Glycan-Modified Apoptotic Melanoma-Derived Extracellular Vesicles as Antigen Source for Anti-Tumor Vaccination

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
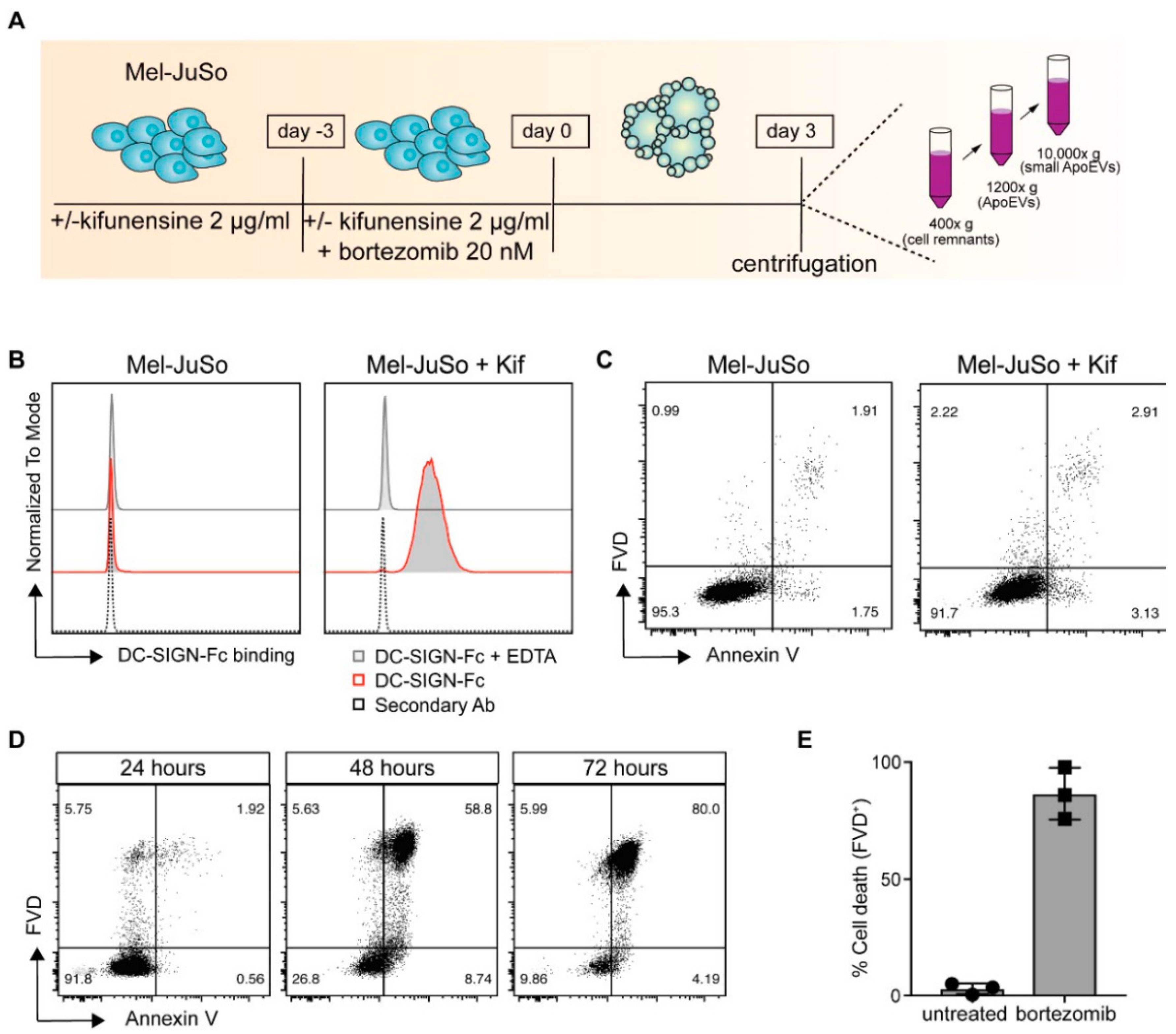
2.1. Kifunensine-Treated Melanoma Cells Express High-Mannose Carbohydrate Structures on Their Cell Surface
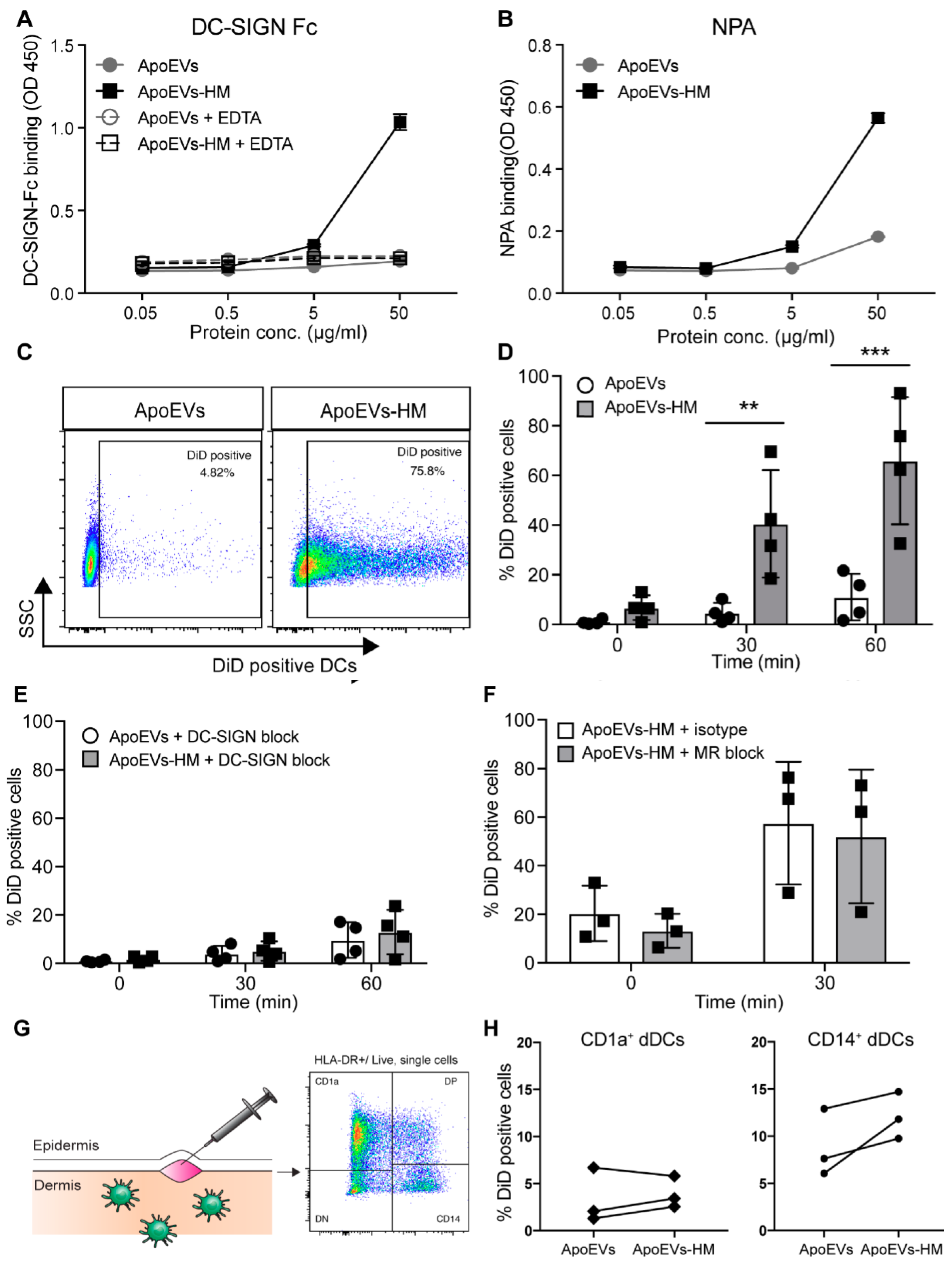
2.2. Glycan Modification Results in ApoEVs with DC-SIGN Binding Properties
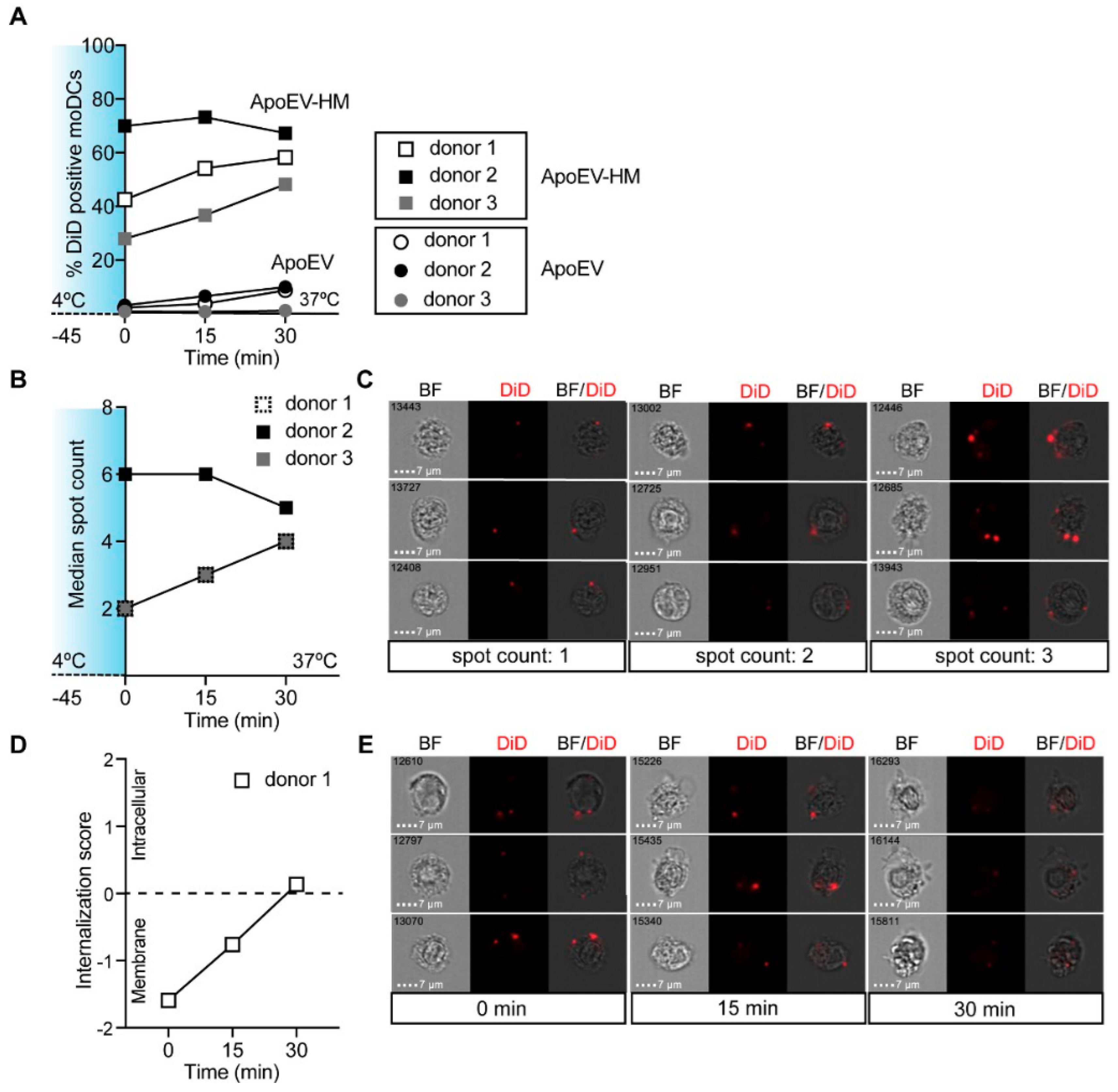
2.3. High-Mannose Expressing ApoEVs Are Internalized via DC-SIGN by moDCs
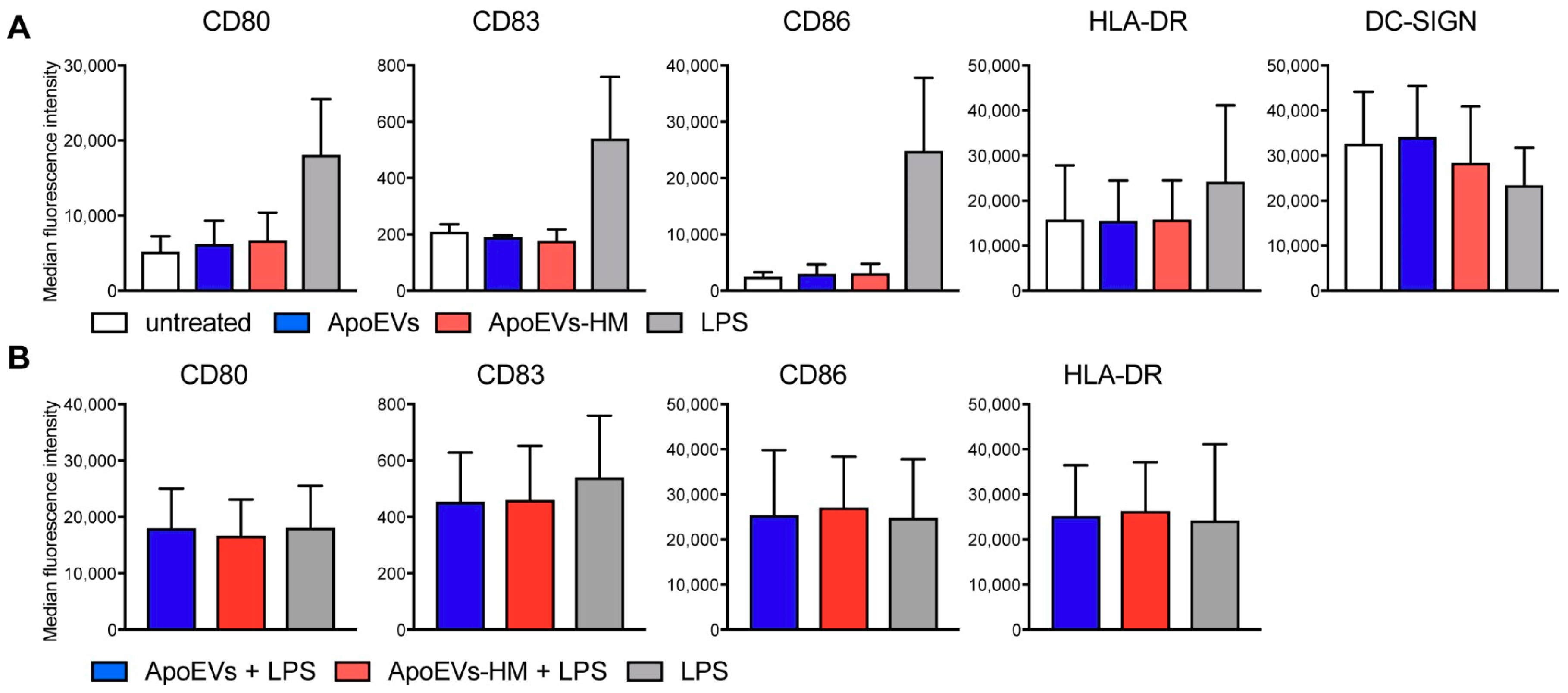
2.4. Mel-JuSo-Derived ApoEVs Do Not Mature moDCs
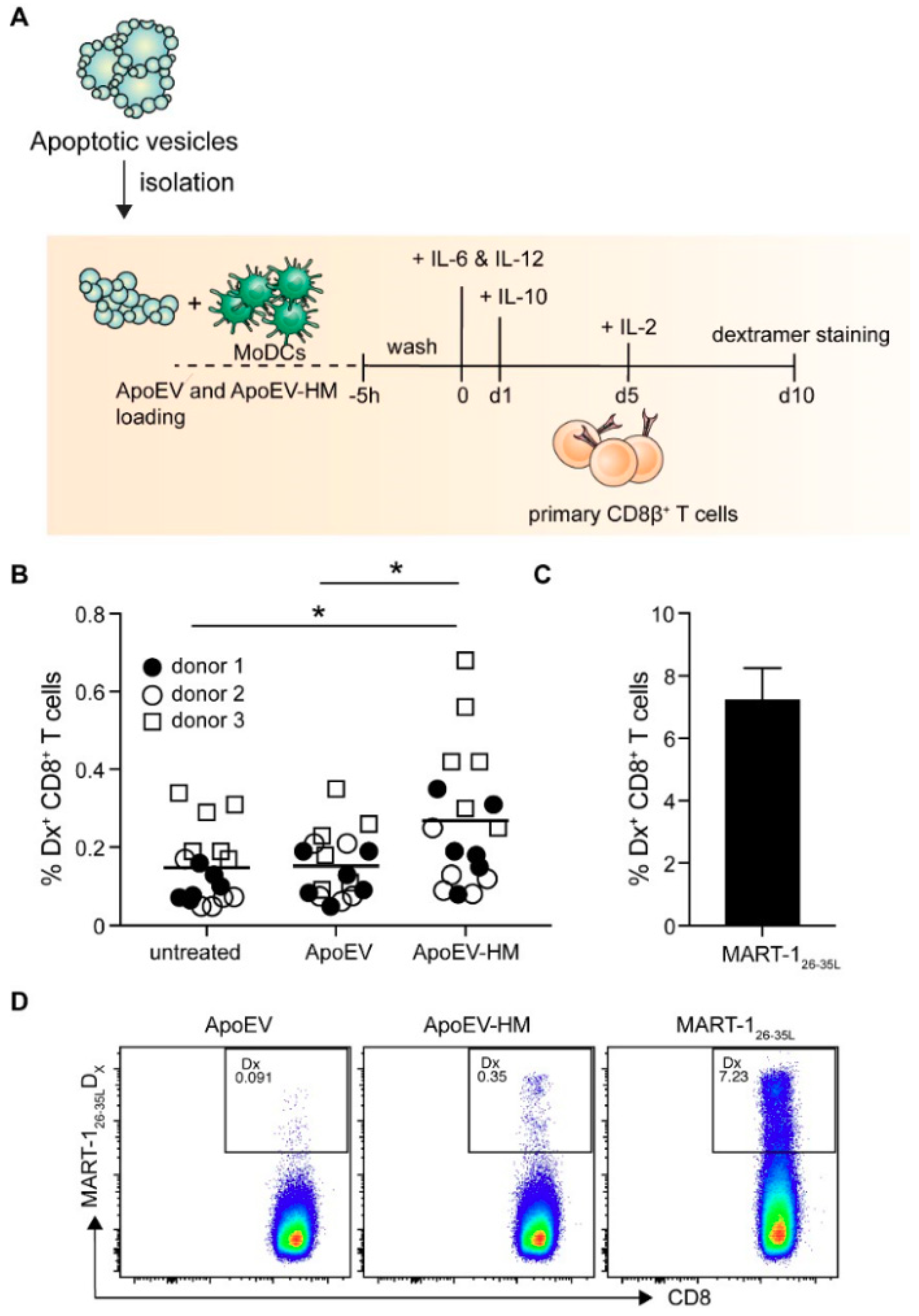
2.5. ApoEV-HM-Loaded moDCs Induce MART-1-Specific CD8+ T Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines, Lentiviral Transduction, Glycan Modification, and Apoptosis Induction
4.3. ApoEV Isolation Protocol
4.4. Monocyte-Derived DCs
4.5. Lectin Binding Assays
4.6. Binding and Uptake of ApoEVs
4.7. In Situ Dermal Dendritic Cell Targeting
4.8. MoDC Maturation Assay
4.9. Mixed Leukocyte Reaction (MLR) and Cytokine Secretion
4.10. Induction of MART-1-Specific CD8+ Effector T Cells
4.11. Imaging Flow Cytometry
4.12. Western Blot
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wolchock, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Van Dinther, D.; Stolk, D.A.; Van De Ven, R.; Van Kooyk, Y.; De Gruijl, T.D.; Den Haan, J.M.M. Targeting C-type lectin receptors: A high-carbohydrate diet for dendritic cells to improve cancer vaccines. J. Leukoc. Biol. 2017, 102, 1017–1034. [Google Scholar] [CrossRef]
- Unger, W.W.J.; Mayer, C.T.; Engels, S.; Hesse, C.; Perdicchio, M.; Puttur, F.; Streng-ouwehand, I.; Litjens, M.; Kalay, H.; Berod, L.; et al. Antigen targeting to dendritic cells combined with transient regulatory T cell inhibition results in long-term tumor regression. Oncoimmunology 2015, 4, e970462. [Google Scholar] [CrossRef]
- Fehres, C.M.; Kalay, H.; Bruijns, S.C.M.; Musaafir, S.A.M.; Ambrosini, M.; van Bloois, L.; van Vliet, S.J.; Storm, G.; Garcia-Vallejo, J.J.; van Kooyk, Y. Cross-presentation through langerin and DC-SIGN targeting requires different formulations of glycan-modified antigens. J. Control. Release 2015, 203, 67–76. [Google Scholar] [CrossRef]
- Van Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient Targeting of Protein Antigen to the Dendritic Cell Receptor DEC-205 in the Steady State Leads to Antigen Presentation on Major Histocompatibility Complex Class I Products and Peripheral CD8+ T Cell Tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In Vivo Targeting of Antigens to Maturing Dendritic Cells via the DEC-205 Receptor Improves T Cell Vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Vallejo, J.J.; Unger, W.W.J.; Kalay, H.; van Kooyk, Y. Glycan-based DC-SIGN targeting vaccines to enhance antigen cross-presentation. Oncoimmunology 2013, 2, e23040. [Google Scholar] [CrossRef] [PubMed]
- Engering, A.; Van Vliet, S.J.; Hebeda, K.; Jackson, D.G.; Prevo, R.; Singh, S.K.; Geijtenbeek, T.B.H.; Van Krieken, H.; Van Kooyk, Y. Dynamic Populations of Dendritic Cell-Specific ICAM-3 Grabbing Nonintegrin-Positive Immature Dendritic Cells and Liver/Lymph Node-Specific ICAM-3 Grabbing Nonintegrin-Positive Endothelial Cells in the Outer Zones of the Paracortex of Human Lymph Nodes. Am. J. Pathol. 2004, 164, 1587–1595. [Google Scholar] [CrossRef] [Green Version]
- Fehres, C.M.; Garcia-Vallejo, J.J.; Unger, W.W.J.; van Kooyk, Y. Skin-resident antigen-presenting cells: Instruction manual for vaccine development. Front. Immunol. 2013, 4, 157. [Google Scholar] [CrossRef]
- Fehres, C.M.; Van Beelen, A.J.; Bruijns, S.C.M.; Ambrosini, M.; Kalay, H.; Van Bloois, L.; Unger, W.W.J.; Garcia-vallejo, J.J.; Storm, G.; De Gruijl, T.D.; et al. In situ Delivery of Antigen to DC-SIGN + CD14 + Dermal Dendritic Cells Results in Enhanced CD8 + T-Cell Responses. J. Investig. Dermatol. 2015, 135, 2228–2236. [Google Scholar] [CrossRef] [Green Version]
- Appelmelk, B.J.; van Die, I.; van Vliet, S.J.; Vandenbroucke-Grauls, C.M.J.E.; Geijtenbeek, T.B.H.; van Kooyk, Y. Cutting Edge: Carbohydrate Profiling Identifies New Pathogens That Interact with Dendritic Cell-Specific ICAM-3-Grabbing Nonintegrin on Dendritic Cells. J. Immunol. 2003, 170, 1635–1639. [Google Scholar] [CrossRef] [Green Version]
- Aarnoudse, C.A.; Bax, M.; Sánchez-Hernández, M.; García-Vallejo, J.J.; Van Kooyk, Y. Glycan modification of the tumor antigen gp100 targets DC-SIGN to enhance dendritic cell induced antigen presentation to T cells. Int. J. Cancer 2008, 122, 839–846. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2014, 160, 48–61. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; Van De Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nat. Publ. Gr. 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Ruben, J.M.; Van Den Ancker, W.; Bontkes, H.J.; Westers, T.M.; Hooijberg, E.; Ossenkoppele, G.J.; De Gruijl, T.D.; Van De Loosdrecht, A.A. Apoptotic blebs from leukemic cells as a preferred source of tumor-associated antigen for dendritic cell-based vaccines. Cancer Immunol. Immunother. 2014, 63, 335–345. [Google Scholar] [CrossRef]
- Ruben, J.M.; Bontkes, H.J.; Westers, T.M.; Hooijberg, E.; Ossenkoppele, G.J.; van de Loosdrecht, A.A.; de Gruijl, T.D. In situ loading of skin dendritic cells with apoptotic bleb-derived antigens for the induction of tumor-directed immunity. Oncoimmunology 2014, 4, 1–10. [Google Scholar]
- Atkin-Smith, G.K.; Poon, I.K.H. Disassembly of the Dying: Mechanisms and Functions. Trends Cell Biol. 2017, 27, 151–162. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Herrmann, M. Therapeutic Prospects of extracellular vesicles in Cancer Treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef]
- Caruso, S.; Poon, I.K.H. Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Front. Immunol. 2018, 9, 1486. [Google Scholar] [Green Version]
- Muhsin-Sharafaldine, M.-R.; Mclellan, A.D. Tumor-Derived Apoptotic vesicles: With Death They Do Part. Front. Immunol. 2018, 9, 957. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Elbein, A.D.; Tropea, J.E.; Mitchell, M.; Kaushal, G.P. Kifunensine, a Potent Inhibitor Mannosidase I * of the Glycoprotein Processing. J. Biol. Chem. 1990, 265, 15599–15605. [Google Scholar] [PubMed]
- Geijtenbeek, T.B.H.; Torensma, R.; Van Vliet, S.J.; Van Duijnhoven, G.C.F.; Adema, G.J.; Van Kooyk, Y.; Figdor, C.G. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 2000, 100, 575–585. [Google Scholar] [CrossRef]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Livia, M.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Garg, A.D.; More, S.; et al. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Horrevorts, S.K.; Duinkerken, S.; Bloem, K.; Secades, P.; Kalay, H.; Musters, R.J.; van Vliet, S.J.; García-Vallejo, J.J.; van Kooyk, Y. Toll-like receptor 4 triggering promotes cytosolic routing of DC-SIGN-targeted antigens for presentation on MHC class I. Front. Immunol. 2018, 9, 1231. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Hangalapura, B.N.; Oosterhoff, D.; Aggarwal, S.; Wijnands, P.G.J.T.B.; van de Ven, R.; Santegoets, S.J.A.M.; van den Tol, M.P.; Hooijberg, E.; Pereboev, A.; van de Eertwegh, A.J.M.; et al. Selective transduction of dendritic cells in human lymph nodes and superior induction of high-avidity melanoma-reactive cytotoxic t cells by a cd40-targeted adenovirus. J. Immunother. 2010, 33, 706–715. [Google Scholar] [CrossRef]
- Fransen, J.H.; Hilbrands, L.B.; Ruben, J.; Stoffels, M.; Adema, G.J.; Van Der Vlag, J.; Berden, J.H. Mouse dendritic cells matured by ingestion of apoptotic blebs induce T cells to produce interleukin-17. Arthritis Rheum. 2009, 60, 2304–2313. [Google Scholar] [CrossRef] [PubMed]
- Fehr, E.M.; Spoerl, S.; Heyder, P.; Herrmann, M.; Bekeredjian-Ding, I.; Blank, N.; Lorenz, H.M.; Schiller, M. Apoptotic-cell-derived membrane vesicles induce an alternative maturation of human dendritic cells which is disturbed in SLE. J. Autoimmun. 2013, 40, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Berda-Haddad, Y.; Robert, S.; Salers, P.; Zekraoui, L.; Farnarier, C.; Dinarello, C.A.; Dignat-George, F.; Kaplanski, G. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1. Proc. Natl. Acad. Sci. USA 2011, 108, 20684–20689. [Google Scholar] [CrossRef] [PubMed]
- Municio, C.; Hugo, E.; Alvarez, Y.; Alonso, S.; Blanco, L.; Fernández, N.; Sánchez Crespo, M. Apoptotic cells enhance IL-10 and reduce IL-23 production in human dendritic cells treated with zymosan. Mol. Immunol. 2011, 49, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Wallet, M.A.; Yi, Z.; Huang, Y.; Henderson, M.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Baldwin, A.S.; Tisch, R.M. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-κB activation in dendritic cells. Blood 2007, 109, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.M.; Lucas, M.; Simpson, C.; Lamb, J.; Savill, J.; Lacy-Hulbert, A. Inhibitory Effects of Apoptotic Cell Ingestion upon Endotoxin-Driven Myeloid Dendritic Cell Maturation. J. Immunol. 2002, 168, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Muhsin-Sharafaldine, M.-R.; Saunderson, S.C.; Dunn, A.C.; Faed, J.M.; Kleffmann, T.; McLellan, A.D. Procoagulant and immunogenic properties of melanoma exosomes, microvesicles and apoptotic vesicles. Oncotarget 2016, 7, 56279–56294. [Google Scholar] [CrossRef]
- Verdegaal, E.M.E.; De Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; Van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; Van Der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.H.; Van Duijnhoven, G.C.F.; Van Vliet, S.J.; Krieger, E.; Vriend, G.; Figdor, C.G.; Van Kooyk, Y. Identification of different binding sites in the dendritic cell-specific receptor DC-SIGN for intercellular adhesion molecule 3 and HIV-1. J. Biol. Chem. 2002, 277, 11314–11320. [Google Scholar] [CrossRef]
- Yssel, H.; De Vries, J.E.; Koken, M.; Van Blitterswijk, W.; Spits, H. Serum-Free Medium for Generation and Propagation of Functional Human Cytotoxic and Helper T cell clones. J. Immunol. Methods 1984, 72, 219–227. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horrevorts, S.K.; Stolk, D.A.; van de Ven, R.; Hulst, M.; van Het Hof, B.; Duinkerken, S.; Heineke, M.H.; Ma, W.; Dusoswa, S.A.; Nieuwland, R.; et al. Glycan-Modified Apoptotic Melanoma-Derived Extracellular Vesicles as Antigen Source for Anti-Tumor Vaccination. Cancers 2019, 11, 1266. https://doi.org/10.3390/cancers11091266
Horrevorts SK, Stolk DA, van de Ven R, Hulst M, van Het Hof B, Duinkerken S, Heineke MH, Ma W, Dusoswa SA, Nieuwland R, et al. Glycan-Modified Apoptotic Melanoma-Derived Extracellular Vesicles as Antigen Source for Anti-Tumor Vaccination. Cancers. 2019; 11(9):1266. https://doi.org/10.3390/cancers11091266
Chicago/Turabian StyleHorrevorts, Sophie K., Dorian A. Stolk, Rieneke van de Ven, Myrthe Hulst, Bert van Het Hof, Sanne Duinkerken, Marieke H. Heineke, Wenbin Ma, Sophie A. Dusoswa, Rienk Nieuwland, and et al. 2019. "Glycan-Modified Apoptotic Melanoma-Derived Extracellular Vesicles as Antigen Source for Anti-Tumor Vaccination" Cancers 11, no. 9: 1266. https://doi.org/10.3390/cancers11091266
APA StyleHorrevorts, S. K., Stolk, D. A., van de Ven, R., Hulst, M., van Het Hof, B., Duinkerken, S., Heineke, M. H., Ma, W., Dusoswa, S. A., Nieuwland, R., Garcia-Vallejo, J. J., van de Loosdrecht, A. A., de Gruijl, T. D., van Vliet, S. J., & van Kooyk, Y. (2019). Glycan-Modified Apoptotic Melanoma-Derived Extracellular Vesicles as Antigen Source for Anti-Tumor Vaccination. Cancers, 11(9), 1266. https://doi.org/10.3390/cancers11091266