Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer

 , ,
, ,
Abstract
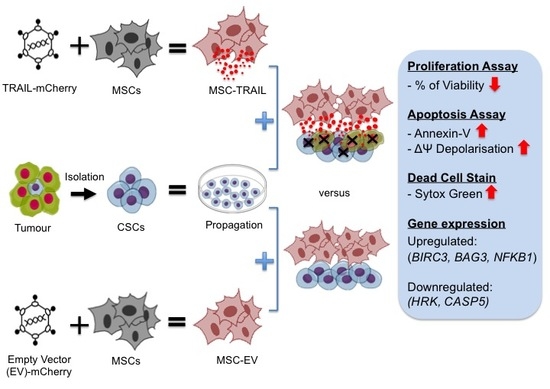
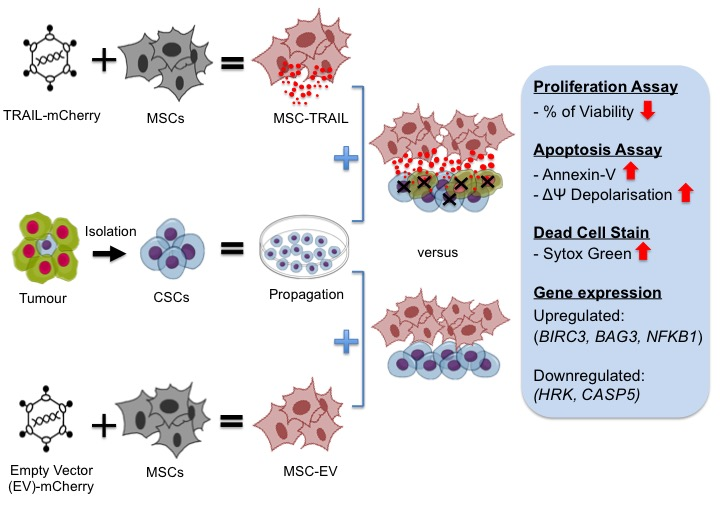{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
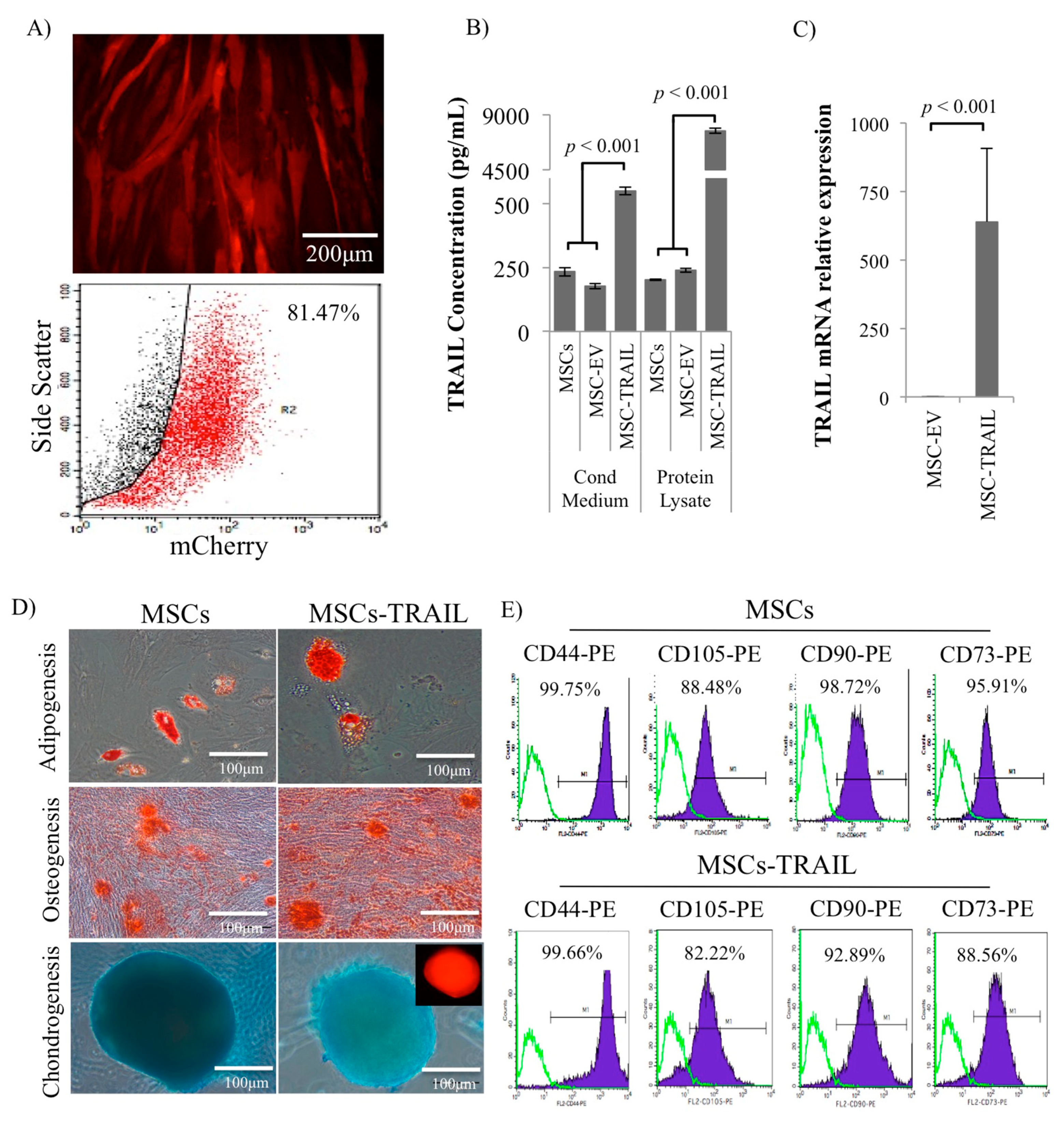
2.1. Characterization of MSC-TRAIL
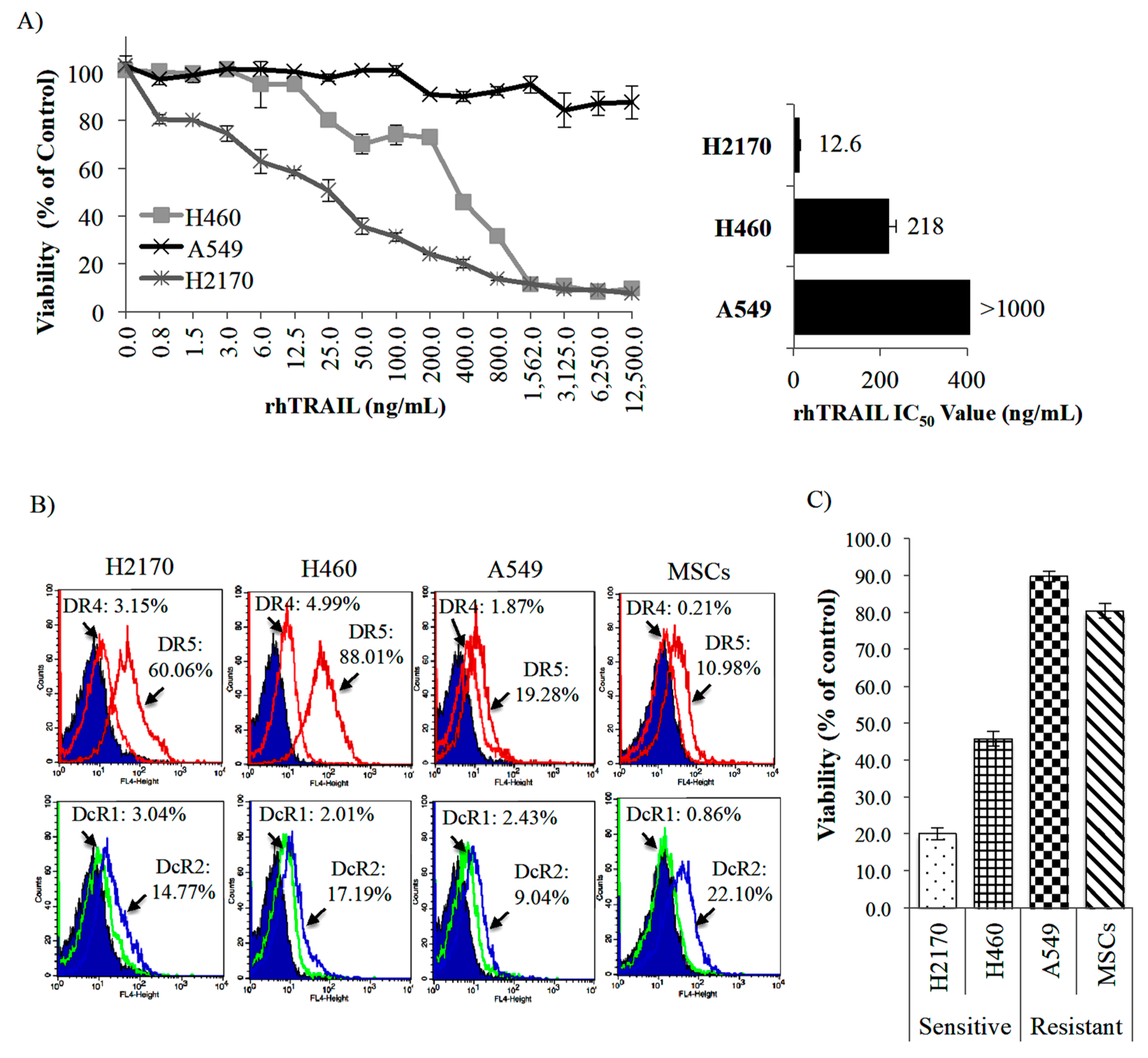
2.2. Targeting of NSCLC Cell Lines by Recombinant Human (rh) TRAIL (rhTRAIL) and Its TRAIL Receptors Expression
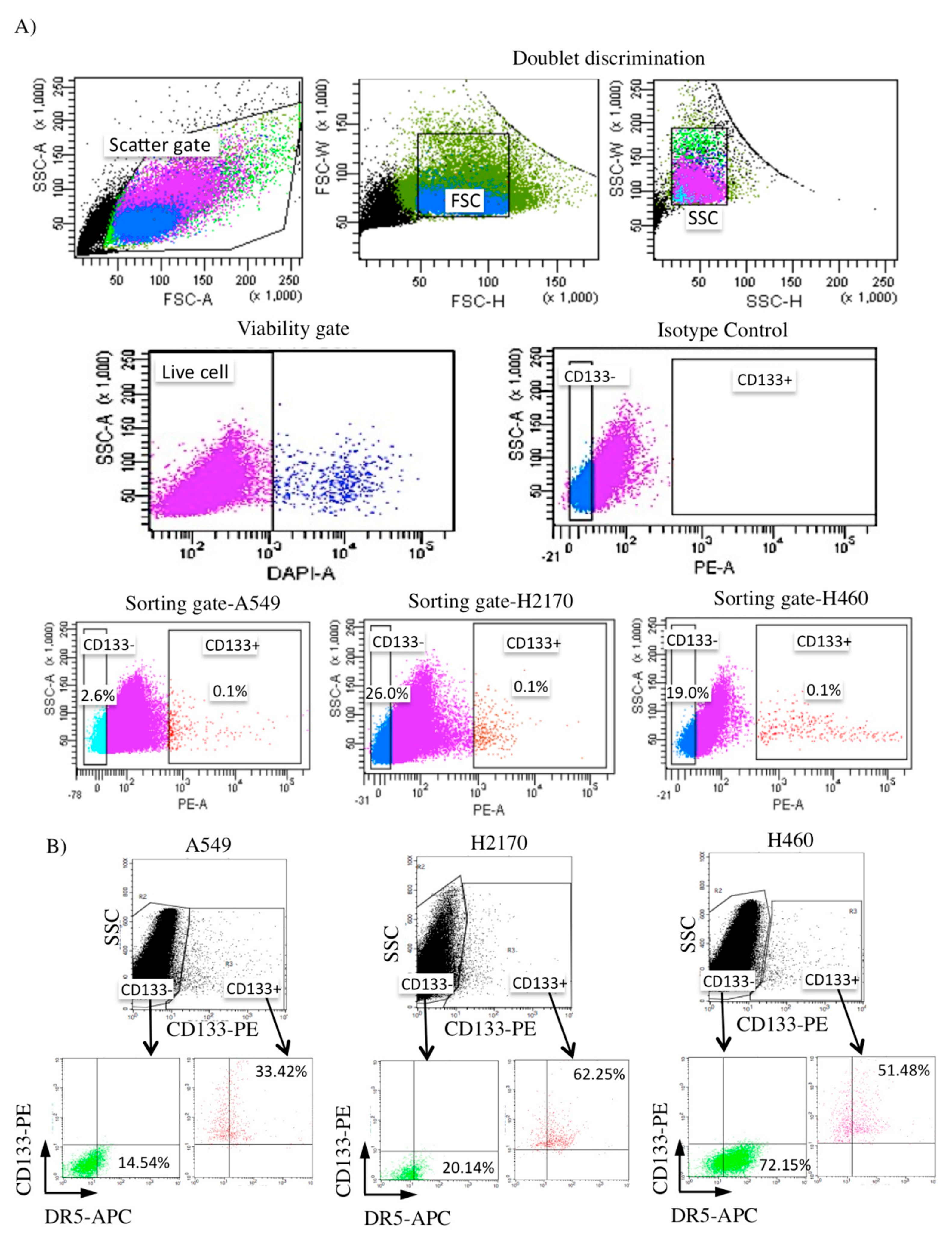
2.3. Isolation of CD133+ from NSCLC Cell Lines and Its DR5 Receptor Expression
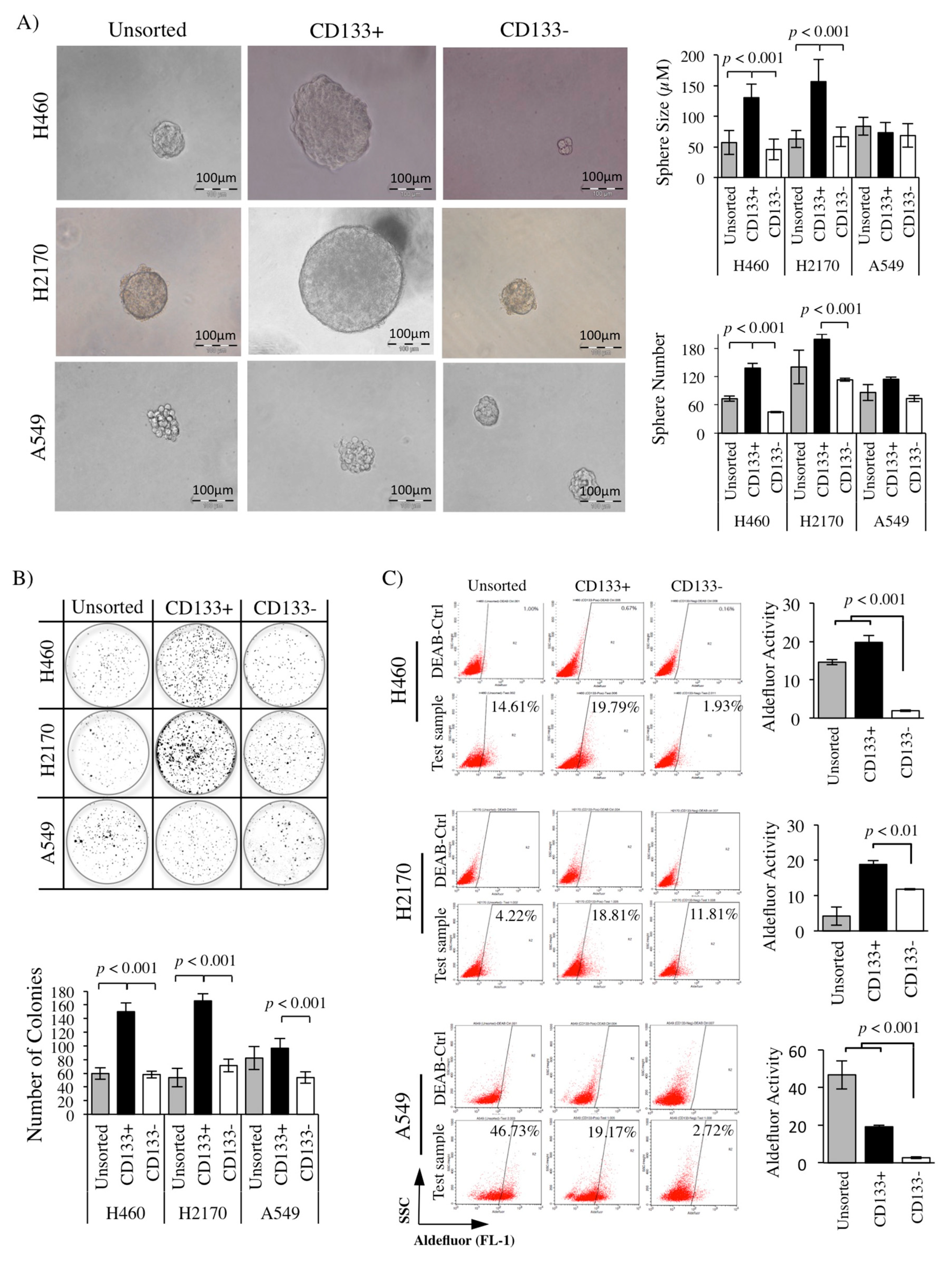
2.4. Greater Sphere Formation and Clonogenicity in CD133+ NSCLC-Derived CSCs
2.5. High ALDH Activity Detected in the CD133+ NSCLC-Derived CSCs
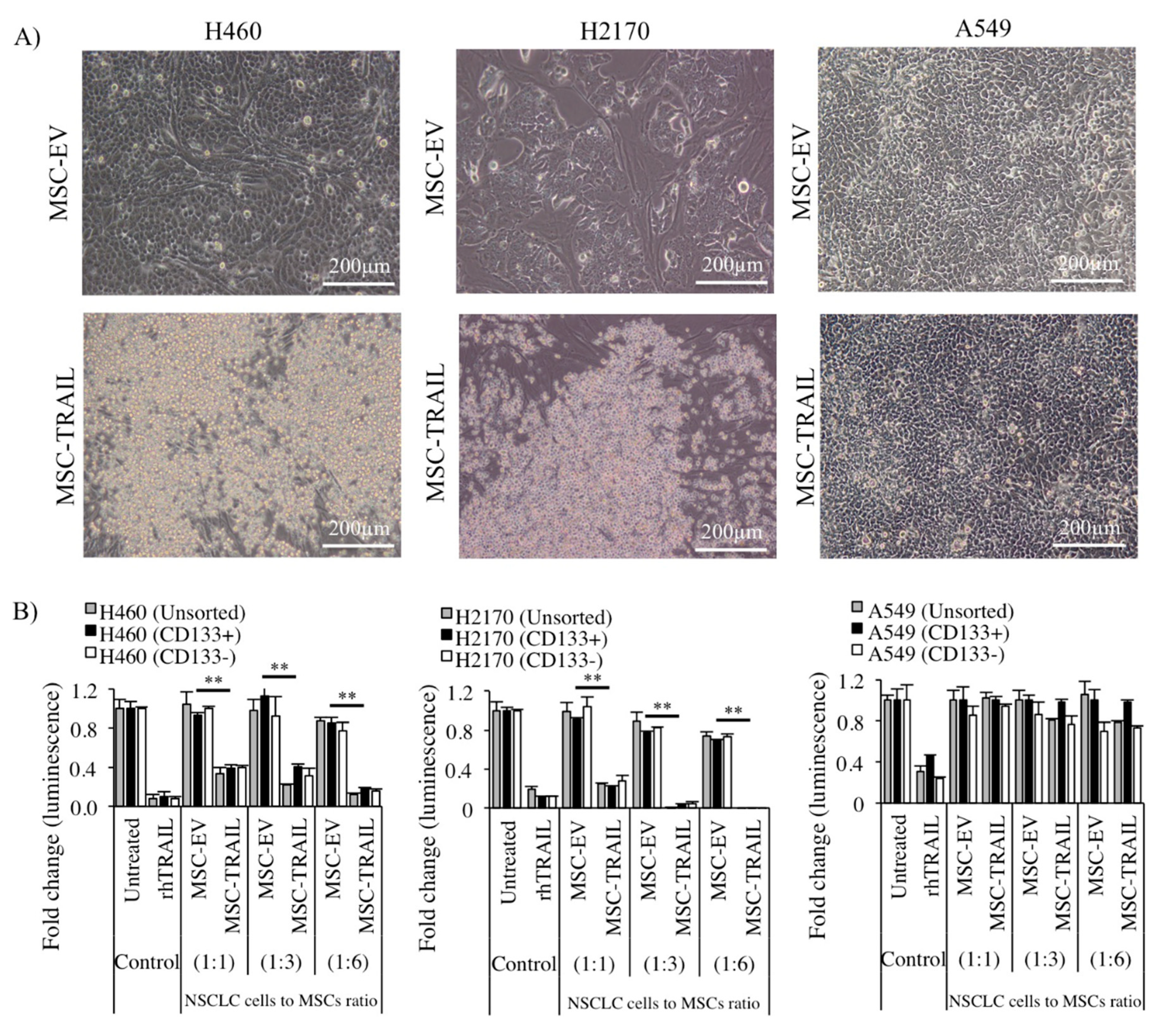
2.6. Inhibition of CD133+ CSC Proliferation by MSC-TRAIL
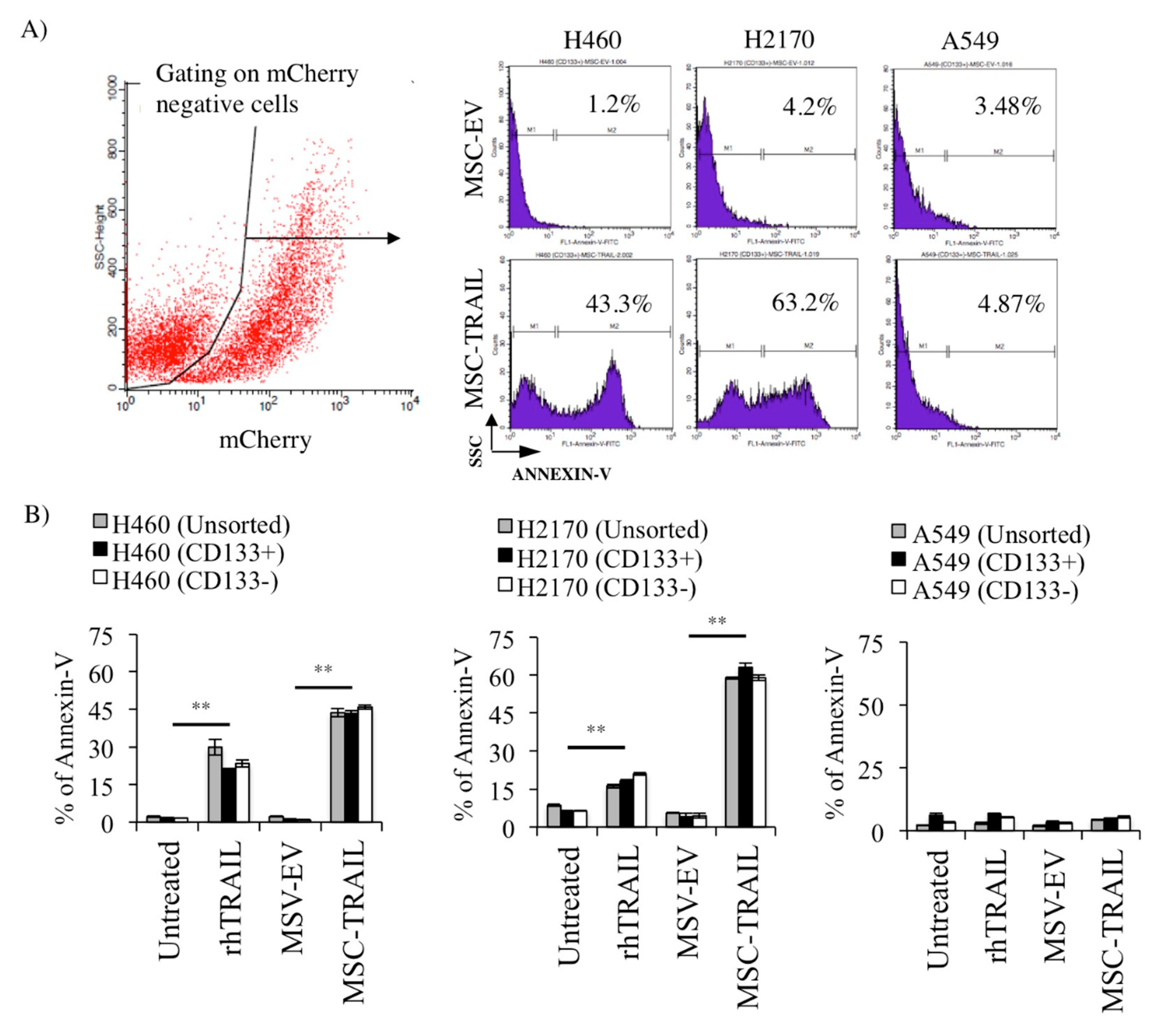
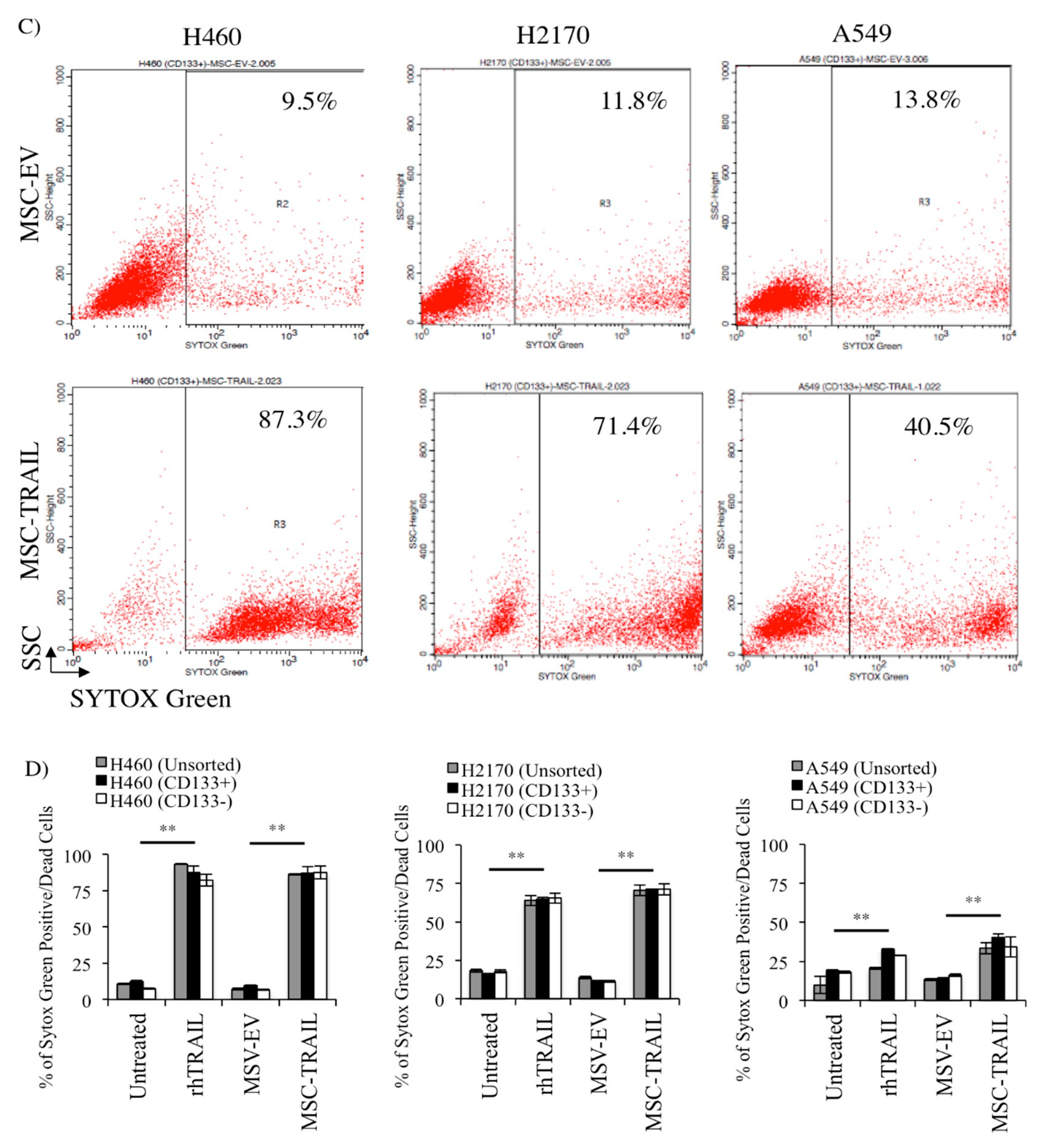
2.7. MSC-TRAIL Induced Annexin V Expression in CD133+ CSCs
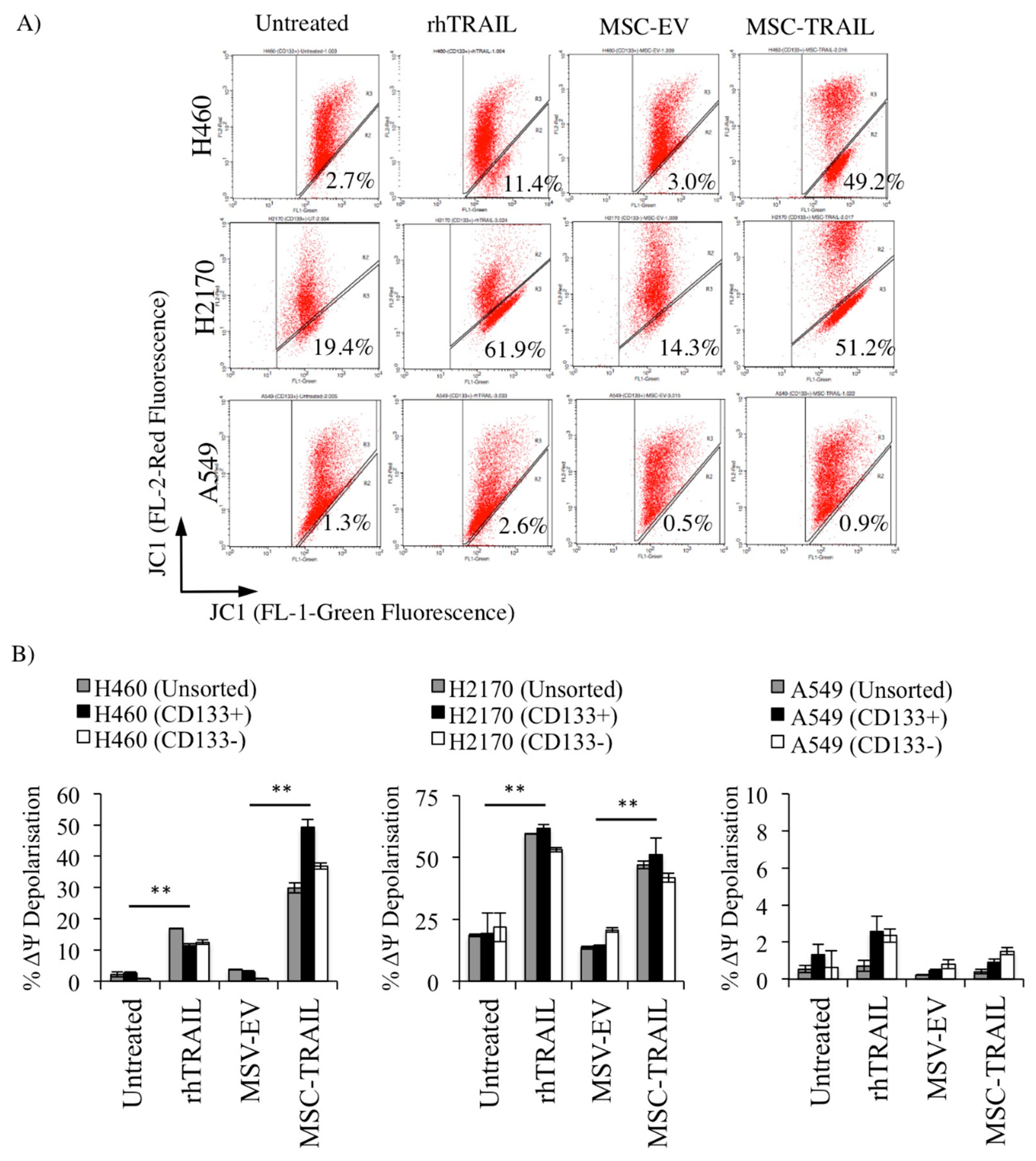
2.8. MSC-TRAIL Induced Intrinsic Apoptosis in CD133+ NSCLC-Derived CSCs
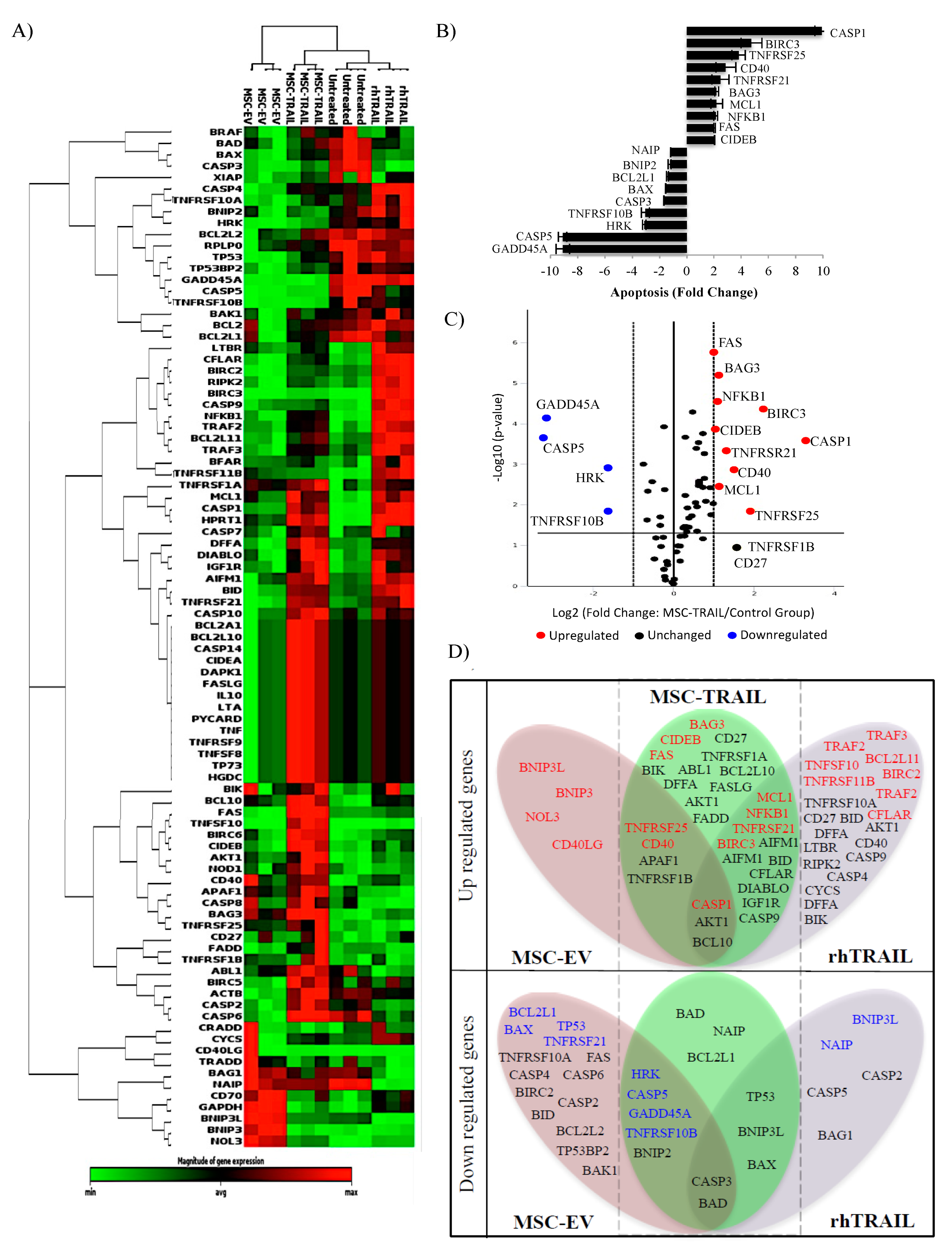
2.9. Specific Apoptotic Molecules Regulated in CD133+ H460-Derived CSCs by MSC-TRAIL
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Lentivirus Production and MSC Transduction
4.3. Characterizations of MSC-TRAIL
4.4. Isolation of CD133+ NSCLC-Derived CSCs
4.5. TRAIL Agonist (DR4 and DR5) and Decoy (DcR1 and DcR2) Receptors Expression in NSCLC Cell Lines
4.6. CSC Characterization (Sphere Formation and Clonogenic Assays)
4.7. Aldehyde Dehydrogenase (ALDH) Activity (Aldefluor Assay)
4.8. Cell Proliferation/MTS Assay
4.9. Luciferase Assay
4.10. Apoptosis and Dead Cell Analysis
4.11. Mitochondria Membrane Potential (ΔΨ)
4.12. RT2 Profiler PCR Array
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Mountain, C.F.; Lukeman, J.M.; Hammar, S.P.; Chamberlain, D.W.; Coulson, W.F.; Page, D.L.; Victor, T.A.; Weiland, L.H.; Lung Cancer Study Group Pathology Committee. Lung Cancer Classification: The Relationship of Disease Extent and Cell Type to Survival in a Clinical Trials Population. J. Surg. Oncol. 1987, 35, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Mascaux, C.; Tomasini, P.; Greillier, L.; Barlesi, F. Personalised Medicine for Nonsmall Cell Lung Cancer. Eur. Respir. Rev. 2017, 26, 170066. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, J.; Sun, S.; Li, F.; Cao, W.; Wang, Y.U.; Ma, Z.; Yu, Z. Mesenchymal Stem Cells Expressing Interleukin-18 Suppress Breast Cancer Cells in Vitro. Exp. Ther. Med. 2015, 9, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Chen, Y.; Lu, L.; Hu, X.; Shao, C.; Zhang, Y.; Zhou, X.; Zhou, Y.; Wu, L.; Liu, R.; et al. Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells Producing IL15 Eradicate Established Pancreatic Tumor in Syngeneic Mice. Mol. Cancer Ther. 2014, 13, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Studeny, M.; Marini, F.C.; Champlin, R.E.; Zompetta, C.; Fidler, I.J.; Andreeff, M. Bone Marrow-Derived Mesenchymal Stem Cells as Vehicles for Interferon-β Delivery into Tumors. Cancer Res. 2002, 62, 3603–3608. [Google Scholar] [PubMed]
- Hakkarainen, T.; Sarkioja, M.; Lehenkari, P.; Miettinen, S.; Ylikomi, T.; Suuronen, R.; Desmond, R.A.; Kanerva, A.; Hemminki, A. Human Mesenchymal Stem Cells Lack Tumor Tropism but Enhance the Antitumor Activity of Oncolytic Adenoviruses in Orthotopic Lung and Breast Tumors. Hum. Gene Ther. 2007, 18, 627–641. [Google Scholar] [CrossRef]
- Hoyos, V.; Del Bufalo, F.; Yagyu, S.; Ando, M.; Dotti, G.; Suzuki, M.; Bouchier-Hayes, L.; Alemany, R.; Brenner, M.K. Mesenchymal Stromal Cells for Linked Delivery of Oncolytic and Apoptotic Adenoviruses to Non-Small-Cell Lung Cancers. Mol. Ther. 2015, 23, 1497–1506. [Google Scholar] [CrossRef]
- Schwartz, R.N.; Stover, L.; Dutcher, J.P. Managing Toxicities of High-Dose Interleukin-2. Oncology 2002, 16, 11–20. [Google Scholar]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12-Associated Toxicity and Interferon-Gamma Production. Blood 1997, 90, 2541–2548. [Google Scholar]
- Chernajovsky, Y.; Layward, L.; Lemoine, N. Fighting Cancer with Oncolytic Viruses. BMJ 2006, 332, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-S.; Wang, X.-W.; He, W.; Ma, X.-T.; Wang, H.-Y.; Han, M.; Li, B.-H. TRAIL Inhibits Platelet-Induced Colorectal Cancer Cell Invasion. J. Int. Med. Res. 2019, 47, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Pan, G.; Weddle, J.J.; Dixit, V.M.; Cavenee, W.K.; Huang, H.J. Increased Death Receptor 5 Expression by Chemotherapeutic Agents in Human Gliomas Causes Synergistic Cytotoxicity with Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Vitro and in Vivo. Cancer Res. 2000, 60, 847–853. [Google Scholar] [PubMed]
- Voortman, J.; Resende, T.P.; Abou El Hassan, M.A.; Giaccone, G.; Kruyt, F.A. TRAIL Therapy in Non-Small Cell Lung Cancer Cells: Sensitization to Death Receptor-Mediated Apoptosis by Proteasome Inhibitor Bortezomib. Mol. Cancer Ther. 2007, 6, 2103–2112. [Google Scholar] [CrossRef] [PubMed]
- Gura, T. How TRAIL Kills Cancer Cells, but Not Normal Cells. Science 1997, 277, 768. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.K.; Harris, L.A.; Xie, D.; Deforge, L.; Totpal, K.; Bussiere, J.; Fox, J.A. Preclinical Studies to Predict the Disposition of Apo2L/Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Humans: Characterization of in Vivo Efficacy, Pharmacokinetics, and Safety. J. Pharmacol. Exp. Ther. 2001, 299, 31–38. [Google Scholar] [PubMed]
- Perlstein, B.; Finniss, S.A.; Miller, C.; Okhrimenko, H.; Kazimirsky, G.; Cazacu, S.; Lee, H.K.; Lemke, N.; Brodie, S.; Umansky, F.; et al. TRAIL Conjugated to Nanoparticles Exhibits Increased Anti-Tumor Activities in Glioma Cells and Glioma Stem Cells in Vitro and in Vivo. Neuro Oncol. 2013, 15, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, M.A.; Cochran, M.C.; Eisenbrey, J.R.; Oum, K.L. Cellular Signal Transduction Can Be Induced by TRAIL Conjugated to Microcapsules. J. Biomed. Mater. Res. A 2012, 100, 2602–2611. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Gerspach, J.; Pfizenmaier, K. Engineering Death Receptor Ligands for Cancer Therapy. Cancer Lett. 2013, 332, 163–174. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal Activity of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudifar, N.; Doran, P.M. Mesenchymal Stem Cells Derived from Human Adipose Tissue. Methods Mol. Biol. 2015, 1340, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Trivanovic, D.; Kocic, J.; Mojsilovic, S.; Krstic, A.; Ilic, V.; Djordjevic, I.O.; Santibanez, J.F.; Jovcic, G.; Terzic, M.; Bugarski, D. Mesenchymal Stem Cells Isolated from Peripheral Blood and Umbilical Cord Wharton’s Jelly. Srp. Arh. Celok. Lek. 2013, 141, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.L.; Troyer, D.L. Stem Cells in the Umbilical Cord. Stem Cell Rev. Rep. 2006, 2, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Gnecchi, M.; Melo, L.G. Bone Marrow-Derived Mesenchymal Stem Cells: Isolation, Expansion, Characterization, Viral Transduction, and Production of Conditioned Medium. Methods Mol. Biol. 2009, 482, 281–294. [Google Scholar] [CrossRef]
- Samsonraj, R.M.; Raghunath, M.; Nurcombe, V.; Hui, J.H.; van Wijnen, A.J.; Cool, S.M. Concise Review: Multifaceted Characterization of Human Mesenchymal Stem Cells for Use in Regenerative Medicine. Stem Cells Transl. Med. 2017, 6, 2173–2185. [Google Scholar] [CrossRef]
- Lohan, P.; Treacy, O.; Griffin, M.D.; Ritter, T.; Ryan, A.E. Anti-Donor Immune Responses Elicited by Allogeneic Mesenchymal Stem Cells and Their Extracellular Vesicles: Are We Still Learning? Front. Immunol. 2017, 8, 1626. [Google Scholar] [CrossRef]
- Nauta, A.J.; Westerhuis, G.; Kruisselbrink, A.B.; Lurvink, E.G.A.; Willemze, R.; Fibbe, W.E. Donor-Derived Mesenchymal Stem Cells Are Immunogenic in an Allogeneic Host and Stimulate Donor Graft Rejection in a Nonmyeloablative Setting. Blood 2006, 108, 2114–2120. [Google Scholar] [CrossRef]
- Eliopoulos, N.; Stagg, J.; Lejeune, L.; Pommey, S.; Galipeau, J. Allogeneic Marrow Stromal Cells Are Immune Rejected by MHC Class I- and Class II-Mismatched Recipient Mice. Blood 2005, 106, 4057–4065. [Google Scholar] [CrossRef]
- Gatza, E.; Choi, S.W. Approaches for the Prevention of Graft-versus-Host Disease Following Hematopoietic Cell Transplantation. Int. J. Hematol. Oncol. 2015, 4, 113–126. [Google Scholar] [CrossRef]
- Bader, P.; Kuci, Z.; Bakhtiar, S.; Basu, O.; Bug, G.; Dennis, M.; Greil, J.; Barta, A.; Kallay, K.M.; Lang, P.; et al. Effective Treatment of Steroid and Therapy-Refractory Acute Graft-versus-Host Disease with a Novel Mesenchymal Stromal Cell Product (MSC-FFM). Bone Marrow Transplant. 2018, 53, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Mareschi, K.; Castiglia, S.; Rustichelli, D.; Mandese, A.; Migliore, E.; Fagioli, F. In Vitro Mesenchymal Progenitor Cell Expansion Is a Predictor of Transplant-Related Mortality and Acute GvHD III-IV After Bone Marrow Transplantation in Univariate Analysis: A Large Single-Center Experience. J. Pediatr. Hematol. Oncol. 2019, 41, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Cesen Mazic, M.; Girandon, L.; Knezevic, M.; Avcin, S.L.; Jazbec, J. Treatment of Severe Steroid-Refractory Acute-Graft-vs.-Host Disease With Mesenchymal Stem Cells-Single Center Experience. Front. Bioeng. Biotechnol. 2018, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, B.; Mafi, R.; Mafi, P.; Khan, W.S. The Regulation of Differentiation of Mesenchymal Stem-Cells into Skeletal Muscle: A Look at Signalling Molecules Involved in Myogenesis. Curr. Stem Cell Res. Ther. 2018, 13, 384–407. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, K.; Sen, D. Mesenchymal Stem Cells as a Source of Dopaminergic Neurons: A Potential Cell Based Therapy for Parkinson’s Disease. Curr. Stem Cell Res. Ther. 2017, 12, 326–347. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, X.; Qian, H.; Zhu, W.; Sun, X.; Hu, J.; Zhou, H.; Chen, Y. Mesenchymal Stern Cells from Adult Human Bone Marrow Differentiate into a Cardiomyocyte Phenotype In Vitro. Exp. Biol. Med. 2004, 229, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Lam, E.W.F.; Soeiro, I.; Tisato, V.; Bonnet, D.; Dazzi, F. Mesenchymal Stem Cells Inhibit Proliferation and Apoptosis of Tumor Cells: Impact on in Vivo Tumor Growth. Leukemia 2006, 21, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, A.Y.; Pati, S.; Anderson, S.A.; Reid, W.; Elshal, M.F.; Rovira, I.I.; Nguyen, A.T.; Malide, D.; Combs, C.A.; Hall, G.; et al. Human Mesenchymal Stem Cells Exert Potent Antitumorigenic Effects in a Model of Kaposi’s Sarcoma. J. Exp. Med. 2006, 203, 1235–1247. [Google Scholar] [CrossRef]
- Fakiruddin, K.S.; Ghazalli, N.; Lim, M.N.; Zakaria, Z.; Abdullah, S. Mesenchymal Stem Cell Expressing TRAIL as Targeted Therapy against Sensitised Tumour. Int. J. Mol. Sci. 2018, 19, 2188. [Google Scholar] [CrossRef]
- Cafforio, P.; Viggiano, L.; Mannavola, F.; Pellè, E.; Caporusso, C.; Maiorano, E.; Felici, C.; Silvestris, F. PIL6-TRAIL-Engineered Umbilical Cord Mesenchymal/Stromal Stem Cells Are Highly Cytotoxic for Myeloma Cells Both in Vitro and in Vivo. Stem Cell Res. Ther. 2017, 8, 206. [Google Scholar] [CrossRef]
- Han, J.; Hwang, H.S.; Na, K. TRAIL-Secreting Human Mesenchymal Stem Cells Engineered by a Non-Viral Vector and Photochemical Internalization for Pancreatic Cancer Gene Therapy. Biomaterials 2018, 182, 259–268. [Google Scholar] [CrossRef]
- Choi, S.A.; Hwang, S.; Wang, K.; Cho, B.; Phi, J.H.; Lee, J.Y.; Jung, H.W.; Lee, D.; Kim, S. Therapeutic Efficacy and Safety of TRAIL—Producing Human Adipose Tissue–Derived Mesenchymal Stem Cells against Experimental Brainstem Glioma. Neuro Oncol. 2011, 13, 61–69. [Google Scholar] [CrossRef]
- Moniri, M.R.; Sun, X.-Y.; Rayat, J.; Dai, D.; Ao, Z.; He, Z.; Verchere, C.B.; Dai, L.-J.; Warnock, G.L. TRAIL-Engineered Pancreas-Derived Mesenchymal Stem Cells: Characterization and Cytotoxic Effects on Pancreatic Cancer Cells. Cancer Gene Ther. 2012, 19, 652–658. [Google Scholar] [CrossRef]
- Mohr, A.; Albarenque, S.M.; Deedigan, L.; Yu, R.; Reidy, M.; Fulda, S.; Zwacka, R.M. Targeting of XIAP Combined with Systemic Mesenchymal Stem Cell-Mediated Delivery of STRAIL Ligand Inhibits Metastatic Growth of Pancreatic Carcinoma Cells. Stem Cells 2010, 28, 2109–2120. [Google Scholar] [CrossRef]
- Grisendi, G.; Bussolari, R.; Cafarelli, L.; Petak, I.; Rasini, V.; Veronesi, E.; De Santis, G.; Spano, C.; Tagliazzucchi, M.; Barti-Juhasz, H.; et al. Adipose-Derived Mesenchymal Stem Cells as Stable Source of Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand Delivery for Cancer Therapy. Cancer Res. 2010, 70, 3718–3729. [Google Scholar] [CrossRef]
- Loebinger, M.R.; Eddaoudi, A.; Davies, D.; Janes, S.M. Mesenchymal Stem Cell Delivery of TRAIL Can Eliminate Metastatic Cancer. Cancer Res. 2009, 69, 4134–4142. [Google Scholar] [CrossRef]
- Kamalabadi-Farahani, M.; Vasei, M.; Ahmadbeigi, N.; Ebrahimi-Barough, S.; Soleimani, M.; Roozafzoon, R. Anti-Tumour Effects of TRAIL-Expressing Human Placental Derived Mesenchymal Stem Cells with Curcumin-Loaded Chitosan Nanoparticles in a Mice Model of Triple Negative Breast Cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, S1011–S1021. [Google Scholar] [CrossRef]
- Reagan, M.R.; Seib, F.P.; McMillin, D.W.; Sage, E.K.; Mitsiades, C.S.; Janes, S.M.; Ghobrial, I.M.; Kaplan, D.L. Stem Cell Implants for Cancer Therapy: TRAIL-Expressing Mesenchymal Stem Cells Target Cancer Cells In Situ. J. Breast Cancer 2012, 15, 273–282. [Google Scholar] [CrossRef]
- Mohr, A.; Chu, T.; Brooke, G.N.; Zwacka, R.M. MSC.STRAIL Has Better Efficacy than MSC.FL-TRAIL and in Combination with AKTi Blocks Pro-Metastatic Cytokine Production in Prostate Cancer Cells. Cancers 2019, 11, 568. [Google Scholar] [CrossRef]
- Loebinger, M.R.; Sage, E.K.; Davies, D.; Janes, S.M. TRAIL-Expressing Mesenchymal Stem Cells Kill the Putative Cancer Stem Cell Population. Br. J. Cancer 2010, 103, 1692–1697. [Google Scholar] [CrossRef]
- Hong, I.S.; Lee, H.Y.; Nam, J.S. Cancer Stem Cells: The “Achilles” Heel’ of Chemo-Resistant Tumors. Recent Pat. Anticancer Drug Discov. 2014, 10, 2–22. [Google Scholar] [CrossRef]
- Leung, E.L.-H.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.-C.; Cheng, L.C.; Sihoe, A.D.-L.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-Small Cell Lung Cancer Cells Expressing CD44 Are Enriched for Stem Cell-like Properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A. Aldehyde Dehydrogenase 1 Is a Tumor Stem Cell-Associated Marker in Lung Cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Zakaria, N.; Yusoff, N.M.; Zakaria, Z.; Lim, M.N.; Baharuddin, P.J.N.; Fakiruddin, K.S.; Yahaya, B. Human Non-Small Cell Lung Cancer Expresses Putative Cancer Stem Cell Markers and Exhibits the Transcriptomic Profile of Multipotent Cells. BMC Cancer 2015, 15. [Google Scholar] [CrossRef]
- Woo, T.; Okudela, K.; Mitsui, H.; Yazawa, T.; Ogawa, N.; Tajiri, M.; Yamamoto, T.; Rino, Y.; Kitamura, H.; Masuda, M. Prognostic Value of CD133 Expression in Stage I Lung Adenocarcinomas. Int. J. Clin. Exp. Pathol. 2010, 4, 32–42. [Google Scholar]
- Qi, X.; Yu, D.; Jia, B.; Jin, C.; Liu, X.; Zhao, X.; Zhang, G. Targeting CD133(+) Laryngeal Carcinoma Cells with Chemotherapeutic Drugs and SiRNA against ABCG2 Mediated by Thermo/PH-Sensitive Mesoporous Silica Nanoparticles. Tumour Biol. 2016, 37, 2209–2217. [Google Scholar] [CrossRef]
- Huang, X.; Huang, J.; Leng, D.; Yang, S.; Yao, Q.; Sun, J.; Hu, J. Gefitinib-Loaded DSPE-PEG2000 Nanomicelles with CD133 Aptamers Target Lung Cancer Stem Cells. World J. Surg. Oncol. 2017, 15, 167. [Google Scholar] [CrossRef]
- Mi, Y.; Huang, Y.; Deng, J. The Enhanced Delivery of Salinomycin to CD133(+) Ovarian Cancer Stem Cells through CD133 Antibody Conjugation with Poly (Lactic-Co-Glycolic Acid)-Poly (Ethylene Glycol) Nanoparticles. Oncol. Lett. 2018, 15, 6611–6621. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, J.; Sun, J.; Huang, L.; Li, Q. Targeting Lung Cancer Initiating Cells by All-Trans Retinoic Acid-Loaded Lipid-PLGA Nanoparticles with CD133 Aptamers. Exp. Ther. Med. 2018, 16, 4639–4649. [Google Scholar] [CrossRef]
- Li, Y.; Shi, S.; Ming, Y.; Wang, L.; Li, C.; Luo, M.; Li, Z.; Li, B.; Chen, J. Specific Cancer Stem Cell-Therapy by Albumin Nanoparticles Functionalized with CD44-Mediated Targeting. J. Nanobiotechnol. 2018, 16, 99. [Google Scholar] [CrossRef]
- Ning, S.-T.; Lee, S.-Y.; Wei, M.-F.; Peng, C.-L.; Lin, S.Y.-F.; Tsai, M.-H.; Lee, P.-C.; Shih, Y.-H.; Lin, C.-Y.; Luo, T.-Y.; et al. Targeting Colorectal Cancer Stem-Like Cells with Anti-CD133 Antibody-Conjugated SN-38 Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 17793–17804. [Google Scholar] [CrossRef]
- Mandal, T.; Beck, M.; Kirsten, N.; Linden, M.; Buske, C. Targeting Murine Leukemic Stem Cells by Antibody Functionalized Mesoporous Silica Nanoparticles. Sci. Rep. 2018, 8, 989. [Google Scholar] [CrossRef]
- Uboldi, C.; Urban, P.; Gilliland, D.; Bajak, E.; Valsami-Jones, E.; Ponti, J.; Rossi, F. Role of the Crystalline Form of Titanium Dioxide Nanoparticles: Rutile, and Not Anatase, Induces Toxic Effects in Balb/3T3 Mouse Fibroblasts. Toxicol. In Vitro 2016, 31, 137–145. [Google Scholar] [CrossRef]
- Shen, C.; James, S.A.; de Jonge, M.D.; Turney, T.W.; Wright, P.F.A.; Feltis, B.N. Relating Cytotoxicity, Zinc Ions, and Reactive Oxygen in ZnO Nanoparticle-Exposed Human Immune Cells. Toxicol. Sci. 2013, 136, 120–130. [Google Scholar] [CrossRef]
- Selck, H.; Handy, R.D.; Fernandes, T.F.; Klaine, S.J.; Petersen, E.J. Nanomaterials in the Aquatic Environment: A European Union-United States Perspective on the Status of Ecotoxicity Testing, Research Priorities, and Challenges Ahead. Environ. Toxicol. Chem. 2016, 35, 1055–1067. [Google Scholar] [CrossRef]
- Khalifeh Soltani, S.; Forogh, B.; Ahmadbeigi, N.; Hadizadeh Kharazi, H.; Fallahzadeh, K.; Kashani, L.; Karami, M.; Kheyrollah, Y.; Vasei, M. Safety and Efficacy of Allogenic Placental Mesenchymal Stem Cells for Treating Knee Osteoarthritis: A Pilot Study. Cytotherapy 2019, 21, 54–63. [Google Scholar] [CrossRef]
- Emadedin, M.; Labibzadeh, N.; Liastani, M.G.; Karimi, A.; Jaroughi, N.; Bolurieh, T.; Hosseini, S.-E.; Baharvand, H.; Aghdami, N. Intra-Articular Implantation of Autologous Bone Marrow-Derived Mesenchymal Stromal Cells to Treat Knee Osteoarthritis: A Randomized, Triple-Blind, Placebo-Controlled Phase 1/2 Clinical Trial. Cytotherapy 2018, 20, 1238–1246. [Google Scholar] [CrossRef]
- Celikkan, F.T.; Mungan, C.; Sucu, M.; Ulus, A.T.; Cinar, O.; Ili, E.G.; Can, A. Optimizing the Transport and Storage Conditions of Current Good Manufacturing Practice -Grade Human Umbilical Cord Mesenchymal Stromal Cells for Transplantation (HUC-HEART Trial). Cytotherapy 2019, 21, 64–75. [Google Scholar] [CrossRef]
- Nowakowski, A.; Drela, K.; Rozycka, J.; Janowski, M.; Lukomska, B. Engineered Mesenchymal Stem Cells as an Anti-Cancer Trojan Horse. Stem Cells Dev. 2016. [Google Scholar] [CrossRef]
- Zhang, J.; Kale, V.; Chen, M. Gene-Directed Enzyme Prodrug Therapy. AAPS J. 2015, 17, 102–110. [Google Scholar] [CrossRef]
- Cavarretta, I.T.; Altanerova, V.; Matuskova, M.; Kucerova, L.; Culig, Z.; Altaner, C. Adipose Tissue–Derived Mesenchymal Stem Cells Expressing Prodrug-Converting Enzyme Inhibit Human Prostate Tumor Growth. Mol. Ther. 2010, 18, 223–231. [Google Scholar] [CrossRef]
- Liu, X.; Hu, J.; Li, Y.; Cao, W.; Wang, Y.; Ma, Z.; Li, F. Mesenchymal Stem Cells Expressing Interleukin-18 Inhibit Breast Cancer in a Mouse Model. Oncol. Lett. 2018, 15, 6265–6274. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Shi, X.; Ding, Y. Genetically Engineered Bone Marrow-Derived Mesenchymal Stem Cells Co-Expressing IFN-Gamma and IL-10 Inhibit Hepatocellular Carcinoma by Modulating MAPK Pathway. J. BUON 2017, 22, 1517–1524. [Google Scholar]
- Xu, G.; Guo, Y.; Seng, Z.; Cui, G.; Qu, J. Bone Marrow-Derived Mesenchymal Stem Cells Co-Expressing Interleukin-18 and Interferon-Beta Exhibit Potent Antitumor Effect against Intracranial Glioma in Rats. Oncol. Rep. 2015, 34, 1915–1922. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Tyler, M.A.; Thaci, B.; Alexiades, N.G.; Han, Y.; Ulasov, I.V.; Lesniak, M.S. A Comparative Study of Neural and Mesenchymal Stem Cell-Based Carriers for Oncolytic Adenovirus in a Model of Malignant Glioma. Mol. Pharm. 2011, 8, 1559–1572. [Google Scholar] [CrossRef]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic Delivery of HER2-Retargeted Oncolytic-HSV by Mesenchymal Stromal Cells Protects from Lung and Brain Metastases. Oncotarget 2015, 6, 34774–34787. [Google Scholar] [CrossRef]
- Choi, S.A.; Lee, C.; Kwak, P.A.; Park, C.-K.; Wang, K.-C.; Phi, J.H.; Lee, J.Y.; Chong, S.; Kim, S.-K. Histone Deacetylase Inhibitor Panobinostat Potentiates the Anti-Cancer Effects of Mesenchymal Stem Cell-Based STRAIL Gene Therapy against Malignant Glioma. Cancer Lett. 2019, 442, 161–169. [Google Scholar] [CrossRef]
- Kuroki, L.M.; Jin, X.; Dmitriev, I.P.; Kashentseva, E.A.; Powell, M.A.; Mutch, D.G.; Dietz, A.B.; Curiel, D.T.; Hawkins, W.G.; Spitzer, D. Adenovirus Platform Enhances Transduction Efficiency of Human Mesenchymal Stem Cells: An Opportunity for Cellular Carriers of Targeted TRAIL-Based TR3 Biologics in Ovarian Cancer. PLoS ONE 2017, 12, e0190125. [Google Scholar] [CrossRef]
- Shamili, F.H.; Bayegi, H.R.; Salmasi, Z.; Sadri, K.; Mahmoudi, M.; Kalantari, M.; Ramezani, M.; Abnous, K. Exosomes Derived from TRAIL-Engineered Mesenchymal Stem Cells with Effective Anti-Tumor Activity in a Mouse Melanoma Model. Int. J. Pharm. 2018, 549, 218–229. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and Expansion of the Tumorigenic Lung Cancer Stem Cell Population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gallagher, M.F.; Ffrench, B.; Gasch, C.; Breen, E.; Gray, S.G.; Nicholson, S.; Leonard, N.; Ryan, R.; Young, V.; et al. Targeting the Cancer Stem Cell Marker, Aldehyde Dehydrogenase 1, to Circumvent Cisplatin Resistance in NSCLC. Oncotarget 2017, 8, 72544–72563. [Google Scholar] [CrossRef]
- Li, D.; Zou, X.-Y.; El-Ayachi, I.; Romero, L.O.; Yu, Z.; Iglesias-Linares, A.; Cordero-Morales, J.F.; Huang, G.T.-J. Human Dental Pulp Stem Cells and Gingival Mesenchymal Stem Cells Display Action Potential Capacity In Vitro after Neuronogenic Differentiation. Stem Cell Rev. Rep. 2019, 15, 67–81. [Google Scholar] [CrossRef]
- Gimble, J.M.; Katz, A.J.; Bunnell, B.A. Adipose-Derived Stem Cells for Regenerative Medicine. Circ. Res. 2007, 100, 1249–1260. [Google Scholar] [CrossRef]
- Kemp, K.C.; Hows, J.; Donaldson, C. Bone Marrow-Derived Mesenchymal Stem Cells. Leuk Lymphoma 2005, 46, 1531–1544. [Google Scholar] [CrossRef]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; van de Water, J.A.J.M.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of Therapeutic Efficacy and Fate of Engineered Human Mesenchymal Stem Cells for Cancer Therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4822–4827. [Google Scholar] [CrossRef]
- Boura, J.S.; Vance, M.; Yin, W.; Madeira, C.; Lobato da Silva, C.; Porada, C.D.; Almeida-Porada, G. Evaluation of Gene Delivery Strategies to Efficiently Overexpress Functional HLA-G on Human Bone Marrow Stromal Cells. Mol. Ther. Methods Clin. Dev. 2014, 2014, 14041. [Google Scholar] [CrossRef]
- Lee, K.; Majumdar, M.K.; Buyaner, D.; Hendricks, J.K.; Pittenger, M.F.; Mosca, J.D. Human Mesenchymal Stem Cells Maintain Transgene Expression during Expansion and Differentiation. Mol. Ther. 2001, 3, 857–866. [Google Scholar] [CrossRef]
- Van Damme, A.; Thorrez, L.; Ma, L.; Vandenburgh, H.; Eyckmans, J.; Dell’Accio, F.; De Bari, C.; Luyten, F.; Lillicrap, D.; Collen, D.; et al. Efficient Lentiviral Transduction and Improved Engraftment of Human Bone Marrow Mesenchymal Cells. Stem Cells 2006, 24, 896–907. [Google Scholar] [CrossRef]
- Jung, P.Y.; Ryu, H.; Rhee, K.-J.; Hwang, S.; Lee, C.G.; Gwon, S.-Y.; Kim, J.; Kim, J.; Yoo, B.-S.; Baik, S.K.; et al. Adipose Tissue-Derived Mesenchymal Stem Cells Cultured at High Density Express IFN-β and TRAIL and Suppress the Growth of H460 Human Lung Cancer Cells. Cancer Lett. 2019, 440–441, 202–210. [Google Scholar] [CrossRef]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly Tumorigenic Lung Cancer CD133+ Cells Display Stem-like Features and Are Spared by Cisplatin Treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Yang, C.-J.; Huang, M.-S.; Yeh, C.-T.; Wu, A.T.H.; Lee, Y.-C.; Lai, T.-C.; Lee, C.-H.; Hsiao, Y.-W.; Lu, J.; et al. Cisplatin Selects for Multidrug-Resistant CD133+ Cells in Lung Adenocarcinoma by Activating Notch Signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer Stem Cell Heterogeneity: Origin and New Perspectives on CSC Targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Szegezdi, E.; O’Reilly, A.; Davy, Y.; Vawda, R.; Taylor, D.L.; Murphy, M.; Samali, A.; Mehmet, H. Stem Cells Are Resistant to TRAIL Receptor-Mediated Apoptosis. J. Cell. Mol. Med. 2009, 13, 4409–4414. [Google Scholar] [CrossRef]
- Franco, A.V.; Zhang, X.D.; Van Berkel, E.; Sanders, J.E.; Zhang, X.Y.; Thomas, W.D.; Nguyen, T.; Hersey, P. The Role of NF-Kappa B in TNF-Related Apoptosis-Inducing Ligand (TRAIL)-Induced Apoptosis of Melanoma Cells. J. Immunol. 2001, 166, 5337–5345. [Google Scholar] [CrossRef]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-Induced Apoptosis by a Family of Signaling and Decoy Receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef]
- Dufour, F.; Rattier, T.; Constantinescu, A.A.; Zischler, L.; Morle, A.; Ben Mabrouk, H.; Humblin, E.; Jacquemin, G.; Szegezdi, E.; Delacote, F.; et al. TRAIL Receptor Gene Editing Unveils TRAIL-R1 as a Master Player of Apoptosis Induced by TRAIL and ER Stress. Oncotarget 2017, 8, 9974–9985. [Google Scholar] [CrossRef]
- Lee, S.-H.; Hyun, S.-K.; Kim, H.-B.; Kang, C.-D.; Kim, S.-H. Potential Role of CD133 Expression in the Susceptibility of Human Liver Cancer Stem-Like Cells to TRAIL. Oncol. Res. 2016, 24, 495–509. [Google Scholar] [CrossRef]
- Signore, M.; Ricci-Vitiani, L.; De Maria, R. Targeting Apoptosis Pathways in Cancer Stem Cells. Cancer Lett. 2013, 332, 374–382. [Google Scholar] [CrossRef]
- Micheau, O. Regulation of TNF-Related Apoptosis-Inducing Ligand Signaling by Glycosylation. Int. J. Mol. Sci. 2018, 19, 715. [Google Scholar] [CrossRef]
- Lee, H.; Oh, Y.; Jeon, Y.-J.; Lee, S.-Y.; Kim, H.; Lee, H.-J.; Jung, Y.-K. DR4-Ser424 O-GlcNAcylation Promotes Sensitization of TRAIL-Tolerant Persisters and TRAIL-Resistant Cancer Cells to Death. Cancer Res. 2019, 79, 2839–2852. [Google Scholar] [CrossRef]
- Dufour, F.; Rattier, T.; Shirley, S.; Picarda, G.; Constantinescu, A.A.; Morlé, A.; Zakaria, A.B.; Marcion, G.; Causse, S.; Szegezdi, E.; et al. N-Glycosylation of Mouse TRAIL-R and Human TRAIL-R1 Enhances TRAIL-Induced Death. Cell Death Differ. 2017, 24, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Busheri, F.; Zadorozhny, V.; Li, T.; Lin, H.; Shawler, D.L.; Fakhrai, H. Pivotal Role of CD38 Biomarker in Combination with CD24, EpCAM, and ALDH for Identification of H460 Derived Lung Cancer Stem Cells. J. Stem Cells 2011, 6, 9–20. [Google Scholar] [PubMed]
- Christgen, M.; Ballmaier, M.; Lehmann, U.; Kreipe, H. Detection of Putative Cancer Stem Cells of the Side Population Phenotype in Human Tumor Cell Cultures. Methods Mol. Biol. 2012, 878, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, C.; Liu, X.; Tang, D.G.; Wang, J. The MicroRNA MiR-34a Inhibits Non-Small Cell Lung Cancer (NSCLC) Growth and the CD44hi Stem-like NSCLC Cells. PLoS ONE 2014, 9, e90022. [Google Scholar] [CrossRef] [PubMed]
- Que, Z.; Luo, B.; Zhou, Z.; Dong, C.; Jiang, Y.; Wang, L.; Shi, Q.; Tian, J. Establishment and Characterization of a Patient-Derived Circulating Lung Tumor Cell Line in Vitro and in Vivo. Cancer Cell Int. 2019, 19, 21. [Google Scholar] [CrossRef]
- Levin, T.G.; Powell, A.E.; Davies, P.S.; Silk, A.D.; Dismuke, A.D.; Anderson, E.C.; Swain, J.R.; Wong, M.H. Characterization of the Intestinal Cancer Stem Cell Marker CD166 in the Human and Mouse Gastrointestinal Tract. Gastroenterology 2010, 139, 2072–2082. [Google Scholar] [CrossRef]
- Luo, W.; Zhang, D.; Ma, S.; Wang, C.; Zhang, Q.; Wang, H.; He, K.; Liu, Z. MiR-27a Is Highly Expressed in H1650 Cancer Stem Cells and Regulates Proliferation, Migration, and Invasion. J. Cancer Res. Ther. 2018, 14, S1004–S1011. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Bender, N.O.; Maciaczyk, D.; Bogiel, T.; Bar, E.E.; Eberhart, C.G.; Nikkhah, G.; Maciaczyk, J. CD133/CD15 Defines Distinct Cell Subpopulations with Differential in Vitro Clonogenic Activity and Stem Cell-Related Gene Expression Profile in in Vitro Propagated Glioblastoma Multiforme-Derived Cell Line with a PNET-like Component. Folia Neuropathol 2012, 50, 357–368. [Google Scholar] [CrossRef]
- Tomuleasa, C.; Soritau, O.; Rus-Ciuca, D.; Pop, T.; Todea, D.; Mosteanu, O.; Pintea, B.; Foris, V.; Susman, S.; Kacso, G.; et al. Isolation and Characterization of Hepatic Cancer Cells with Stem-like Properties from Hepatocellular Carcinoma. J. Gastrointest. Liver Dis. 2010, 19, 61–67. [Google Scholar]
- Reyes, E.E.; Kunovac, S.K.; Duggan, R.; Kregel, S.; Vander Griend, D.J. Growth Kinetics of CD133-Positive Prostate Cancer Cells. Prostate 2013, 73, 724–733. [Google Scholar] [CrossRef]
- Roy, S.; Lu, K.; Nayak, M.K.; Bhuniya, A.; Ghosh, T.; Kundu, S.; Ghosh, S.; Baral, R.; Dasgupta, P.S.; Basu, S. Activation of D2 Dopamine Receptors in CD133+ve Cancer Stem Cells in Non-Small Cell Lung Carcinoma Inhibits Proliferation, Clonogenic Ability, and Invasiveness of These Cells. J. Biol. Chem. 2017, 292, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.-Z.; Zhang, J.-Q.; Liu, L.; Fu, W.-P.; Shu, J.-K.; Feng, J.-G.; Liang, X. Silencing of Btbd7 Inhibited Epithelial-Mesenchymal Transition and Chemoresistance in CD133 (+) Lung Carcinoma A549 Cells. Oncol. Res. 2017, 25, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Han, J.; Zhu, H.; Peng, L.; Chen, Z. MiR181b5p Mediates TGFbeta1-Induced Epithelial-to-Mesenchymal Transition in Non-Small Cell Lung Cancer Stem-like Cells Derived from Lung Adenocarcinoma A549 Cells. Int. J. Oncol. 2017, 51, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Cai, H.; Zhang, Y.; Chang, L.; Cui, Y. MiR-129-5p Inhibits Non-Small Cell Lung Cancer Cell Stemness and Chemoresistance through Targeting DLK1. Biochem. Biophys. Res. Commun. 2017, 490, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Zeng, Z.; Bai, B.; Zhu, J.; Song, Z. The Prognostic Value of CSCs Biomarker CD133 in NSCLC: A Meta-Analysis. Oncotarget 2016, 7, 56526–56539. [Google Scholar] [CrossRef] [PubMed]
- Skirecki, T.; Hoser, G.; Kawiak, J.; Dziedzic, D.; Domagała-Kulawik, J. Flow Cytometric Analysis of CD133- and EpCAM-Positive Cells in the Peripheral Blood of Patients with Lung Cancer. Arch. Immunol. Ther. Exp. 2014, 62, 67–75. [Google Scholar] [CrossRef]
- Zhao, W.; Luo, Y.; Li, B.; Zhang, T. Tumorigenic Lung Tumorospheres Exhibit Stem-like Features with Significantly Increased Expression of CD133 and ABCG2. Mol. Med. Rep. 2016, 14, 2598–2606. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Z.; Wong, S.K.-M.; Tin, V.P.-C.; Ho, K.-Y.; Wang, J.; Sham, M.-H.; Wong, M.P. Lung Cancer Tumorigenicity and Drug Resistance Are Maintained through ALDH(Hi)CD44(Hi) Tumor Initiating Cells. Oncotarget 2013, 4, 1698–1711. [Google Scholar] [CrossRef]
- Broadley, K.W.R.; Hunn, M.K.; Farrand, K.J.; Price, K.M.; Grasso, C.; Miller, R.J.; Hermans, I.F.; McConnell, M.J. Side Population Is Not Necessary or Sufficient for a Cancer Stem Cell Phenotype in Glioblastoma Multiforme. Stem Cells 2011, 29, 452–461. [Google Scholar] [CrossRef]
- Akunuru, S.; Palumbo, J.; Zhai, Q.J.; Zheng, Y. Rac1 Targeting Suppresses Human Non-Small Cell Lung Adenocarcinoma Cancer Stem Cell Activity. PLoS ONE 2011, 6, e16951. [Google Scholar] [CrossRef]
- Gu, H.; Wu, X.-Y.; Fan, R.-T.; Wang, X.; Guo, Y.-Z.; Wang, R. Side Population Cells from Long-Term Passage Non-Small Cell Lung Cancer Cells Display Loss of Cancer Stem Cell-like Properties and Chemoradioresistance. Oncol. Lett. 2016, 12, 2886–2893. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.K.; Kolluri, K.K.; McNulty, K.; Lourenco, S.D.S.; Kalber, T.L.; Ordidge, K.L.; Davies, D.; Gary Lee, Y.C.; Giangreco, A.; Janes, S.M. Systemic but Not Topical TRAIL-Expressing Mesenchymal Stem Cells Reduce Tumour Growth in Malignant Mesothelioma. Thorax 2014, 69, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Sayers, T.J. Targeting the Extrinsic Apoptosis Signaling Pathway for Cancer Therapy. Cancer Immunol. Immunother. 2011, 60, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Barr, M.P.; Gray, S.G.; Hoffmann, A.C.; Hilger, R.A.; Thomale, J.; O’Flaherty, J.D.; Fennell, D.A.; Richard, D.; O’Leary, J.J.; O’Byrne, K.J. Generation and Characterisation of Cisplatin-Resistant Non-Small Cell Lung Cancer Cell Lines Displaying a Stem-like Signature. PLoS ONE 2013, 8, e54193. [Google Scholar] [CrossRef] [PubMed]
- Redjal, N.; Zhu, Y.; Shah, K. Combination of Systemic Chemotherapy with Local Stem Cell Delivered S-TRAIL in Resected Brain Tumors. Stem Cells 2015, 33, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Sargent, R.; Rabson, A. Inhibitor of Apoptosis Protein BIRC3 (API2, CIAP2, AIP1) Is Upregulated by the Non-Canonical NFKB Pathway. Cancer Res. 2007, 67, 5327. [Google Scholar]
- Corazza, N.; Jakob, S.; Schaer, C.; Frese, S.; Keogh, A.; Stroka, D.; Kassahn, D.; Torgler, R.; Mueller, C.; Schneider, P.; et al. TRAIL Receptor-Mediated JNK Activation and Bim Phosphorylation Critically Regulate Fas-Mediated Liver Damage and Lethality. J. Clin. Investig. 2006, 116, 2493–2499. [Google Scholar] [CrossRef]
- Fang, L.; Adkins, B.; Deyev, V.; Podack, E.R. Essential Role of TNF Receptor Superfamily 25 (TNFRSF25) in the Development of Allergic Lung Inflammation. J. Exp. Med. 2008, 205, 1037–1048. [Google Scholar] [CrossRef]
- Zakaria, N.; Mohd Yusoff, N.; Zakaria, Z.; Widera, D.; Yahaya, B.H. Inhibition of NF-ΚB Signaling Reduces the Stemness Characteristics of Lung Cancer Stem Cells. Front. Oncol. 2018, 8, 166. [Google Scholar] [CrossRef]
- Seo, P.W.; Lee, K.Y. The Proteasome Inhibitor MG132 Sensitizes Lung Cancer Cells to TRAIL-Induced Apoptosis by Inhibiting NF-ΚB Activation. Tuberc. Respir. Dis. 2008, 65, 476–486. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.-H.; Lu, Q.; Wang, Y.-J. Bag3 Promotes Resistance to Apoptosis through Bcl-2 Family Members in Non-Small Cell Lung Cancer. Oncol. Rep. 2012, 27, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Murphy, Á.C.; Weyhenmeyer, B.; Noonan, J.; Kilbride, S.M.; Schimansky, S.; Loh, K.P.; Kögel, D.; Letai, A.G.; Prehn, J.H.M.; Murphy, B.M. Modulation of Mcl-1 Sensitizes Glioblastoma to TRAIL-Induced Apoptosis. Apoptosis 2014, 19, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Vallbohmer, D.; Warnecke-Eberz, U.; Hokita, S.; Xi, H.; Brabender, J.; Metzger, R.; Baldus, S.E.; Natsugoe, S.; Aikou, T.; et al. Down-Regulation of Gadd45 Expression Is Associated with Tumor Differentiation in Non-Small Cell Lung Cancer. Anticancer Res. 2006, 26, 2143–2147. [Google Scholar] [PubMed]
- Hirose, T.; Sowa, Y.; Takahashi, S.; Saito, S.; Yasuda, C.; Shindo, N.; Furuichi, K.; Sakai, T. P53-Independent Induction of Gadd45 by Histone Deacetylase Inhibitor: Coordinate Regulation by Transcription Factors Oct-1 and NF-Y. Oncogene 2003, 22, 7762–7773. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.P.; Martins, M.D.; Martins, M.A.T.; Almeida, L.O.; Warner, K.A.; Nor, J.E.; Squarize, C.H.; Castilho, R.M. Targeting Histone Deacetylase and NFkappaB Signaling as a Novel Therapy for Mucoepidermoid Carcinomas. Sci. Rep. 2018, 8, 2065. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, B.; Chen, D.; Setroikromo, R.; Haisma, H.J.; Quax, W.J. Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers 2019, 11, 645. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Aksoy, E.; Cingoz, A.; Senbabaoglu, F.; Seker, F.; Sur-Erdem, I.; Kayabolen, A.; Lokumcu, T.; Sahin, G.N.; Karahuseyinoglu, S.; Bagci-Onder, T. The Pro-Apoptotic Bcl-2 Family Member Harakiri (HRK) Induces Cell Death in Glioblastoma Multiforme. Cell Death Discov. 2019, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, A.; Liu, H.; Ma, Y.; Zhou, Y.; Zang, C.; Habbel, J.-P.; Possinger, K.; Eucker, J. Tumor Cell-Selective Synergism of TRAIL-and ATRA-Induced Cytotoxicity in Breast Cancer Cells. Anticancer Res. 2018, 38, 2669–2682. [Google Scholar] [CrossRef]
- Rahman, M.; Davis, S.R.; Pumphrey, J.G.; Bao, J.; Nau, M.M.; Meltzer, P.S.; Lipkowitz, S. TRAIL Induces Apoptosis in Triple-Negative Breast Cancer Cells with a Mesenchymal Phenotype. Breast Cancer Res. Treat. 2009, 113, 217–230. [Google Scholar] [CrossRef]
- GeneGlobe Data Analysis Center. Available online: https://www.qiagen.com/my/shop/genes-and-pathways/data-analysis-center-overview-page/ (accessed on 11 December 2018).









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakiruddin, K.S.; Lim, M.N.; Nordin, N.; Rosli, R.; Zakaria, Z.; Abdullah, S. Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1261. https://doi.org/10.3390/cancers11091261
Fakiruddin KS, Lim MN, Nordin N, Rosli R, Zakaria Z, Abdullah S. Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer. Cancers. 2019; 11(9):1261. https://doi.org/10.3390/cancers11091261
Chicago/Turabian StyleFakiruddin, Kamal Shaik, Moon Nian Lim, Norshariza Nordin, Rozita Rosli, Zubaidah Zakaria, and Syahril Abdullah. 2019. "Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer" Cancers 11, no. 9: 1261. https://doi.org/10.3390/cancers11091261
APA StyleFakiruddin, K. S., Lim, M. N., Nordin, N., Rosli, R., Zakaria, Z., & Abdullah, S. (2019). Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer. Cancers, 11(9), 1261. https://doi.org/10.3390/cancers11091261