FAK-Copy-Gain Is a Predictive Marker for Sensitivity to FAK Inhibition in Breast Cancer

 , , ,
, , ,  and
and 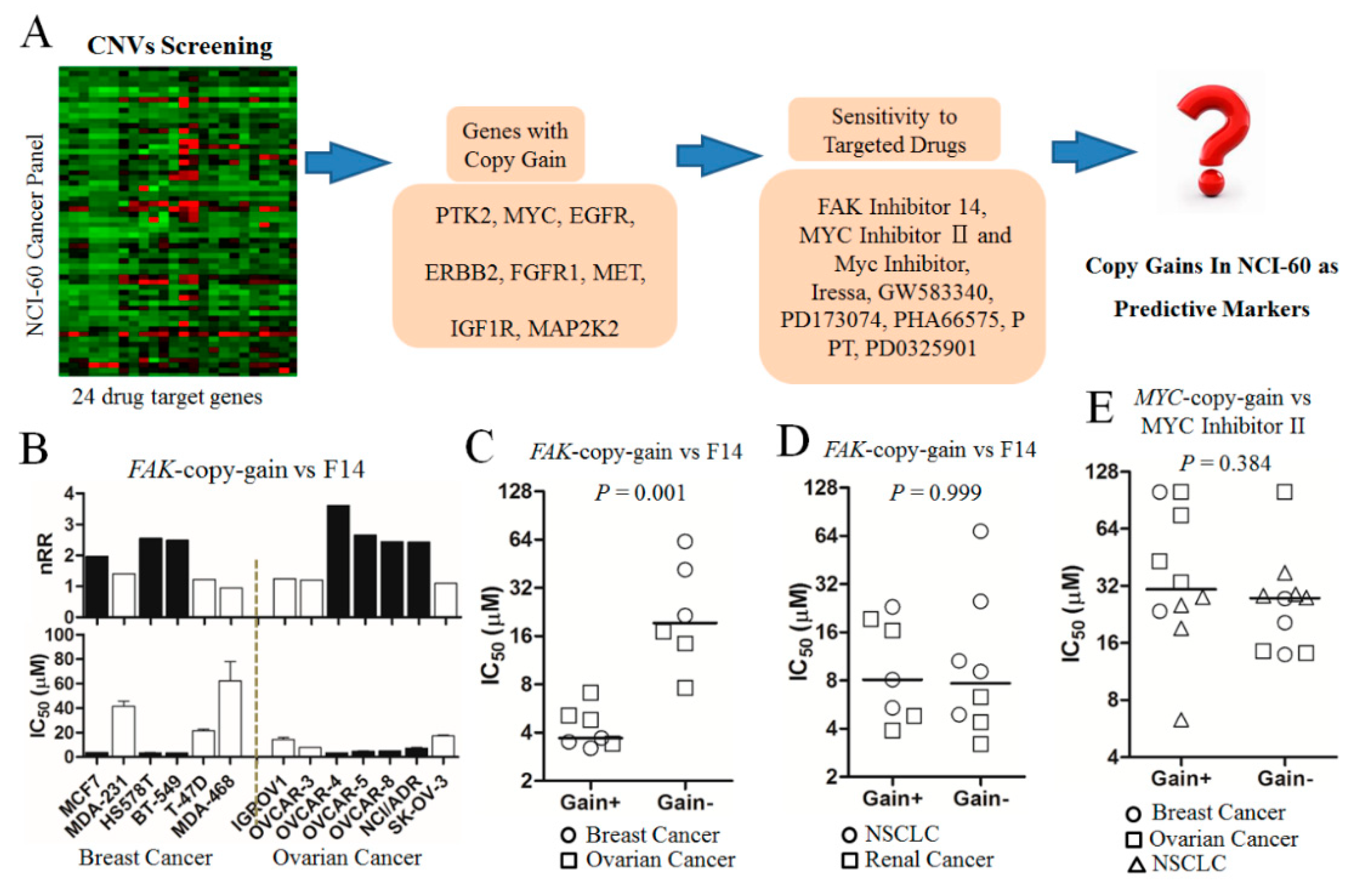{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Drug Sensitivities in Nci-60 Cells with Copy-Gain Drug-Target Genes
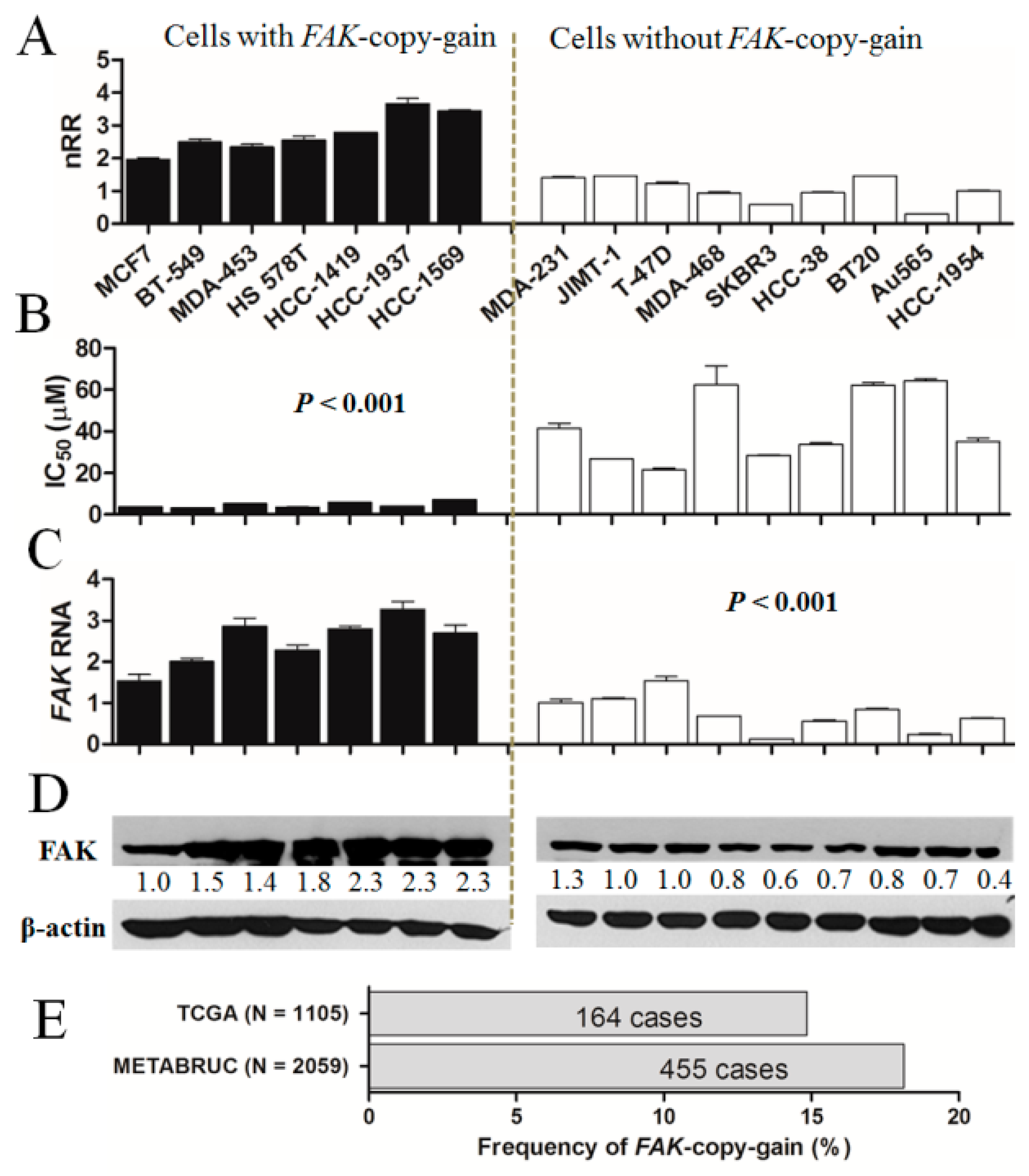
2.2. Significantly Higher Sensitivity to Fak Inhibition in Breast Cancer Cells with FAK-Copy-Gain
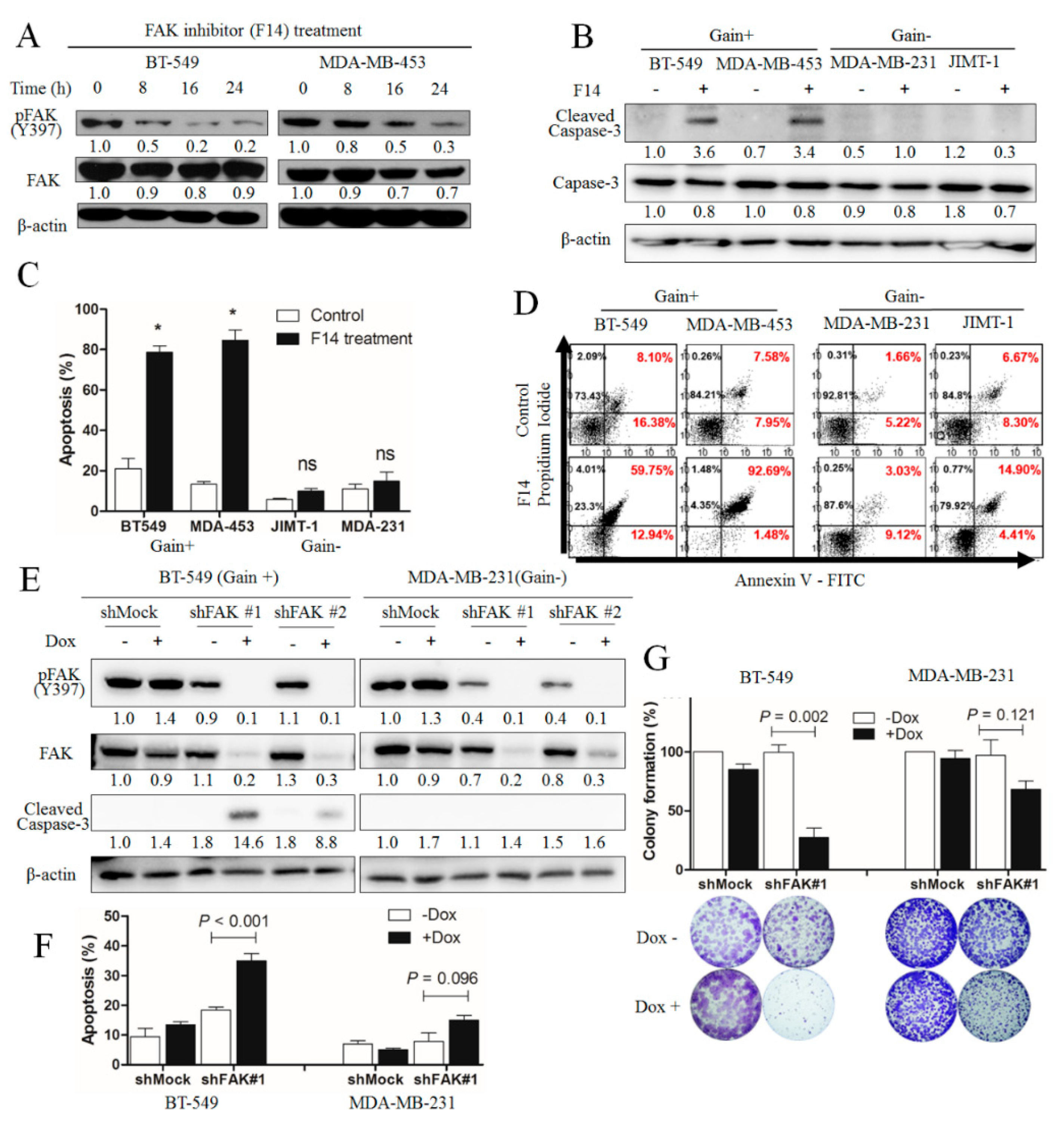
2.3. Apoptotic Effect of F14 in Breast Cancer Cells With FAK-Copy-Gain
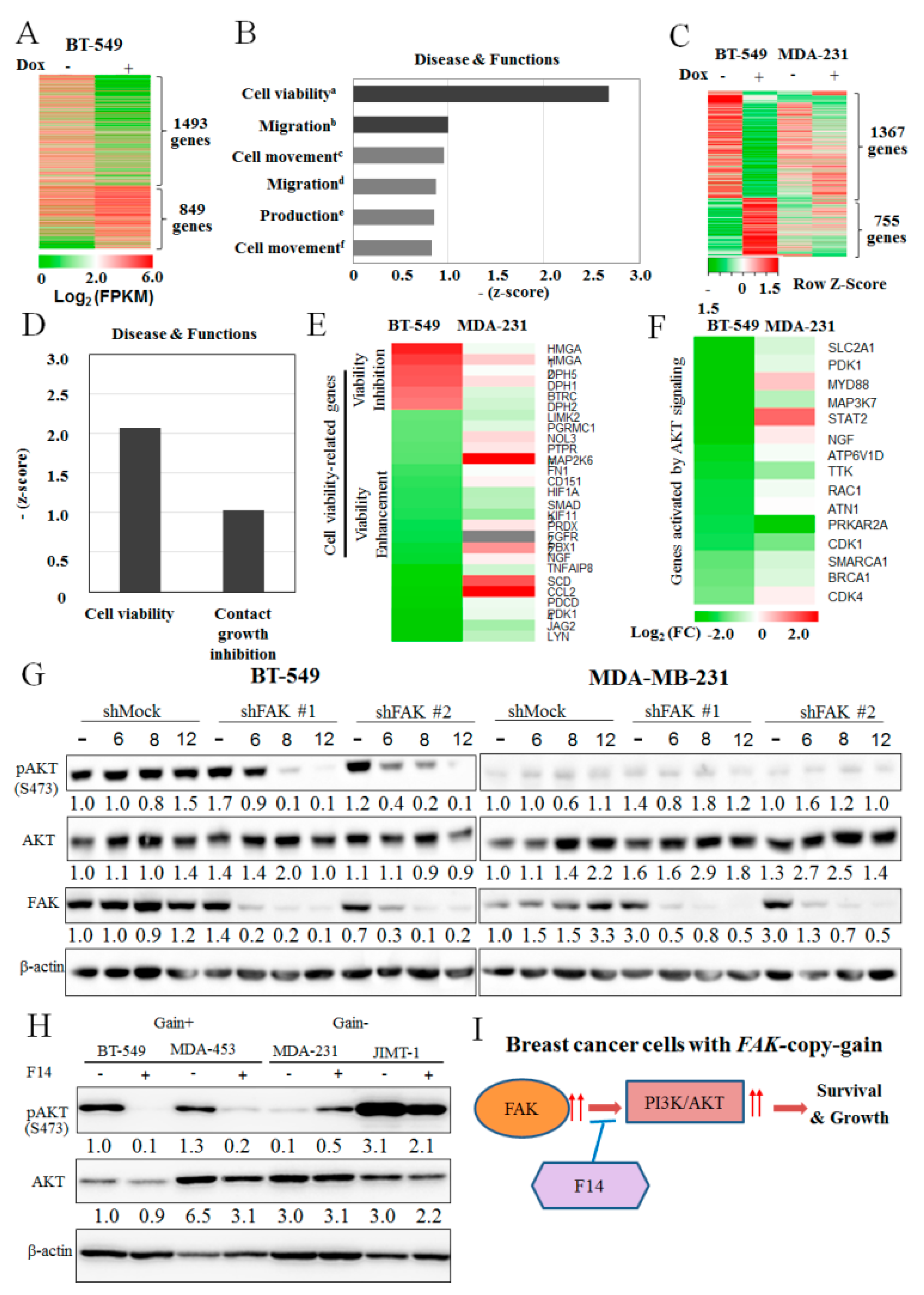
2.4. Downstream Molecular Changes by FAK-Knockdown in FAK-Copy-Gain Cells
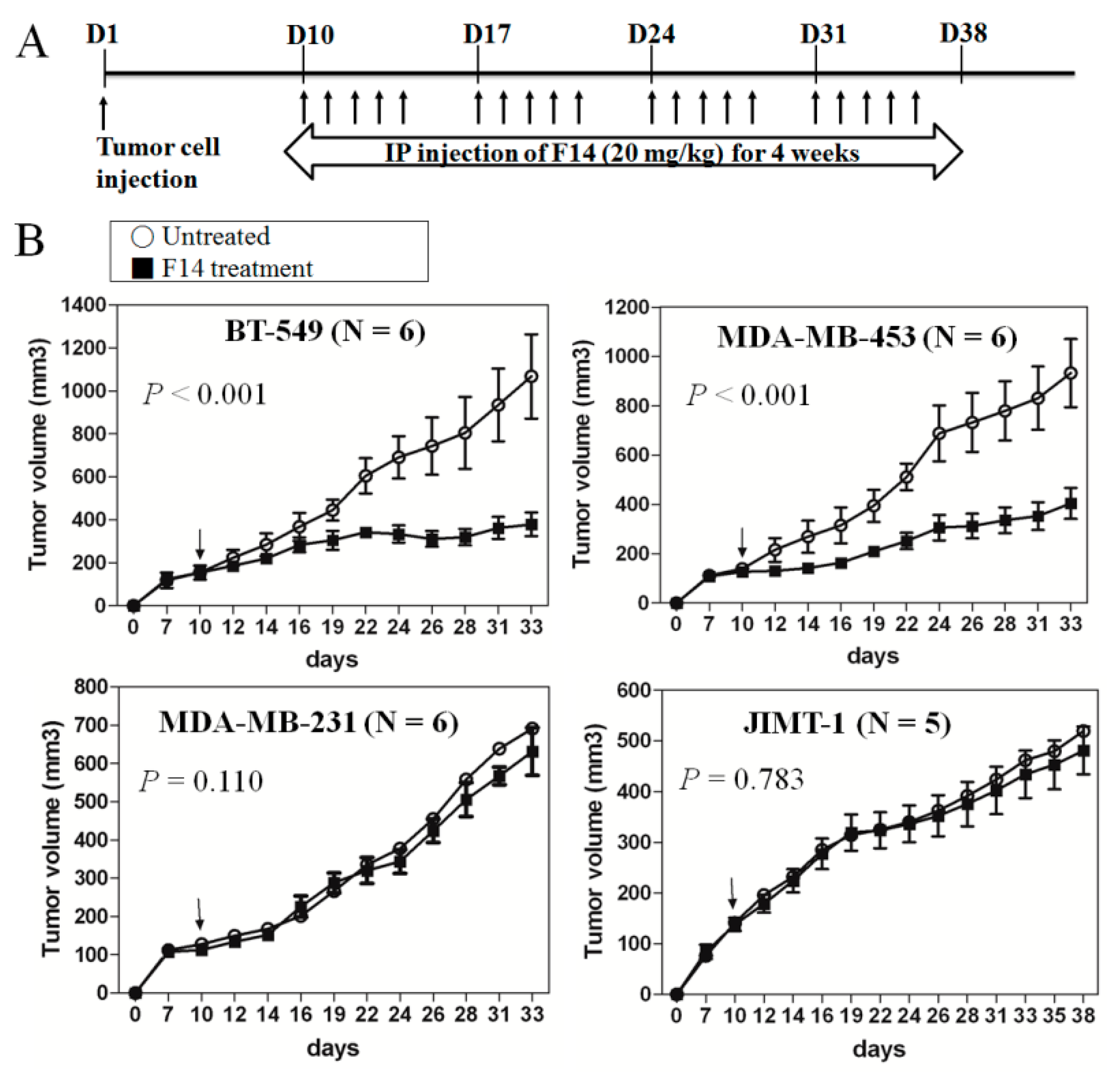
2.5. Effect of F14 On Tumor Progression in a Mouse Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. Determination of Half-Maximal Inhibitory Concentration (IC50)
4.3. Colony-Formation Assay
4.4. Evaluation of FAK-Copy-Gain and Expression of FAK mRNA and Protein
4.5. Acquisition of Databases for the Analysis of the Prevalence of FAK-Copy-Gain in Breast Cancer
4.6. Flow Cytometry Analysis for Apoptosis
4.7. Generation of FAK-Conditional Knockdown Cells
4.8. RNA Sequencing to Search for Genes Inhibited by FAK Knockdown
4.9. Tumor Xenograft Experiment on Nude Mice
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pegram, M.D.; Lipton, A.; Hayes, D.F.; Weber, B.L.; Baselga, J.M.; Tripathy, D.; Baly, D.; Baughman, S.A.; Twaddell, T.; Glaspy, J.A.; et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. J. Clin. Oncol. 1998, 16, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Moroni, M.; Veronese, S.; Carnaghi, C.; Bajetta, E.; Luppi, G.; Sobrero, A.; Barone, C.; Cascinu, S.; Colucci, G.; et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J. Clin. Oncol. 2007, 25, 3238–3245. [Google Scholar] [CrossRef] [PubMed]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Natl. Proc. Acad. Sci. USA 2006, 103, 2316–2321. [Google Scholar] [CrossRef] [PubMed]
- Tanizaki, J.; Okamoto, I.; Okamoto, K.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; Nakagawa, K. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J. Thorac. Oncol. 2011, 6, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yashiro, M.; Matsuoka, T.; Tendo, M.; Shimizu, T.; Miwa, A.; Hirakawa, K. A novel molecular targeting compound as K-samII/FGF-R2 phosphorylation inhibitor, Ki23057, for Scirrhous gastric cancer. Gastroenterology 2006, 131, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zou, L.; Li, Q.; Haibe-Kains, B.; Tian, R.; Li, Y.; Desmedt, C.; Sotiriou, C.; Szallasi, Z.; Iglehart, J.D.; et al. Amplification of LAPTM4B and YWHAZ contributes to chemotherapy resistance and recurrence of breast cancer. Nat. Med. 2010, 16, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Hwang, H.L.; Park, S.Y.; Lee, K.M.; Park, W.C.; Kim, H.S.; Um, T.H.; Hong, Y.J.; Lee, J.K.; Joo, S.Y.; et al. Simple and versatile molecular method of copy-number measurement using cloned competitors. PLoS ONE 2013, 8, e69414. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Nyberg, C.; Zheng, M.; Kweh, F.; Magis, A.; Ostrov, D.; Cance, W.G. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 2008, 51, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. Targeting FAK in human cancer: From finding to first clinical trials. Front. Biosci. 2014, 19, 687–706. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nature reviews. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Ylagan, L.; Miller, A.; Hughes, M.; Wilson, J.; Wang, D.; Brese, E.; Bshara, W.; Edge, S.; Morrison, C.; et al. High focal adhesion kinase expression in breast carcinoma is associated with lymphovascular invasion and triple-negative phenotype. BMC Cancer 2014, 14, 769. [Google Scholar] [CrossRef] [PubMed]
- Yom, C.K.; Noh, D.Y.; Kim, W.H.; Kim, H.S. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast Cancer Res. Treat. 2011, 128, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Meyer-Schaller, N.; Kalathur, R.K.R.; Ivanek, R.; Fagiani, E.; Schmassmann, P.; Stillhard, P.; Hafliger, S.; Kraut, N.; Schweifer, N.; et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis 2018, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Guan, J.L. Focal adhesion kinase: A prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010, 289, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Yang, X.; Bradham, C.A.; Brenner, D.A.; Baldwin, A.S.; Craven, R.J.; Cance, W.G. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J. Biol. Chem. 2000, 275, 30597–30604. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Xiang, B.; Zent, R.; Quaranta, V.; Pozzi, A.; Arteaga, C.L. Transforming growth factor beta induces clustering of HER2 and integrins by activating Src-focal adhesion kinase and receptor association to the cytoskeleton. Cancer Res. 2009, 69, 475–482. [Google Scholar] [CrossRef]
- Reiske, H.R.; Kao, S.C.; Cary, L.A.; Guan, J.L.; Lai, J.F.; Chen, H.C. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J. Biol. Chem. 1999, 274, 12361–12366. [Google Scholar] [CrossRef]
- Del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Xia, H.; Nho, R.S.; Kahm, J.; Kleidon, J.; Henke, C.A. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J. Biol. Chem. 2004, 279, 33024–33034. [Google Scholar] [CrossRef]
- Imaizumi, M.; Nishimura, M.; Takeuchi, S.; Murase, M.; Hamaguchi, M. Role of tyrosine specific phosphorylation of cellular proteins, especially EGF receptor and p125FAK in human lung cancer cells. Lung Cancer 1997, 17, 69–84. [Google Scholar] [CrossRef]
- Lark, A.L.; Livasy, C.A.; Calvo, B.; Caskey, L.; Moore, D.T.; Yang, X.; Cance, W.G. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: Immunohistochemistry and real-time PCR analyses. Clin. Cancer Res. 2003, 9, 215–222. [Google Scholar]
- Park, J.H.; Lee, B.L.; Yoon, J.; Kim, J.; Kim, M.A.; Yang, H.K.; Kim, W.H. Focal adhesion kinase (FAK) gene amplification and its clinical implications in gastric cancer. Hum. Pathol. 2010, 41, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)] decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth In Vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.G.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Mak, G.; Soria, J.C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; McWhirter, E.; Welch, S.; Wang, L.; Vovell, S.; Stayner, L.-A.; Ali, S.; Malpage, A.; Makepeace, B.; Ramachandran, M.; et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma (PDAC) (MOBILITY-002 Trial, NCT02428270). J. Clin. Oncol. 2018, 36, 409. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Kim, D.K.; Shin, J.; Bae, S.H.; Kim, H.Y.; Won, B.; Kim, J.K.; Youn, H.D.; Kim, S.T.; Kang, S.W.; et al. OCT4 directly regulates stemness and extracellular matrix-related genes in human germ cell tumours. Biochem. Biophys. Res. Commun. 2018, 503, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, D.K.; Bae, S.H.; Gwak, H.; Jeon, J.H.; Kim, J.K.; Lee, B.I.; You, H.J.; Shin, D.H.; Kim, Y.H.; et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp. Mol. Med. 2018, 50, 137. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-H.; Kim, H.-K.; Kim, H.Y.; Gawk, H.; Bae, S.-H.; Sim, H.W.; Kang, E.-K.; Seoh, J.-Y.; Jang, H.; Hong, K.-M. FAK-Copy-Gain Is a Predictive Marker for Sensitivity to FAK Inhibition in Breast Cancer. Cancers 2019, 11, 1288. https://doi.org/10.3390/cancers11091288
Kim Y-H, Kim H-K, Kim HY, Gawk H, Bae S-H, Sim HW, Kang E-K, Seoh J-Y, Jang H, Hong K-M. FAK-Copy-Gain Is a Predictive Marker for Sensitivity to FAK Inhibition in Breast Cancer. Cancers. 2019; 11(9):1288. https://doi.org/10.3390/cancers11091288
Chicago/Turabian StyleKim, Young-Ho, Hyun-Kyoung Kim, Hee Yeon Kim, HyeRan Gawk, Seung-Hyun Bae, Hye Won Sim, Eun-Kyung Kang, Ju-Young Seoh, Hyonchol Jang, and Kyeong-Man Hong. 2019. "FAK-Copy-Gain Is a Predictive Marker for Sensitivity to FAK Inhibition in Breast Cancer" Cancers 11, no. 9: 1288. https://doi.org/10.3390/cancers11091288
APA StyleKim, Y.-H., Kim, H.-K., Kim, H. Y., Gawk, H., Bae, S.-H., Sim, H. W., Kang, E.-K., Seoh, J.-Y., Jang, H., & Hong, K.-M. (2019). FAK-Copy-Gain Is a Predictive Marker for Sensitivity to FAK Inhibition in Breast Cancer. Cancers, 11(9), 1288. https://doi.org/10.3390/cancers11091288