with a semiconductor-based massive parallel sequencing approach: feasibility, costs, and performance compared with conventional sequencing. J. Mol. Diagn. 2013, 15, 765–775. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics and Features | All | AMP | OCA |
|---|---|---|---|
| n | 517 | 285 | 232 |
| age [y] | 61.5 (4–84) | 62.1 (6–84) | 60.4 (4–82) |
| % females | 45.7 | 38.2 | 54.7 |
| n different entities | 21 | 18 | 14 |
| tumor cell content [%] | 70 (10–95) | 80 (10–95) | 70 (10–95) |
| Turnaround time [days] | 9 (3-35) | 6 (3-44) | |
| RNA [ng/µL] | 35.5 (2.4–880) | 41.5 (2.4–880) | 29.3 (2.6–499) |
| % fusion positive | 6.58 | 8.42 | 4.31 |
| Unique fragments > 150,000 | 465,740 (21,498–3,974,814) | ||
| Unique fragments > 10% | 8.6 (0.7–48.4) | ||
| Average unique RNA start sites | 171.88 (18.6–405.3) | ||
| per GSPS control > 50 | |||
| On target deduplication ratio < 40 | 12.2 (2.0–144.4) | ||
| Usable reads | 579,492 (37,591–13,897,936) | ||
| Sample ID | Fusion | Entity | Reads | Panel | Validation Successful with Any Method * |
|---|---|---|---|---|---|
| AMP-1 | AXL::CAPN15 (A19C2) | Gastric adenocarcinoma | 6 | AMP | no |
| AMP-2 | BRD3::NUTM1 (B11N2) | NUT-midline carcinoma of the lung | 2968 | AMP | |
| AMP-3 | ETV6::NTRK3 (E5N15) | MASC | 15,730 | AMP | |
| AMP-4 | FGFR2::INA (F17I2) | Pancreatic blastoma | >100 | AMP | |
| AMP-5 | GATM::RAF1 (G2R8) | Neuroendocrine tumor of the pancreas | 4186 | AMP | |
| AMP-6 | GPBP1L1::MAST2 (G6M4) | Adenocarcinoma, pancreatobiliary type | 6 | AMP | no |
| AMP-7 | HNRNPA2B1::ETV1 (H9E6) | Acinar adenocarcinoma of the prostate | >11,194 | AMP | |
| AMP-8 | MTMR::MAML2 (M2M1) | Cholangiocarcinoma | 5 | AMP | no |
| AMP-9 | MYB::NFIB (M11N9) | Adenoid cystic carcinoma | >100 | AMP | |
| AMP-10 | MYB::NFIB (M12N9) | Adenoid cystic carcinoma | >1800 | AMP | |
| AMP-11 | MYB::NFIB (M12N9) | Adenoid cystic carcinoma | >580 | AMP | |
| AMP-12 | MYB::NFIB (M13N9) | Adenoid cystic carcinoma | >6000 | AMP | |
| AMP-13 | MYB::NFIB (M13N9) | Adenoid cystic carcinoma | >100 | AMP | |
| AMP-14 | MYB::NFIB (M14N10) | Adenoid cystic carcinoma | >310 | AMP | |
| AMP-15 | MYB::NFIB (M14N10) | Adenoid cystic carcinoma | >5873 | AMP | |
| AMP-16 | PTPRK::RSPO3 (P1R2) | Adenocarcinoma of the hepatopancreatic ampulla, pancreato biliary type | >800 | AMP | |
| AMP-17 | SND1::BRAF (S9B9) | Pancreatic ductal adenocarcinoma | >3800 | AMP | |
| AMP-18 | TMPRSS2::ERG (T1E2) | Acinar adenocarcinoma of the prostate | >974 | AMP | |
| AMP-19 | TMPRSS2::ERG (T1E2) | Acinar adenocarcinoma of the prostate | >15,022 | AMP | |
| AMP-20 | TMPRSS2::ERG (T2E4) | Acinar adenocarcinoma of the prostate | >3432 | AMP | |
| AMP-21 | TMPRSS2::ERG (T2E4) | Acinar adenocarcinoma of the prostate | >177 | AMP | |
| AMP-22 | TMPRSS2::ERG (T1E5) | Acinar adenocarcinoma of the prostate | >11,745 | AMP | |
| AMP-23 | TMPRSS2::ERG (T2E4) | Acinar adenocarcinoma of the prostate | >2666 | AMP | |
| AMP-24 | TMPRSS2::ERG (T1E4) | Acinar adenocarcinoma of the prostate | >1207 | AMP | |
| AMP-25 | TMPRSS2::ERG (T1E4) | Adenocarcinoma of the prostate | >2000 | AMP | |
| AMP-26 | VTCN1::NRG1 (V2N2) | Cholangiocarcinoma | >12,000 | AMP | |
| AMP-27 | WHSC1L1::FGFR1 (W1F2) | Adenocarcinoma, pancreatobiliary type | >100 | AMP | |
| OCA-1 | BRD4::NUTM1 (B11N2) | CUP | >102,528 | OCA | |
| OCA-2 | EML4::ALK (E20A20) | CUP | 893 | OCA | |
| OCA-3 | EML4::ALK (E6A20) | NSCLC | >6200 | OCA | |
| OCA-4 | ESR1::QK1 (E2Q5) | CUP | 2857 | OCA | |
| OCA-5 | FNDC3B::PIK3CA (F3P2) | Gallbladder carcinoma | 73 | OCA | no |
| OCA-6 | KIF5B::RET (K15R12) | CUP | 744 | OCA | no |
| OCA-7 | RNF130::SEPT14 (R3S10) | CUP | >2153 | OCA | |
| OCA-8 | SND1::BRAF (S9B9) | Pancreatic ductal adenocarcinoma | 85,887 | OCA | |
| OCA-9 | TBL1XR1::PIK3CA (T1P2) | Chordoma | 531 | OCA | no |
| OCA-10 | TBL1XR1::PIK3CA (T1P2) | Colloid carcinoma of the pancreas | 973 | OCA | |
| OCA-11 | TMPRSS2::ERG (T1E4) | CUP | 35,966 | OCA | |
| OCA-12 | TRIM24::BRAF (T9B11) | CUP | 341,016 | OCA | |
| OCA-13 | WHSC1L1::FGFR1 (W1F2) | Anaplastic carcinoma of the thyroid | >345 | OCA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirchner, M.; Neumann, O.; Volckmar, A.-L.; Stögbauer, F.; Allgäuer, M.; Kazdal, D.; Budczies, J.; Rempel, E.; Brandt, R.; Talla, S.B.; et al. RNA-Based Detection of Gene Fusions in Formalin-Fixed and Paraffin-Embedded Solid Cancer Samples. Cancers 2019, 11, 1309. https://doi.org/10.3390/cancers11091309
Kirchner M, Neumann O, Volckmar A-L, Stögbauer F, Allgäuer M, Kazdal D, Budczies J, Rempel E, Brandt R, Talla SB, et al. RNA-Based Detection of Gene Fusions in Formalin-Fixed and Paraffin-Embedded Solid Cancer Samples. Cancers. 2019; 11(9):1309. https://doi.org/10.3390/cancers11091309
Chicago/Turabian StyleKirchner, Martina, Olaf Neumann, Anna-Lena Volckmar, Fabian Stögbauer, Michael Allgäuer, Daniel Kazdal, Jan Budczies, Eugen Rempel, Regine Brandt, Suranand Babu Talla, and et al. 2019. "RNA-Based Detection of Gene Fusions in Formalin-Fixed and Paraffin-Embedded Solid Cancer Samples" Cancers 11, no. 9: 1309. https://doi.org/10.3390/cancers11091309