Lithocholic Acid, a Metabolite of the Microbiome, Increases Oxidative Stress in Breast Cancer

 , ,
, ,  ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines
2.3. Proliferation Assay
2.4. Real-Time Quantitative PCR (RT-qPCR)
2.5. SDS-PAGE and Western Blotting
2.6. Determination of Lipid Peroxidation
2.7. Transfections
2.8. ABTS Decoloration Assay
2.9. Database Screening
2.10. Tissue Microarray, Immunohistochemistry, and Analysis
2.11. Animal Study
2.12. Statistical Analysis
3. Results
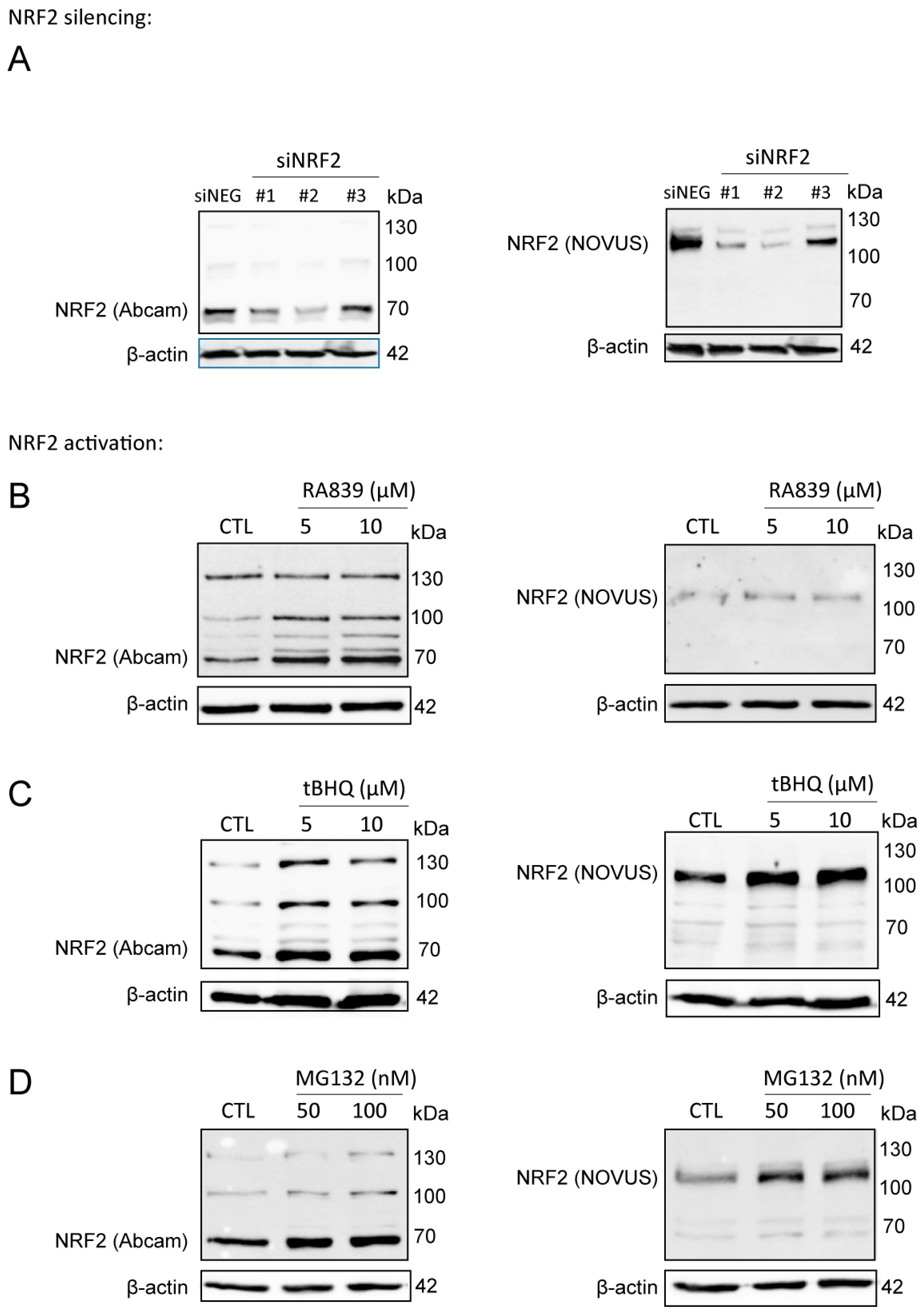
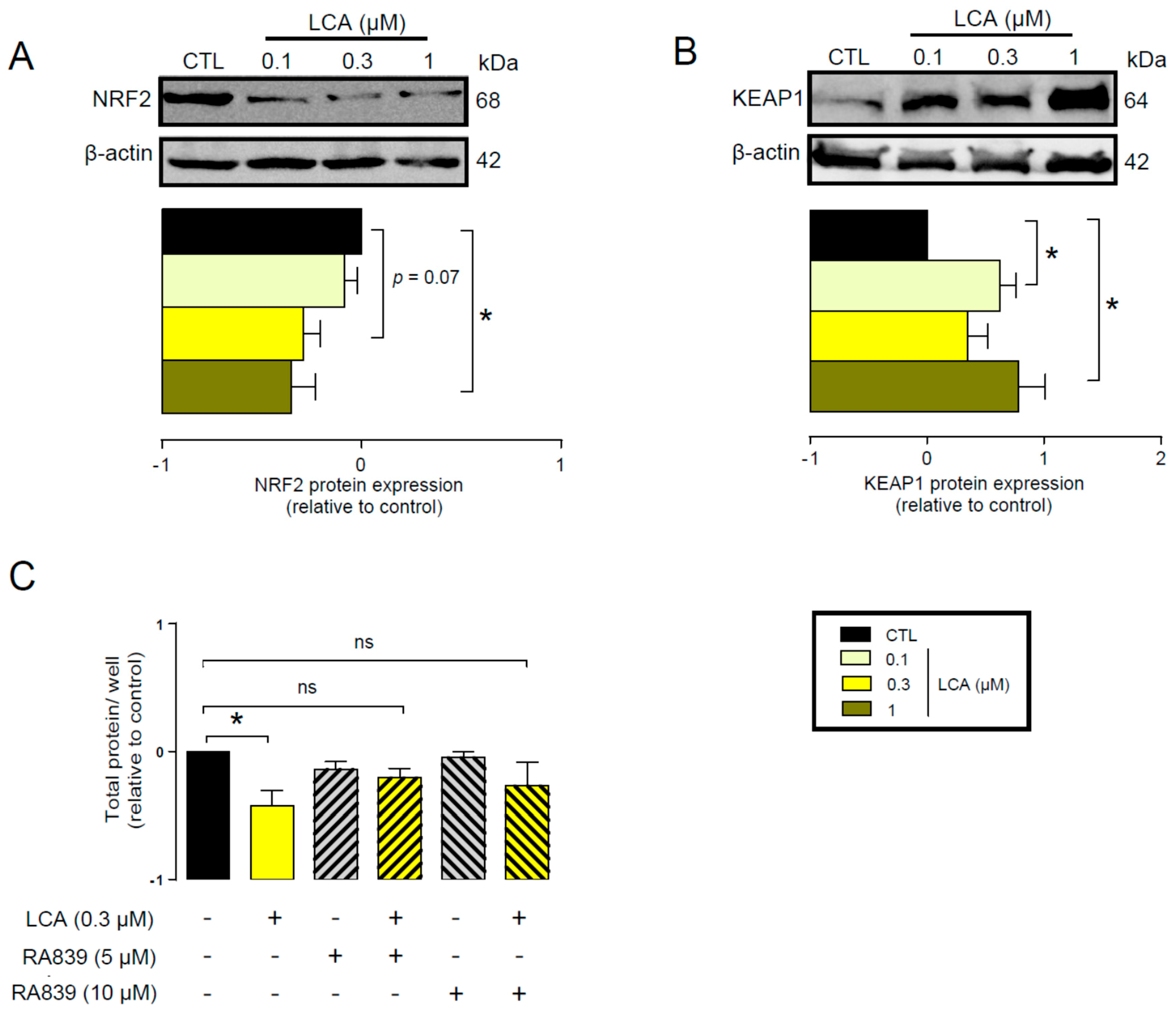
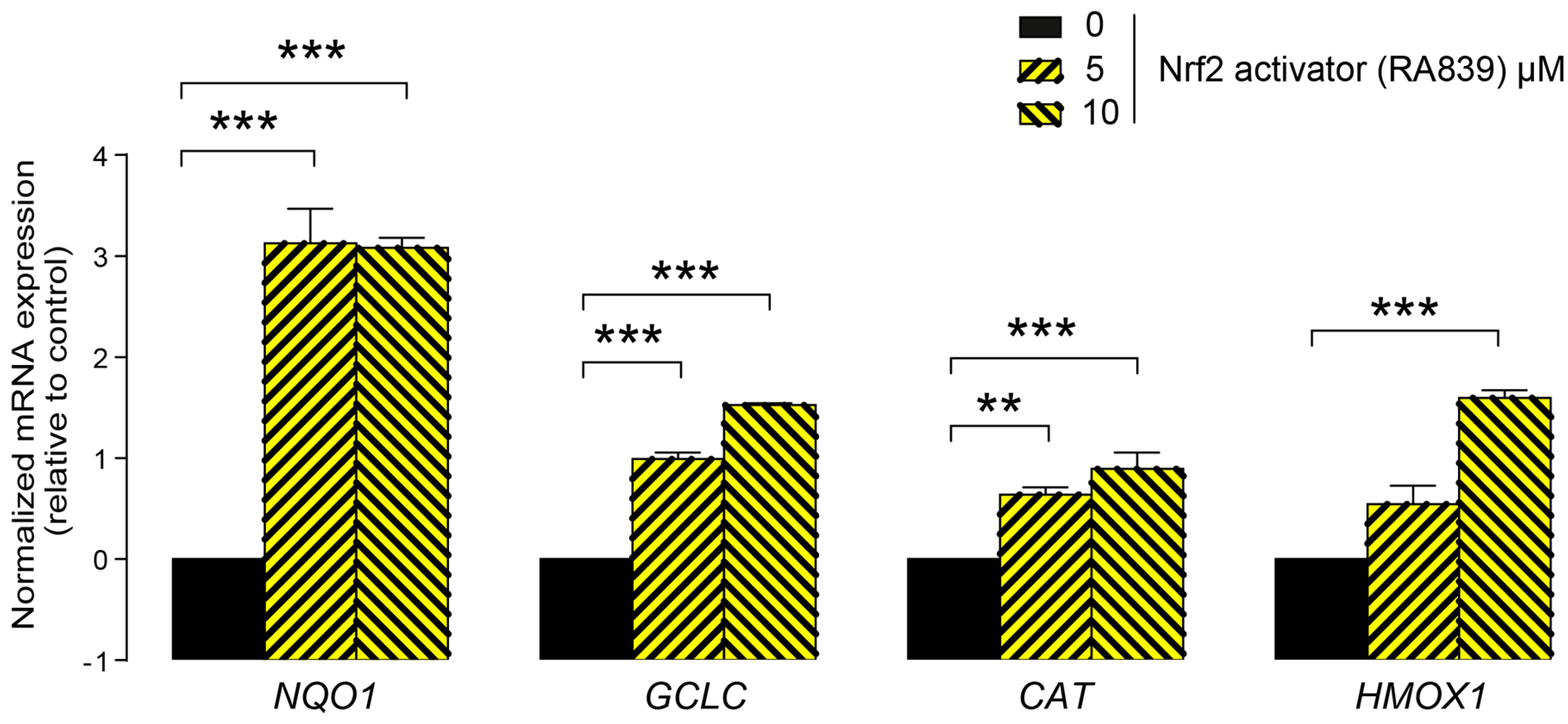
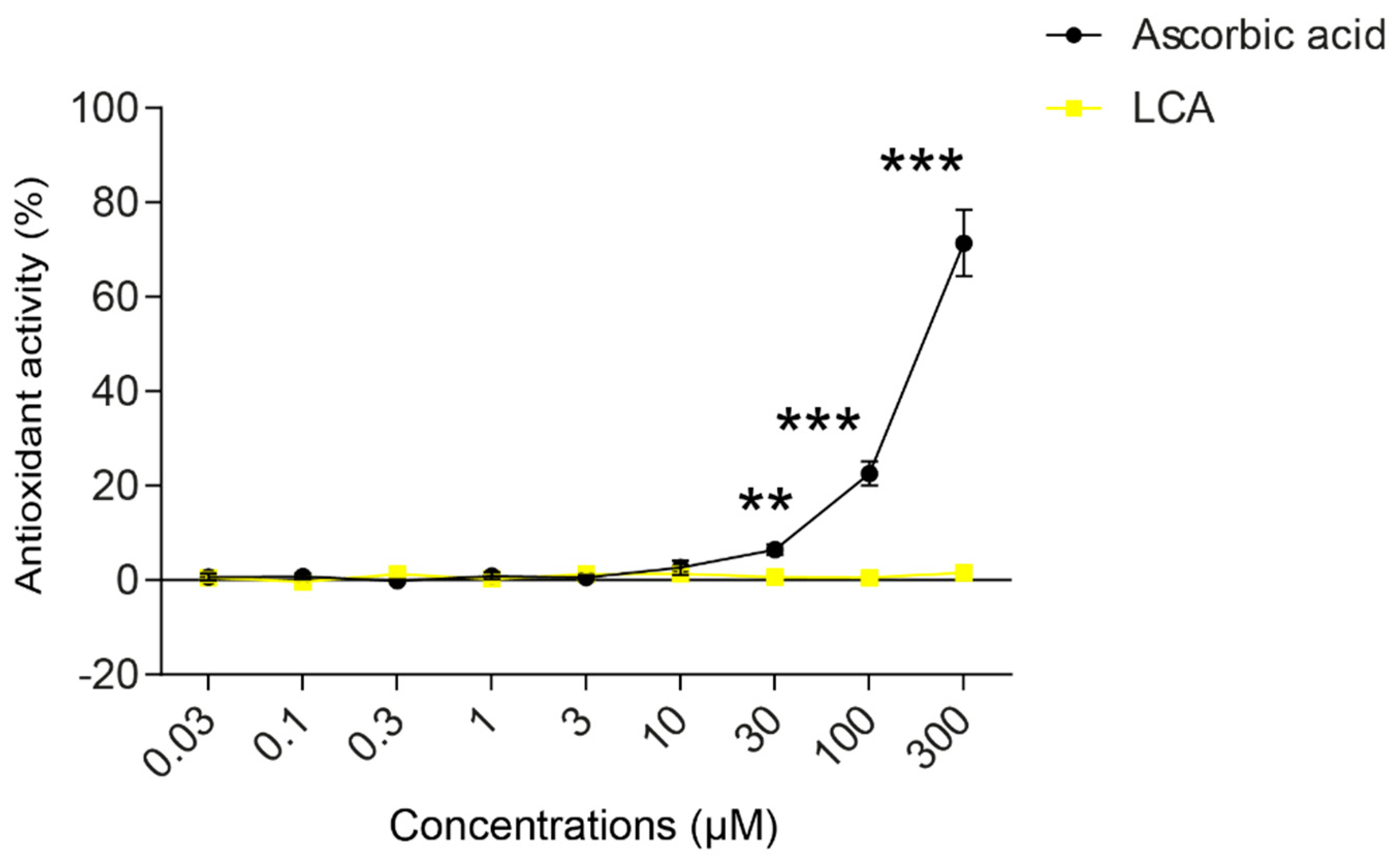
3.1. Lithocholic Acid Inhibited NRF2 Activation
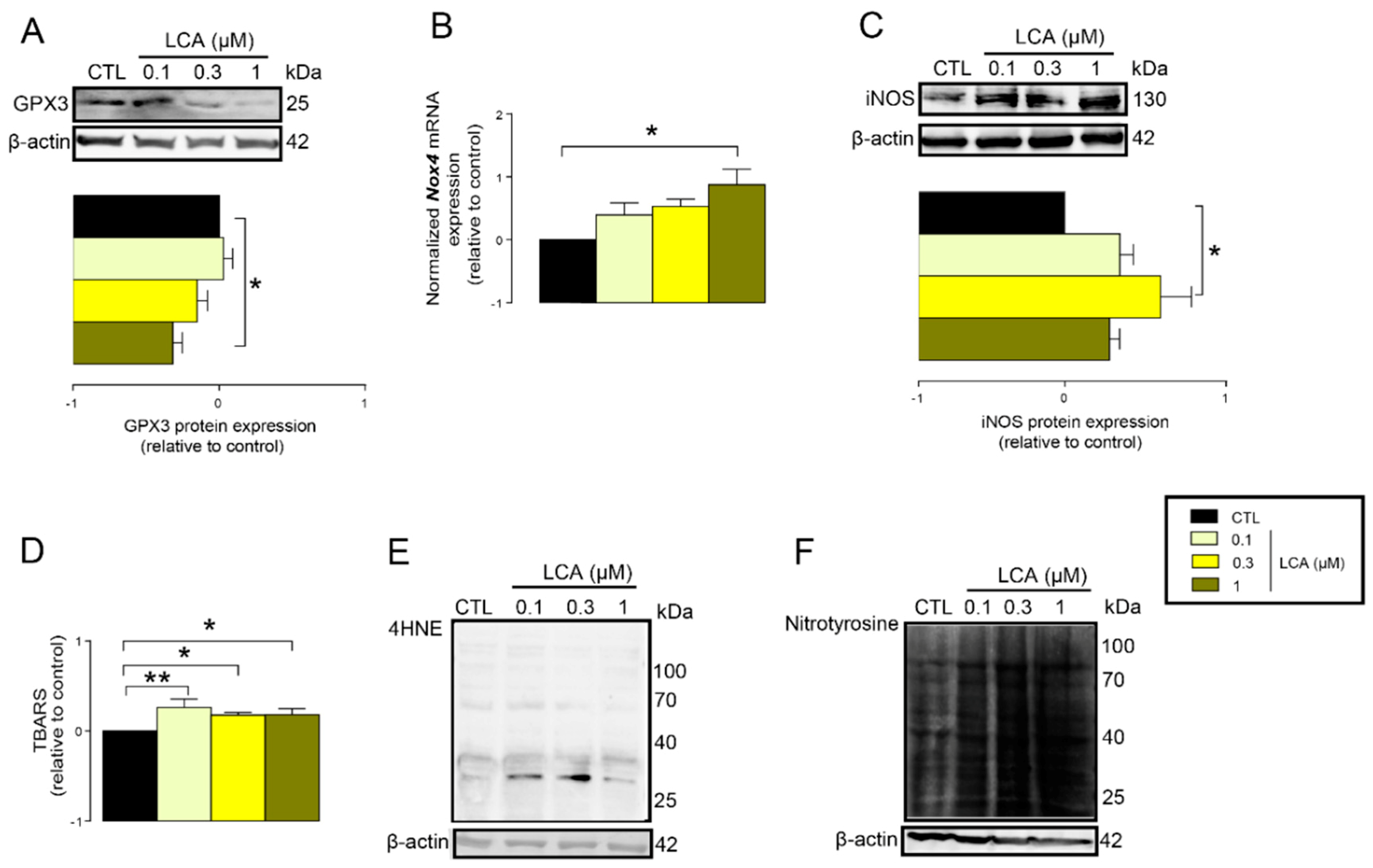
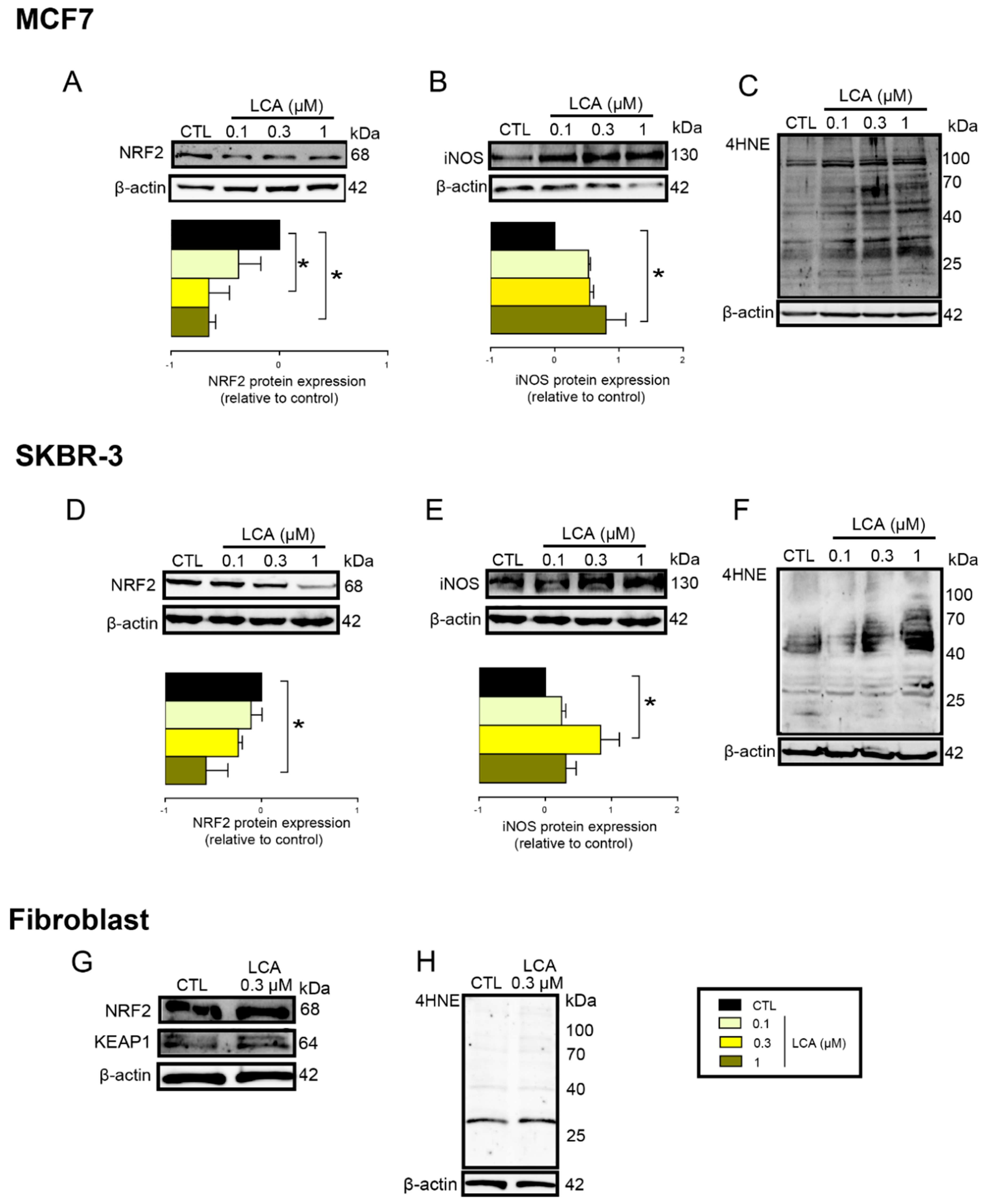
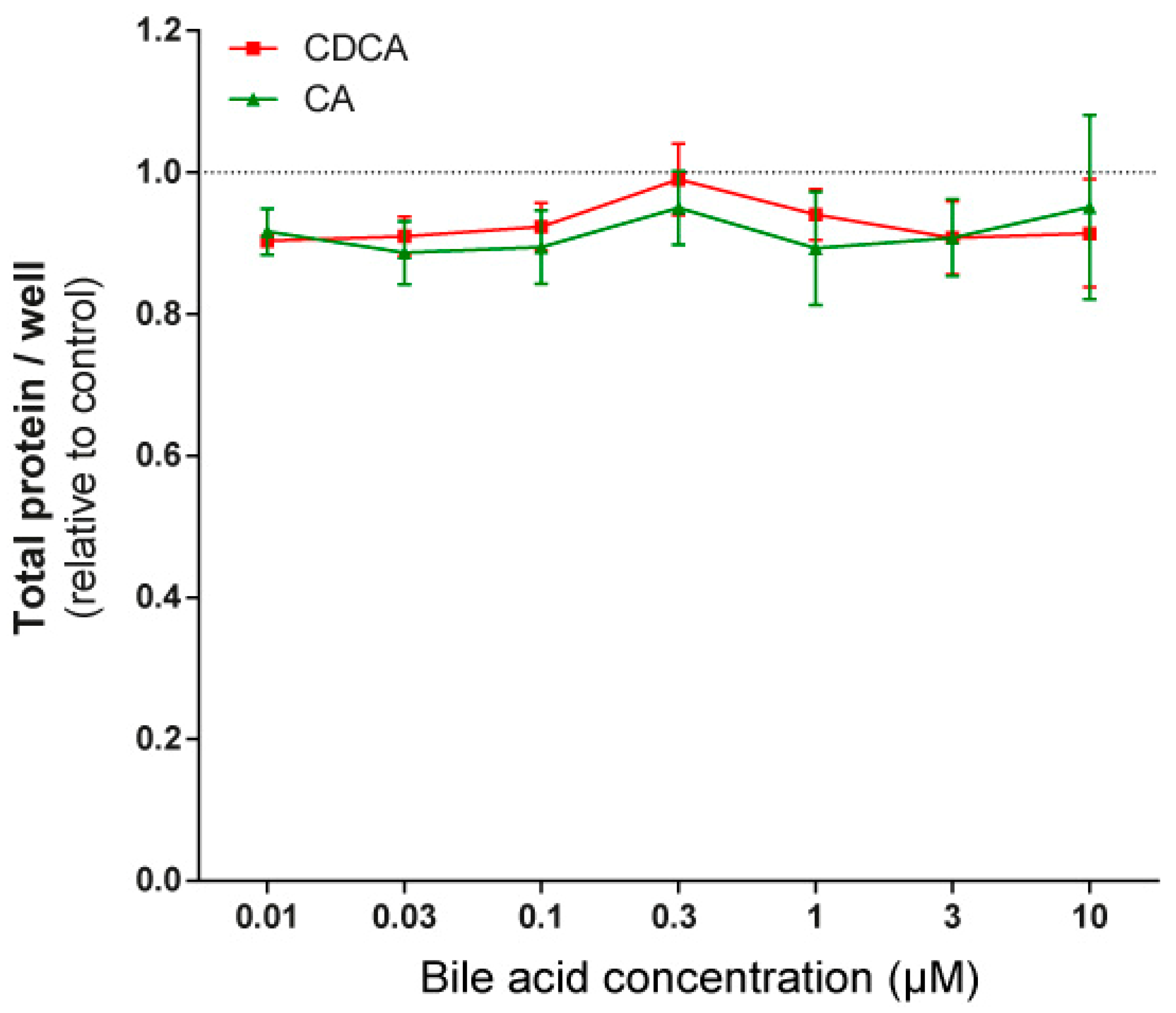
3.2. LCA Treatment Induced Oxidative Stress by Reducing NRF2 Expression
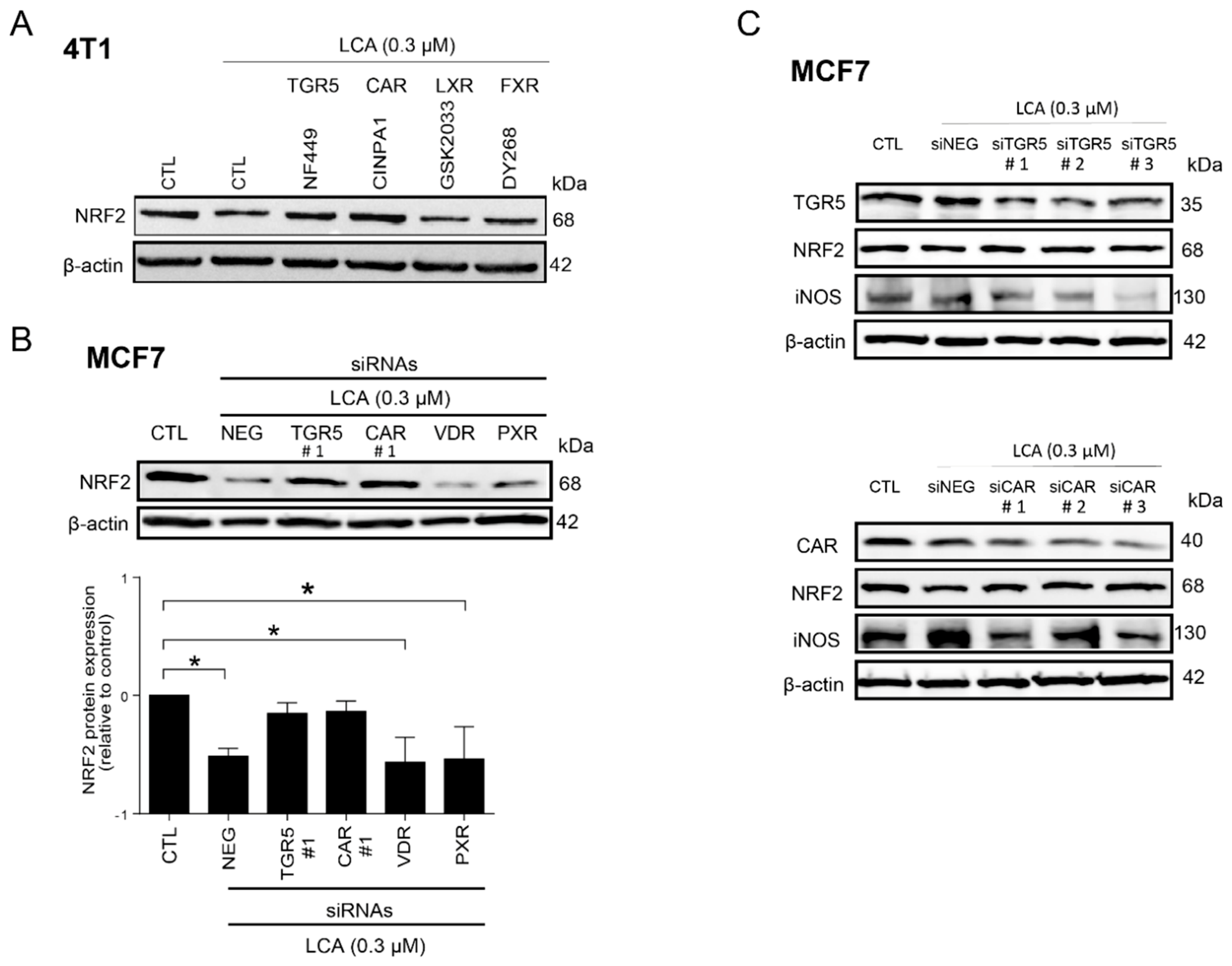
3.3. LCA-Elicited Oxidative Stress Was Mediated by TGR5 and Partially by CAR Receptor
3.4. LCA Supplementation Suppressed Antioxidant Defense in an Animal Model of Breast Cancer
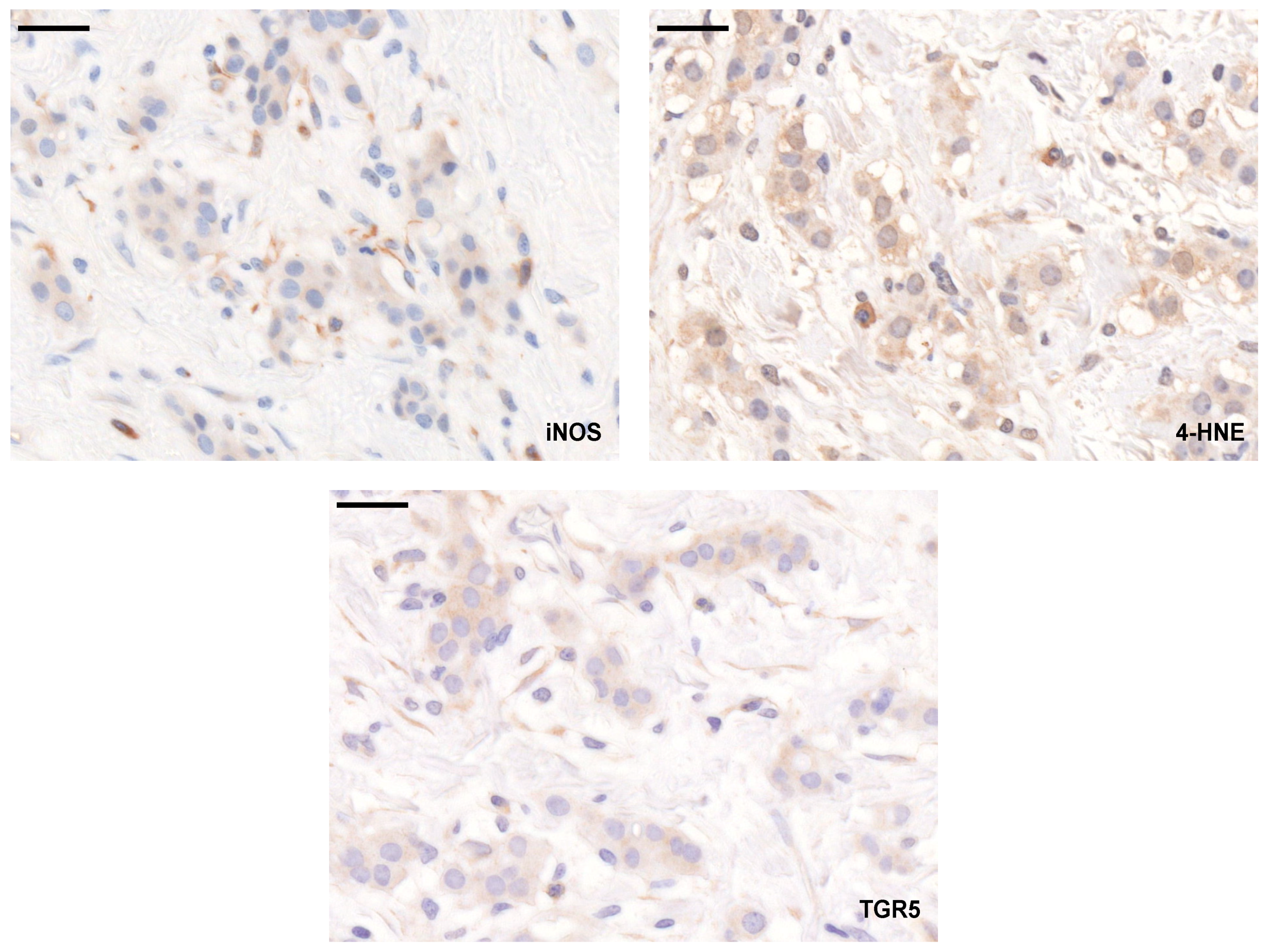
3.5. Elements of the LCA-Elicited Anticancer Pathway Correlated with Stage, Grade, and Receptor Status of the Disease
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
Abbreviations
| CAR | constitutive androstane receptor |
| CAT | catalase |
| CA | cholic acid |
| CDCA | chenodeoxycholic acid |
| ER | estrogen receptor |
| FXR | farnesoid X-activated receptor |
| GCLC | glutamate–cysteine ligase catalytic subunit |
| GSH | reduced glutathione |
| GPX2 | glutathione peroxidase 2 |
| GPX3 | glutathione peroxidase 3 |
| Her2 | Human epidermal growth factor receptor 2/herceptin receptor/erbB receptor |
| HMOX1 | heme oxygenase 1 |
| iNOS | inducible NO synthase |
| KEAP1 | Kelch like ECH associating protein 1 |
| LCA | lithocholic acid |
| LXR | liver X nuclear receptor |
| Mib1 | molecular immunology borstel-1 |
| NOX4 | NADPH oxidase 4 |
| NAC | N-acetylcysteine |
| nNOS | neuronal NOS |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NRF2 | nuclear factor, erythroid 2-like 2 |
| PR | progesterone receptor |
| PXR | pregnane X receptor |
| SOD1 | superoxide dismutase 1 |
| SOD2 | superoxide dismutase 2 |
| SOD3 | superoxide dismutase 3 |
| SRB | sulphorhodamine B |
| TBA | thiobarbituric acid |
| TBARS | thiobarbituric acid reactive substances |
| TGR5/GPBAR1 | G protein-coupled bile acid receptor 1/Takeda G-protein coupled receptor |
| TNBC | triple negative breast cancer |
| VDR | vitamin D receptor |
| 4HNE | 4-hydroxynonenal |
References
- Goedert, J.J.; Hua, X.; Bielecka, A.; Okayasu, I.; Milne, G.L.; Jones, G.S.; Fujiwara, M.; Sinha, R.; Wan, Y.; Xu, X.; et al. Postmenopausal Breast Cancer and Oestrogen Associations with the Iga-Coated and Iga-Noncoated Faecal Microbiota. Br. J. Cancer 2018, 23, 435. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Porporato, P.; Dewulf, E.M.; Verrax, J.; Neyrinck, A.M.; Martin, J.C.; Scott, K.P.; Calderon, P.B.; Feron, O.; Muccioli, G.G.; et al. Gut Microbiota-Derived Propionate Reduces Cancer Cell Proliferation in the Liver. Br. J. Cancer 2012, 107, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Goedert, J.J.; Jones, G.; Hua, X.; Xu, X.; Yu, G.; Flores, R.; Falk, R.T.; Gail, M.H.; Shi, J.; Ravel, J.; et al. Investigation of the Association between the Fecal Microbiota and Breast Cancer in Postmenopausal Women: A Population-Based Case-Control Pilot Study. J. Natl. Cancer Inst. 2015, 107, 147. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, B.J.; Feigelson, H.S.; Flores, R.; Gail, M.H.; Xu, X.; Ravel, J.; Goedert, J.J. Associations of the Fecal Microbiome with Urinary Estrogens and Estrogen Metabolites in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2014, 99, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal Microbial Determinants of Fecal and Systemic Estrogens and Estrogen Metabolites: A Cross-Sectional Study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef]
- Xuan, C.; Shamonki, J.M.; Chung, A.; Dinome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial Dysbiosis Is Associated with Human Breast Cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef]
- Hieken, T.J.; Chen, J.; Hoskin, T.L.; Walther-Antonio, M.; Johnson, S.; Ramaker, S.; Xiao, J.; Radisky, D.C.; Knutson, K.L.; Kalari, K.R.; et al. The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci. Rep. 2016, 6, 30751. [Google Scholar] [CrossRef]
- Chan, A.A.; Bashir, M.; Rivas, M.N.; Duvall, K.; Sieling, P.A.; Pieber, T.R.; Vaishampayan, P.A.; Love, S.M.; Lee, D.J. Characterization of the Microbiome of Nipple Aspirate Fluid of Breast Cancer Survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [Green Version]
- Fulbright, L.E.; Ellermann, M.; Arthur, J.C. The Microbiome and the Hallmarks of Cancer. PLoS Pathog. 2017, 13, e1006480. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and Anticancer Immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Jobin, C. The Microbiome and Cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and Malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Miko, E.; Vida, A.; Bai, P. Translational Aspects of the Microbiome-to Be Exploited. Cell Biol Toxicol. 2016, 32, 153–156. [Google Scholar] [CrossRef]
- Miko, E.; Kovacs, T.; Sebo, E.; Toth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabo, J.; Mehes, G.; Bai, P. Microbiome-Microbial Metabolome-Cancer Cell Interactions in Breast Cancer-Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell. 2018, 33, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Kovács, T.; Mikó, E.; Vida, A.; Sebő, É.; Toth, J.; Csonka, T.; Boratkó, A.; Ujlaki, G.; Lente, G.; Kovács, P.; et al. Cadaverine, a Metabolite of the Microbiome, Reduces Breast Cancer Aggressiveness through Trace Amino Acid Receptors. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Miko, E.; Vida, A.; Kovacs, T.; Ujlaki, G.; Trencsenyi, G.; Marton, J.; Sari, Z.; Kovacs, P.; Boratko, A.; Hujber, Z.; et al. Lithocholic Acid, a Bacterial Metabolite Reduces Breast Cancer Cell Proliferation and Aggressiveness. Biochim. Biophys. Acta 2018, 1859, 958–974. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The Role of Short-Chain Fatty Acids in Health and Disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar]
- Luu, T.H.; Bard, J.M.; Carbonnelle, D.; Chaillou, C.; Huvelin, J.M.; Bobin-Dubigeon, C.; Nazih, H. Lithocholic Bile Acid Inhibits Lipogenesis and Induces Apoptosis in Breast Cancer Cells. Cell Oncol. (Dordr) 2018, 41, 13–24. [Google Scholar] [CrossRef]
- Goldberg, A.A.; Beach, A.; Davies, G.F.; Harkness, T.A.; Leblanc, A.; Titorenko, V.I. Lithocholic Bile Acid Selectively Kills Neuroblastoma Cells, While Sparing Normal Neuronal Cells. Oncotarget 2011, 2, 761–782. [Google Scholar] [CrossRef]
- Bai, P.; Goedert, J.J.; Kovács, T.; Mikó, E.; Vida, A. Methods for Diagnosing Breast Cancer; WIPO: Ginevra, Switzerland, 2018. [Google Scholar]
- Tang, X.; Lin, C.C.; Spasojevic, I.; Iversen, E.S.; Chi, J.T.; Marks, J.R. A Joint Analysis of Metabolomics and Genetics of Breast Cancer. Breast Cancer Res. 2014, 16, 415. [Google Scholar] [CrossRef]
- Goldberg, A.A.; Titorenko, V.I.; Beach, A.; Sanderson, J.T. Bile Acids Induce Apoptosis Selectively in Androgen-Dependent and -Independent Prostate Cancer Cells. Peer J. 2013, 1, e122. [Google Scholar] [CrossRef]
- Gafar, A.A.; Draz, H.M.; Goldberg, A.A.; Bashandy, M.A.; Bakry, S.; Khalifa, M.A.; AbuShair, W.; Titorenko, V.I.; Sanderson, J.T. Lithocholic Acid Induces Endoplasmic Reticulum Stress, Autophagy and Mitochondrial Dysfunction in Human Prostate Cancer Cells. Peer J. 2016, 4, e2445. [Google Scholar] [CrossRef]
- Tnm Classification of Malignant Tumours; Brierly, J.D. (Ed.) John Wiley and Sons: Oxford, UK, 2017. [Google Scholar]
- Badve, S.S.; Beitsch, P.D.; Bose, S.; Byrd, D.R.; Chen, V.W.; Connolly, J.L.; Dogan, B.; D’Orsi, C.J.; Stephen, E.B.; Giuliano, A.; et al. Breast. In Ajcc Cancer Staging Manual, 8th ed.; AJCC: Chicago, IL, USA, 2018. [Google Scholar]
- Elston, C.W.; Ellis, I.O. Pathological Prognostic Factors in Breast Cancer. I. The Value of Histological Grade in Breast Cancer: Experience from a Large Study with Long-Term Follow-Up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Bloom, H.J.; Richardson, W.W. Histological Grading and Prognosis in Breast Cancer; a Study of 1409 Cases of Which 359 Have Been Followed for 15 Years. Br. J. Cancer 1957, 11, 359–377. [Google Scholar] [CrossRef]
- Senkus, E.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rutgers, E.; Zackrisson, S.; Cardoso, F. “Primary Breast Cancer: Esmo Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up”. Ann. Oncol. 2015, 26, 8–30. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.D.; Cai, Q.; Shu, X.O.; Nechuta, S.J. The Role of Biomarkers of Oxidative Stress in Breast Cancer Risk and Prognosis: A Systematic Review of the Epidemiologic Literature. J. Womens Health (Larchmt) 2017, 26, 467–482. [Google Scholar] [CrossRef] [Green Version]
- Nourazarian, A.R.; Kangari, P.; Salmaninejad, A. Roles of Oxidative Stress in the Development and Progression of Breast Cancer. Asian Pac. J. Cancer Prev. 2014, 15, 4745–4751. [Google Scholar] [CrossRef]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of Oxidative Stress and the Microenvironment in Breast Cancer Development and Progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. Nrf2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Fuentes-Broto, L.; Martinez-Ballarin, E.; Miana-Mena, J.; Berzosa, C.; Piedrafita, E.; Cebrian, I.; Reiter, R.J.; Garcia, J.J. Lipid and Protein Oxidation in Hepatic Homogenates and Cell Membranes Exposed to Bile Acids. Free Radic. Res. 2009, 43, 1080–1089. [Google Scholar] [CrossRef]
- Fuentes-Broto, L.; Miana-Mena, F.J.; Piedrafita, E.; Berzosa, C.; Martinez-Ballarin, E.; Garcia-Gil, F.A.; Reiter, R.J.; Garcia, J.J. Melatonin Protects against Taurolithocholic-Induced Oxidative Stress in Rat Liver. J. Cell Biochem. 2010, 110, 1219–1225. [Google Scholar] [CrossRef]
- Chen, P.; Li, J.; Fan, X.; Zeng, H.; Deng, R.; Li, D.; Huang, M.; Bi, H. Oleanolic Acid Attenuates Obstructive Cholestasis in Bile Duct-Ligated Mice, Possibly Via Activation of Nrf2-Mrps and Fxr Antagonism. Eur. J. Pharmacol. 2015, 765, 131–139. [Google Scholar] [CrossRef]
- Liu, J.; Wu, K.C.; Lu, Y.F.; Ekuase, E.; Klaassen, C.D. Nrf2 Protection against Liver Injury Produced by Various Hepatotoxicants. Oxid. Med. Cell Longev. 2013, 2013, 305861. [Google Scholar] [CrossRef]
- Raju, U.; Levitz, M.; Javitt, N.B. Bile Acids in Human Breast Cyst Fluid: The Identification of Lithocholic Acid. J. Clin. Endocrinol. Metab. 1990, 70, 1030–1034. [Google Scholar] [CrossRef]
- Fodor, T.; Szanto, M.; Abdul-Rahman, O.; Nagy, L.; Der, A.; Kiss, B.; Bai, P. Combined Treatment of Mcf-7 Cells with Aicar and Methotrexate, Arrests Cell Cycle and Reverses Warburg Metabolism through Amp-Activated Protein Kinase (Ampk) and Foxo1. PLoS ONE 2016, 11, e0150232. [Google Scholar] [CrossRef]
- Szántó, M.; Brunyánszki, A.; Márton, J.; Vámosi, G.; Nagy, L.; Fodor, T.; Kiss, B.; Virag, L.; Gergely, P.; Bai, P. Deletion of Parp-2 Induces Hepatic Cholesterol Accumulation and Decrease in Hdl Levels. Biochem. Biophys. Acta Mol. Basis Dis. 2014, 1842, 594–602. [Google Scholar] [CrossRef]
- Nagy, L.; Marton, J.; Vida, A.; Kis, G.; Bokor, E.; Kun, S.; Gonczi, M.; Docsa, T.; Toth, A.; Antal, M.; et al. Glycogen Phosphorylase Inhibition Improves Beta Cell Function. Br. J. Pharmacol. 2018, 175, 301–319. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. Imagej2: Imagej for the Next Generation of Scientific Image Data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Lau, A.; Tian, W.; Whitman, S.A.; Zhang, D.D. The Predicted Molecular Weight of Nrf2: It Is What It Is Not. Antioxid Redox Signal. 2013, 18, 91–93. [Google Scholar] [CrossRef] [Green Version]
- Venkatraman, G.; Benesch, M.G.; Tang, X.; Dewald, J.; McMullen, T.P.; Brindley, D.N. Lysophosphatidate Signaling Stabilizes Nrf2 and Increases the Expression of Genes Involved in Drug Resistance and Oxidative Stress Responses: Implications for Cancer Treatment. FASEB J. 2015, 29, 772–785. [Google Scholar] [CrossRef]
- Mabley, J.G.; Pacher, P.; Bai, P.; Wallace, R.; Goonesekera, S.; Virag, L.; Southan, G.J.; Szabo, C. Suppression of Intestinal Polyposis in Apcmin/+ Mice by Targeting the Nitric Oxide or Poly(Adp-Ribose) Pathways. Mutat. Res. 2004, 548, 107–116. [Google Scholar] [CrossRef]
- Hegedus, C.; Lakatos, P.; Kiss-Szikszai, A.; Patonay, T.; Gergely, S.; Gregus, A.; Bai, P.; Hasko, G.; Szabo, E.; Virag, L. Cytoprotective Dibenzoylmethane Derivatives Protect Cells from Oxidative Stress-Induced Necrotic Cell Death. Pharmacol. Res. 2013, 72, 25–34. [Google Scholar] [CrossRef]
- Lanczky, A.; Nagy, A.; Bottai, G.; Munkacsy, G.; Szabo, A.; Santarpia, L.; Gyorffy, B. Mirpower: A Web-Tool to Validate Survival-Associated Mirnas Utilizing Expression Data from 2178 Breast Cancer Patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Csonka, T.; Murnyák, B.; Szepesi, R.; Bencze, J.; Bognár, L.; Klekner, Á.; Hortobágyi, T. Assessment of Candidate Immunohistochemical Prognostic Markers of Meningioma Recurrence. Folia Neuropathol. 2016, 54, 114–126. [Google Scholar] [CrossRef]
- Stenger, M. Calculating H-Score. Available online: http://www.ascopost.com/issues/april-10-2015/calculating-h-score/ (accessed on 7 January 2019).
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal Research: Reporting in Vivo Experiments: The Arrive Guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for Reporting Experiments Involving Animals: The Arrive Guidelines. Br. J. Pharmacol 2010, 160, 1573–1576. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 1 December 2018).
- Aryal, B.; Rao, V.A. Specific Protein Carbonylation in Human Breast Cancer Tissue Compared to Adjacent Healthy Epithelial Tissue. PLoS ONE 2018, 13, e0194164. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, Pathophysiology and Development of Therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Ingman, W.V. The Gut Microbiome: A New Player in Breast Cancer Metastasis. Cancer Res. 2019, 79, 3539–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchta Rosean, C.; Bostic, R.R.; Ferey, J.C.M.; Feng, T.Y.; Azar, F.N.; Tung, K.S.; Dozmorov, M.G.; Smirnova, E.; Bos, P.D.; Rutkowski, M.R. Preexisting Commensal Dysbiosis Is a Host-Intrinsic Regulator of Tissue Inflammation and Tumor Cell Dissemination in Hormone Receptor-Positive Breast Cancer. Cancer Res. 2019, 79, 3662–3675. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Putluri, V.; Ambati, C.R.; Dorsey, T.H.; Putluri, N.; Ambs, S. Liver- and Microbiome-Derived Bile Acids Accumulate in Human Breast Tumors and Inhibit Growth and Improve Patient Survival. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- NCBI_GEO_Profiles. Tgr5 in Breast Cancer. Available online: https://www.ncbi.nlm.nih.gov/geoprofiles/70190573 (accessed on 12 August 2018).
- Zhang, H.S.; Du, G.Y.; Zhang, Z.G.; Zhou, Z.; Sun, H.L.; Yu, X.Y.; Shi, Y.T.; Xiong, D.N.; Li, H.; Huang, Y.H. Nrf2 Facilitates Breast Cancer Cell Growth Via Hif1a-Mediated Metabolic Reprogramming. Int. J. Biochem. Cell Biol. 2018, 95, 85–92. [Google Scholar] [CrossRef]
- Dermit, M.; Casado, P.; Rajeeve, V.; Wilkes, E.H.; Foxler, D.E.; Campbell, H.; Critchlow, S.; Sharp, T.V.; Gribben, J.G.; Unwin, R.; et al. Oxidative Stress Downstream of Mtorc1 but Not Akt Causes a Proliferative Defect in Cancer Cells Resistant to Pi3k Inhibition. Oncogene 2017, 36, 2762–2774. [Google Scholar] [CrossRef]
- Lisek, K.; Walerych, D.; del Sal, G. Mutant P53-Nrf2 Axis Regulates the Proteasome Machinery in Cancer. Mol. Cell Oncol. 2017, 4, e1217967. [Google Scholar] [CrossRef]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Kovacs, K.; Polgar, Z.; Regdon, Z.; Szabo, E.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virag, L. Redox Control of Cancer Cell Destruction. Redox Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of Inducible Nitric Oxide Synthase (Inos) Expression on Triple Negative Breast Cancer Outcome and Activation of Egfr and Erk Signaling Pathways. Oncotarget 2017, 8, 80568–80588. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Krishnan, A.; Sivasithambaram, N.D. Significance of Twist and Inos Expression in Human Breast Carcinoma. Mol. Cell Biochem. 2016, 412, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Girotti, A.W. Upregulation of Nitric Oxide in Tumor Cells as a Negative Adaptation to Photodynamic Therapy. Lasers Surg Med. 2018, 50, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Davila, A.; Liu, L.; Chellappa, K.; Redpath, P.; Nakamaru-Ogiso, E.; Paolella, L.M.; Zhang, Z.; Migaud, M.E.; Rabinowitz, J.D.; Baur, J.A. Nicotinamide Adenine Dinucleotide Is Transported into Mammalian Mitochondria. Elife 2018, 7, 33246. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.M.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of Triple Negative Breast Cancer and the Impact of Inducible Nitric Oxide Synthase on Tumor Biology and Patient Outcomes. Crit. Rev. Oncog. 2016, 21, 333–351. [Google Scholar] [CrossRef]
- Davila-Gonzalez, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; et al. “Pharmacological Inhibition of Nos Activates Ask1/Jnk Pathway Augmenting Docetaxel-Mediated Apoptosis in Triple-Negative Breast Cancer”. Clin. Cancer Res. 2018, 24, 21152–21162. [Google Scholar] [CrossRef]
- Pons, D.G.; Nadal-Serrano, M.; Torrens-Mas, M.; Valle, A.; Oliver, J.; Roca, P. Ucp2 Inhibition Sensitizes Breast Cancer Cells to Therapeutic Agents by Increasing Oxidative Stress. Free Radic. Biol. Med. 2015, 86, 67–77. [Google Scholar] [CrossRef]
- Onodera, Y.; Motohashi, H.; Takagi, K.; Miki, Y.; Shibahara, Y.; Watanabe, M.; Ishida, T.; Hirakawa, H.; Sasano, H.; Yamamoto, M.; et al. Nrf2 Immunolocalization in Human Breast Cancer Patients as a Prognostic Factor. Endocr Relat. Cancer. 2014, 21, 241–252. [Google Scholar] [CrossRef]
- Hartikainen, J.M.; Tengstrom, M.; Winqvist, R.; Jukkola-Vuorinen, A.; Pylkas, K.; Kosma, V.M.; Soini, Y.; Mannermaa, A. Keap1 Genetic Polymorphisms Associate with Breast Cancer Risk and Survival Outcomes. Clin. Cancer Res. 2015, 21, 1591–1601. [Google Scholar] [CrossRef]
- Hartikainen, J.M.; Tengstrom, M.; Kosma, V.M.; Kinnula, V.L.; Mannermaa, A.; Soini, Y. Genetic Polymorphisms and Protein Expression of Nrf2 and Sulfiredoxin Predict Survival Outcomes in Breast Cancer. Cancer Res. 2012, 72, 5537–5546. [Google Scholar] [CrossRef]
- Al Azhary, N.M.; Kamel, M.M.; Ismail, Y.M.; Mahmoud, A.A.; Radwan, E.M. The Role of Genetic Polymorphisms in Nrf2 and P73 in Egyptian Women with Breast Cancer. Asian Pac. J. Cancer Prev. 2016, 17, 4945–4949. [Google Scholar]
- Syu, J.P.; Chi, J.T.; Kung, H.N. Nrf2 Is the Key to Chemotherapy Resistance in Mcf7 Breast Cancer Cells under Hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Choi, B.H.; Ku, S.K.; Kwak, M.K. High Cd44 Expression Mediates P62-Associated Nfe2l2/Nrf2 Activation in Breast Cancer Stem Cell-Like Cells: Implications for Cancer Stem Cell Resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef]
- Hsu, W.T.; Huang, C.Y.; Yen, C.Y.T.; Cheng, A.L.; Hsieh, P.C.H. The Her2 Inhibitor Lapatinib Potentiates Doxorubicin-Induced Cardiotoxicity through Inos Signaling. Theranostics 2018, 8, 3176–3188. [Google Scholar] [CrossRef]
- Velicer, C.M.; Heckbert, S.R.; Lampe, J.W.; Potter, J.D.; Robertson, C.A.; Taplin, S.H. Antibiotic Use in Relation to the Risk of Breast Cancer. JAMA 2004, 291, 827–835. [Google Scholar] [CrossRef]
- Velicer, C.M.; Heckbert, S.R.; Rutter, C.; Lampe, J.W.; Malone, K. Association between Antibiotic Use Prior to Breast Cancer Diagnosis and Breast Tumour Characteristics (United States). Cancer Causes Control. 2006, 17, 307–313. [Google Scholar] [CrossRef]
- Wirtz, H.S.; Buist, D.S.; Gralow, J.R.; Barlow, W.E.; Gray, S.; Chubak, J.; Yu, O.; Bowles, E.J.; Fujii, M.; Boudreau, D.M. Frequent Antibiotic Use and Second Breast Cancer Events. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1588–1599. [Google Scholar] [CrossRef]
- Tamim, H.M.; Hanley, J.A.; Hajeer, A.H.; Boivin, J.F.; Collet, J.P. Risk of Breast Cancer in Relation to Antibiotic Use. Pharmacoepidemiol. Drug Saf. 2008, 17, 144–150. [Google Scholar] [CrossRef]
- Satram-Hoang, S.; Moran, E.M.; Anton-Culver, H.; Burras, R.W.; Heimann, T.M.; Boggio, I.; Dykstra-Long, G.R.; Wood, P.A.; Zulka, R.; Hufnagel, G.; et al. A Pilot Study of Male Breast Cancer in the Veterans Affairs Healthcare System. J. Environ. Pathol. Toxicol. Oncol. 2010, 29, 235–244. [Google Scholar] [CrossRef]
- Kirkup, B.; McKee, A.; Makin, K.; Paveley, J.; Caim, S.; Alcon-Giner, C.; Leclaire, C.; Dalby, M.; le Gall, G.; Andrusaite, A.; et al. Perturbation of the Gut Microbiota by Antibiotics Results in Accelerated Breast Tumour Growth and Metabolic Dysregulation. bioRxiv 2019. [Google Scholar] [CrossRef]
- Chen, M.C.; Chen, Y.L.; Wang, T.W.; Hsu, H.P.; Lai, M.D. Membrane Bile Acid Receptor Tgr5 Predicts Good Prognosis in Ampullary Adenocarcinoma Patients with Hyperbilirubinemia. Oncol Rep. 2016, 36, 1997–2008. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | MURINE Forward Primer (5′–3′) | Murine Reverse Primer (5′–3′) |
|---|---|---|
| CAT | CCTTCAAGTTGGTTAATGCAGA | CAAGTTTTTGATGCCCTGGT |
| GCLC | GATTCGGGATGGGCAACT | AAAGGTATCTTGCCTCAGATATGC |
| GPX2 | GTTCTCGGCTTCCCTTGC | TTCAGGATCTCCTCGTTCTGA |
| GPX3 | GGCTTCCCTTCCAACCAA | CCCACCTGGTCGAACATACT |
| HMOX1 | AGGCTAAGACCGCCTTCCT | TGTGTTCCTCTGTCAGCATCA |
| iNOS | GAAGTGCAAAGTCTCAGACATGG | GATTCTGGAACATTCTGTGCTGTC |
| NOX4 | GCAGATTTACTCTGTGTGTTGCAT | TCCCATCTGTTTGACTGAGGT |
| NQO1 | AGCGTTCGGTATTACGATCC | AGTACAATCAGGGCTCTTCTCG |
| NRF2 | CATCAGGCCCAGTCCCTCAAT | CAGCGGTAGTATAÓCAGCCAGCT |
| SOD1 | CCATCAGTATGGGGACAATACA | GGTCTCCAACATGCCTCTCT |
| SOD2 | TGCTCTAATCAGGACCCATTG | GTAGTAAGCGTGCTCCCACAC |
| SOD3 | CTCTTGGGAGAGCCTGACA | GCCAGTAGCAAGCCGTAGAA |
| 36B4 | AGATTCGGGATATGCTGTTGG | AAAGCCTGGAAGAAGGAGGTC |
| Antibody Symbol | Vendor | Dilution |
|---|---|---|
| NRF2 | Abcam (ab31163) | 1:1000 |
| NRF2 | Novus (NBP1-32822 | 1:1000 |
| KEAP1 | Cell Signaling (8047) | 1:1000 |
| GPX3 | Abcam (ab104448) | 1:1000 |
| iNOS | Novus (NB300-605) | 1:1000 |
| 4HNE | Abcam (ab46545) | 1:1000 |
| Nitrotyrosine | Millipore (06-284) | 1:1000 |
| TGR5/GPBAR1 | Novus (NBP2-23669) | 1:1000 |
| CAR | Abcam (ab186869) | 1:1000 |
| ACTIN | Sigma-Aldrich (A3854) | 1:20000 |
| Antibody Symbol | Vendor | Antigen Retrieval | Dilution | Detection |
|---|---|---|---|---|
| NRF2 | Abcam (ab31163) | Ventana BenchMark ULTRA/Roche Cell Conditioning 1 (CC1) 40 min, 95 °C | 1:100 | OptiView DAB IHC Detection kit/Roche |
| iNOS | ThermoFisher Scientific (PA5-16855) | Ventana BenchMark ULTRA/Roche Cell Conditioning 1 (CC1) 20 min, 95 °C | 1:100 | UltraView Universal DAB Detection kit/Roche |
| 4HNE | Abcam (ab46545) | Ventana BenchMark ULTRA/Roche Cell Conditioning 1 (CC1) 20 min, 95 °C | 1:1000 | UltraView Universal DAB Detection kit/Roche |
| TGR5 | GeneTEX (GTX100026, Hsinchu City, Taiwan) | pressure cooker (Avair) in 0.1 m citrate buffer at pH 6 | 1:1000 | EnVision Flex (K8000, Dako, Santa Clara, CA, USA) |
| CAR (207007_a_at) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 0.7 | 3.00 × 10−10 |
| Grade 1, N = 345 | 1.15 | 0.61 |
| Grade 2, N = 901 | 0.88 | 0.3 |
| Grade 3, N = 903 | 0.72 | 0.0036 |
| ER(+), N = 3082 | 0.81 | 0.0013 |
| ER(−), N = 869 | 0.62 | 9.20 × 10−6 |
| PR(+), N = 589 | 0.72 | 0.063 |
| PR(−), N = 549 | 0.9 | 0.49 |
| Lymph(+), N = 1133 | 0.97 | 0.73 |
| Lymph(−), N = 2020 | 0.99 | 0.93 |
| HER2(+), N = 252 | 0.69 | 0.1 |
| HER2(−), N = 800 | 1.13 | 0.37 |
| ER(+), PR(+), N = 577 | 0.79 | 0.19 |
| ER(+), PR(+), Lymph node (+),N = 344 | 0.99 | 0.97 |
| ER(+), PR(+), Lymph node (−),N = 228 | 0.5 | 0.037 |
| ER(−), PR(−), N = 298 | 0.74 | 0.14 |
| ER(−), PR(−), Lymph node(+),N = 127 | 0.76 | 0.3 |
| ER(−), PR(−), Lymph node(−),N = 167 | 0.97 | 0.93 |
| ER(−), PR(−), HER2(−),N = 198 | 0.93 | 0.77 |
| Basal subtype, N = 618 | 0.68 | 0.0025 |
| Luminal A, N = 1933 | 0.76 | 0.0015 |
| Luminal B, N = 1149 | 0.77 | 0.0069 |
| ER(+), HER2(+), N = 156 | 1.21 | 0.53 |
| ER(−), HER2(+), N = 96 | 0.61 | 0.12 |
| TGR5 (1552501_at) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 0.91 | 0.25 |
| Grade 1, N = 345 | 0.41 | 0.12 |
| Grade 2, N = 901 | 1.21 | 0.46 |
| Grade 3, N = 903 | 0.77 | 0.1 |
| ER(+), N = 3082 | 1 | 0.98 |
| ER(−), N = 869 | 0.74 | 0.025 |
| PR(+), N = 589 | 1.21 | 0.33 |
| PR(−), N = 549 | 0.96 | 0.81 |
| Lymph(+), N = 1133 | 0.98 | 0.9 |
| Lymph(−), N = 2020 | 0.91 | 0.61 |
| HER2(+), N = 252 | 1.12 | 0.68 |
| HER2(−), N = 800 | 0.91 | 0.51 |
| ER(+), PR(+), N = 577 | 1.18 | 0.4 |
| ER(+), PR(+), Lymph node(+),N = 344 | 1.39 | 0.19 |
| ER(+), PR(+), Lymph node(−),N = 228 | 0.76 | 0.4 |
| ER(−), PR(−), N = 298 | 0.96 | 0.86 |
| ER(−), PR(−), Lymph node(+),N = 127 | 0.96 | 0.89 |
| ER(−), PR(−), Lymph node(−),N = 167 | 1.25 | 0.61 |
| ER(−), PR(−), HER2(−),N = 198 | 0.9 | 0.73 |
| Basal subtype, N = 618 | 0.79 | 0.15 |
| Luminal A, N = 1933 | 1.1 | 0.44 |
| Luminal B, N = 1149 | 0.83 | 0.24 |
| ER(+), HER2(+), N = 156 | 1.07 | 0.88 |
| ER(−), HER2(+), N = 96 | 0.94 | 0.86 |
| NRF2 (201145_at) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 1.23 | 2.00 × 10−4 |
| Grade 1, N = 345 | 1.1 | 0.72 |
| Grade 2, N = 901 | 0.87 | 0.25 |
| Grade 3, N = 903 | 1.06 | 0.63 |
| ER(+), N = 3082 | 1.27 | 2.40 × 10−4 |
| ER(−), N = 869 | 1.04 | 0.71 |
| PR(+), N = 589 | 1.23 | 0.24 |
| PR(−), N = 549 | 1.49 | 0.0075 |
| Lymph(+), N = 1133 | 1.14 | 0.21 |
| Lymph(−), N = 2020 | 1.11 | 0.21 |
| HER2(+), N = 252 | 1.32 | 0.21 |
| HER2(−), N = 800 | 1.34 | 0.031 |
| ER(+), PR(+), N = 577 | 1.27 | 0.19 |
| ER(+), PR(+), Lymph node(+),N = 344 | 1.08 | 0.71 |
| ER(+), PR(+), Lymph node(−),N = 228 | 1.96 | 0.051 |
| ER(−), PR(−), N = 298 | 1.01 | 0.95 |
| ER(−), PR(−), Lymph node(+),N = 127 | 1.23 | 0.45 |
| ER(−), PR(−), Lymph node(−),N = 167 | 0.93 | 0.8 |
| ER(−), PR(−), HER2(−),N = 198 | 0.9 | 0.66 |
| Basal subtype, N = 618 | 1.01 | 0.95 |
| Luminal A, N = 1933 | 1.33 | 0.0011 |
| Luminal B, N = 1149 | 1.26 | 0.017 |
| ER(+), HER2(+), N = 156 | 1.69 | 0.097 |
| ER(−), HER2(+), N = 96 | 1.26 | 0.47 |
| KEAP1 (202417_at) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 0.84 | 0.0015 |
| Grade 1, N = 345 | 0.98 | 0.94 |
| Grade 2, N = 901 | 0.73 | 0.012 |
| Grade 3, N = 903 | 0.93 | 0.54 |
| ER(+), N = 3082 | 0.89 | 0.06 |
| ER(−), N = 869 | 0.92 | 0.41 |
| PR(+), N = 589 | 1.09 | 0.64 |
| PR(−), N = 549 | 0.91 | 0.55 |
| Lymph(+), N = 1133 | 0.76 | 0.006 |
| Lymph(−), N = 2020 | 0.98 | 0.83 |
| HER2(+), N = 252 | 1.14 | 0.57 |
| HER2(−), N = 800 | 0.92 | 0.55 |
| ER(+), PR(+), N = 577 | 1.06 | 0.76 |
| ER(+), PR(+), Lymph node(+),N = 344 | 0.98 | 0.91 |
| ER(+), PR(+), Lymph node(−),N = 228 | 1.38 | 0.33 |
| ER(−), PR(−), N = 298 | 0.95 | 0.82 |
| ER(−), PR(−), Lymph node(+),N = 127 | 1 | 1 |
| ER(−), PR(−), Lymph node(−),N = 167 | 1.12 | 0.72 |
| ER(−), PR(−), HER2(−),N = 198 | 1.07 | 0.8 |
| Basal subtype, N = 618 | 1 | 0.98 |
| Luminal A, N = 1933 | 0.9 | 0.24 |
| Luminal B, N = 1149 | 0.91 | 0.36 |
| ER(+), HER2(+), N = 156 | 0.89 | 0.7 |
| ER(−), HER2(+), N = 96 | 1.18 | 0.61 |
| iNOS (210037_at) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 0.72 | 5.40 × 10−9 |
| Grade 1, N = 345 | 1.21 | 0.48 |
| Grade 2, N = 901 | 0.95 | 0.68 |
| Grade 3, N = 903 | 0.85 | 0.15 |
| ER(+), N = 3082 | 0.8 | 0.00066 |
| ER(−), N = 869 | 0.63 | 1.40 × 10−5 |
| PR(+), N = 589 | 1.34 | 0.099 |
| PR(−), N = 549 | 1.01 | 0.95 |
| Lymph(+), N = 1133 | 1.12 | 0.25 |
| Lymph(−), N = 2020 | 1 | 0.96 |
| HER2(+), N = 252 | 0.79 | 0.3 |
| HER2(−), N = 800 | 1.04 | 0.75 |
| ER(+), PR(+), N = 577 | 1.32 | 0.13 |
| ER(+), PR(+), Lymph node(+),N = 344 | 1.52 | 0.058 |
| ER(+), PR(+), Lymph node(−),N = 228 | 1.04 | 0.9 |
| ER(−), PR(−), N = 298 | 0.90 | 0.59 |
| ER(−), PR(−), Lymph node(+),N = 127 | 1.03 | 0.91 |
| ER(−), PR(−), Lymph node(−),N = 167 | 0.91 | 0.77 |
| ER(−), PR(−), HER2(−),N = 198 | 0.8 | 0.37 |
| Basal subtype, N = 618 | 0.63 | 0.00042 |
| Luminal A, N = 1933 | 0.75 | 0.00082 |
| Luminal B, N = 1149 | 0.77 | 0.0077 |
| ER(+), HER2(+), N = 156 | 1.04 | 0.9 |
| ER(−), HER2(+), N = 96 | 0.92 | 0.79 |
| nNOS (207309_at) | HR (Hazard Ratio) | p-value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 0.71 | 6.70 × 10−10 |
| Grade 1, N = 345 | 0.85 | 0.55 |
| Grade 2, N = 901 | 0.99 | 0.96 |
| Grade 3, N = 903 | 0.91 | 0.41 |
| ER(+), N = 3082 | 0.77 | 5.80 × 10−5 |
| ER(−), N = 869 | 0.68 | 2.80 × 10−4 |
| PR(+), N = 589 | 0.79 | 0.19 |
| PR(−), N = 549 | 1.07 | 0.67 |
| Lymph(+), N = 1133 | 1.08 | 0.47 |
| Lymph(−), N = 2020 | 0.91 | 0.25 |
| HER2(+), N = 252 | 0.7 | 0.11 |
| HER2(−), N = 800 | 0.92 | 0.51 |
| ER(+), PR(+), N = 577 | 0.86 | 0.41 |
| ER(+), PR(+), Lymph node(+),N = 344 | 1.23 | 0.35 |
| ER(+), PR(+), Lymph node(−),N = 228 | 0.34 | 0.0055 |
| ER(−), PR(−), N = 298 | 0.96 | 0.83 |
| ER(−), PR(−), Lymph node(+),N = 127 | 1.07 | 0.8 |
| ER(−), PR(−), Lymph node(−),N = 167 | 1.55 | 0.16 |
| ER(−), PR(−), HER2(−),N = 198 | 1.17 | 0.53 |
| Basal subtype, N = 618 | 0.73 | 0.013 |
| Luminal A, N = 1933 | 0.72 | 0.00019 |
| Luminal B, N = 1149 | 0.69 | 0.00014 |
| ER(+), HER2(+), N = 156 | 1.4 | 0.28 |
| ER(−), HER2(+), N = 96 | 0.48 | 0.024 |
| nNOS (207310_at) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 0.8 | 3.80 × 10−5 |
| Grade 1, N = 345 | 1.27 | 0.37 |
| Grade 2, N = 901 | 1.20 | 0.14 |
| Grade 3, N = 903 | 0.89 | 0.27 |
| ER(+), N = 3082 | 0.84 | 7.70 × 10−3 |
| ER(−), N = 869 | 0.72 | 1.90 × 10−3 |
| PR(+), N = 589 | 1.07 | 0.72 |
| PR(−), N = 549 | 1.04 | 0.8 |
| Lymph(+), N = 1133 | 1 | 0.97 |
| Lymph(−), N = 2020 | 1.09 | 0.34 |
| HER2(+), N = 252 | 0.78 | 0.26 |
| HER2(−), N = 800 | 1 | 0.97 |
| ER(+), PR(+), N = 577 | 1.07 | 0.73 |
| ER(+), PR(+), Lymph node(+),N = 344 | 1.51 | 0.06 |
| ER(+), PR(+), Lymph node(−),N = 228 | 0.59 | 0.13 |
| ER(−), PR(−), N = 298 | 1.18 | 0.41 |
| ER(−), PR(−), Lymph node(+),N = 127 | 1.02 | 0.93 |
| ER(−), PR(−), Lymph node(−),N = 167 | 1.42 | 0.25 |
| ER(−), PR(−), HER2(−),N = 198 | 1.44 | 0.14 |
| Basal subtype, N = 618 | 0.76 | 0.037 |
| Luminal A, N = 1933 | 0.75 | 0.0011 |
| Luminal B, N = 1149 | 0.85 | 0.09 |
| ER(+), HER2(+), N = 156 | 1.15 | 0.65 |
| ER(−), HER2(+), N = 96 | 0.45 | 0.013 |
| NOX4 (236843_at) | HR (Hazard Ratio) | p-Value (Log Rank Test) |
| All Breast Cancers, N = 3951 | 0.75 | 0.00027 |
| Grade 1, N = 345 | 0.76 | 0.6 |
| Grade 2, N = 901 | 0.68 | 0.14 |
| Grade 3, N = 903 | 0.69 | 0.79 |
| ER(+), N = 3082 | 0.7 | 0.00027 |
| ER(−), N = 869 | 0.8 | 0.1 |
| PR(+), N = 589 | 0.71 | 0.082 |
| PR(−), N = 549 | 1.04 | 0.85 |
| Lymph(+), N = 1133 | 0.78 | 0.057 |
| Lymph(−), N = 2020 | 1.08 | 0.71 |
| HER2(+), N = 252 | 1.31 | 0.33 |
| HER2(−), N = 800 | 0.81 | 0.16 |
| ER(+), PR(+), N = 577 | 0.69 | 0.06 |
| ER(+), PR(+), Lymph node(+),N = 344 | 0.73 | 0.21 |
| ER(+), PR(+), Lymph node(−),N = 228 | 0.97 | 0.93 |
| ER(−), PR(−), N = 298 | 1.23 | 0.41 |
| ER(−), PR(−), Lymph node(+),N = 127 | 1.35 | 0.34 |
| ER(−), PR(−), Lymph node(−),N = 167 | 1.07 | 0.87 |
| ER(−), PR(−), HER2(−),N = 198 | 1.25 | 0.49 |
| Basal subtype, N = 618 | 0.72 | 0.046 |
| Luminal A, N = 1933 | 0.71 | 0.0061 |
| Luminal B, N = 1149 | 0.77 | 0.089 |
| ER(+), HER2(+), N = 156 | 1.34 | 0.51 |
| ER(−), HER2(+), N = 96 | 1.5 | 0.25 |
| Probe | Time (Months) | HR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 50 | 100 | 150 | 200 | 250 | |||||
| All cancers | Number at risk | CAR | low | 1998 | 1154 | 486 | 124 | 10 | 2 | 0.7 (0.63–0.79) |
| 207007_at | high | 1953 | 1365 | 589 | 117 | 17 | 1 | |||
| TGR5 | low | 58 | 41 | 13 | 1 | 0.41 (0.13–1.3) | ||||
| 1552501_a_at | high | 50 | 44 | 15 | 4 | |||||
| NRF2 | low | 1977 | 1368 | 624 | 145 | 11 | 0 | 1.23 (1.1–1.37) | ||
| 201146_at | high | 1974 | 1151 | 451 | 96 | 16 | 3 | |||
| KEAP1 | low | 1978 | 1181 | 513 | 131 | 17 | 2 | 0.84 (0.75–0.93) | ||
| 202417_at | high | 1973 | 1338 | 562 | 110 | 10 | 1 | |||
| iNOS | low | 1993 | 1154 | 446 | 107 | 14 | 2 | 0.72 (0.65–0.81) | ||
| 210037_s_at | high | 1958 | 1365 | 629 | 134 | 13 | 1 | |||
| nNOS | low | 1981 | 1128 | 454 | 114 | 14 | 3 | 0.71 (0.64–0.79) | ||
| 207309_at | high | 1970 | 1391 | 621 | 127 | 13 | 0 | |||
| nNOS | low | 2016 | 1185 | 470 | 112 | 15 | 2 | 0.8 (0.71–0.89) | ||
| 207310_s_at | high | 1935 | 1334 | 605 | 129 | 12 | 1 | |||
| NOX4 | low | 885 | 464 | 145 | 32 | 6 | 1 | 0.75 (0.64–0.88) | ||
| 236843_at | high | 879 | 513 | 200 | 36 | 4 | 1 | |||
| ER + | Number at risk | CAR | low | 1561 | 1024 | 443 | 114 | 12 | 3 | 0.81 (0.71–0.92) |
| 207007_at | high | 1521 | 1092 | 476 | 87 | 10 | 0 | |||
| TGR5 | low | 628 | 382 | 149 | 25 | 5 | 1 | 1 (0.83–1.22) | ||
| 1552501_a_at | high | 620 | 399 | 139 | 35 | 5 | 1 | |||
| NRF2 | low | 1541 | 1132 | 525 | 113 | 6 | 0 | 1.27 (1.12–1.44) | ||
| 201146_at | high | 1541 | 984 | 394 | 88 | 16 | 3 | |||
| KEAP1 | low | 1544 | 1011 | 448 | 106 | 14 | 2 | 0.89 (0.78–1.01) | ||
| 202417_at | high | 1538 | 1105 | 471 | 95 | 8 | 1 | |||
| iNOS | low | 1543 | 995 | 393 | 97 | 14 | 2 | 0.8 (0.71–0.91) | ||
| 210037_s_at | high | 1539 | 1121 | 526 | 104 | 8 | 1 | |||
| nNOS | low | 1544 | 986 | 407 | 105 | 17 | 3 | 0.77 (0.68–0.88) | ||
| 207309_at | high | 1538 | 1130 | 512 | 96 | 5 | 0 | |||
| nNOS | low | 1561 | 1013 | 409 | 101 | 12 | 2 | 0.84 (0.74–0.96) | ||
| 207310_s_at | high | 1521 | 1103 | 510 | 100 | 10 | 1 | |||
| NOX4 | low | 639 | 387 | 129 | 28 | 6 | 1 | 0.7 (0.57–0.85) | ||
| 236843_at | high | 609 | 394 | 159 | 32 | 4 | 1 | |||
| Triple negative | Number at risk | CAR | low | 99 | 42 | 13 | 1 | 0 | 0.93 (0.57–1.51) | |
| 207007_at | high | 99 | 41 | 6 | 1 | 0 | ||||
| TGR5 | low | 64 | 30 | 8 | 1 | 0 | 0.9 (0.48–1.68) | |||
| 1552501_a_at | high | 62 | 29 | 9 | 1 | 0 | ||||
| NRF2 | low | 99 | 41 | 5 | 1 | 0 | 0.9 (0.55–1.45) | |||
| 201146_at | high | 99 | 42 | 14 | 1 | 0 | ||||
| KEAP1 | low | 99 | 44 | 12 | 2 | 0 | 1.07 (0.66–1.73) | |||
| 202417_at | high | 99 | 39 | 7 | 0 | 0 | ||||
| iNOS | low | 99 | 44 | 15 | 1 | 0 | 0.8 (0.49–1.3) | |||
| 210037_s_at | high | 99 | 39 | 4 | 1 | 0 | ||||
| nNOS | low | 100 | 44 | 16 | 1 | 0 | 1.77 (0.72–1.9) | |||
| 207309_at | high | 98 | 39 | 3 | 1 | 0 | ||||
| nNOS | low | 102 | 51 | 14 | 2 | 0 | 1.44 (0.89–2.35) | |||
| 207310_s_at | high | 96 | 32 | 5 | 0 | 0 | ||||
| NOX4 | low | 64 | 30 | 8 | 2 | 0 | 1.25 (0.66–2.35) | |||
| 236843_at | high | 62 | 29 | 9 | 0 | 0 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, P.; Csonka, T.; Kovács, T.; Sári, Z.; Ujlaki, G.; Sipos, A.; Karányi, Z.; Szeőcs, D.; Hegedűs, C.; Uray, K.; et al. Lithocholic Acid, a Metabolite of the Microbiome, Increases Oxidative Stress in Breast Cancer. Cancers 2019, 11, 1255. https://doi.org/10.3390/cancers11091255
Kovács P, Csonka T, Kovács T, Sári Z, Ujlaki G, Sipos A, Karányi Z, Szeőcs D, Hegedűs C, Uray K, et al. Lithocholic Acid, a Metabolite of the Microbiome, Increases Oxidative Stress in Breast Cancer. Cancers. 2019; 11(9):1255. https://doi.org/10.3390/cancers11091255
Chicago/Turabian StyleKovács, Patrik, Tamás Csonka, Tünde Kovács, Zsanett Sári, Gyula Ujlaki, Adrien Sipos, Zsolt Karányi, Dóra Szeőcs, Csaba Hegedűs, Karen Uray, and et al. 2019. "Lithocholic Acid, a Metabolite of the Microbiome, Increases Oxidative Stress in Breast Cancer" Cancers 11, no. 9: 1255. https://doi.org/10.3390/cancers11091255
APA StyleKovács, P., Csonka, T., Kovács, T., Sári, Z., Ujlaki, G., Sipos, A., Karányi, Z., Szeőcs, D., Hegedűs, C., Uray, K., Jankó, L., Kiss, M., Kiss, B., Laoui, D., Virág, L., Méhes, G., Bai, P., & Mikó, E. (2019). Lithocholic Acid, a Metabolite of the Microbiome, Increases Oxidative Stress in Breast Cancer. Cancers, 11(9), 1255. https://doi.org/10.3390/cancers11091255