ADAM17: An Emerging Therapeutic Target for Lung Cancer


Abstract
:1. Introduction
2. Lung Cancer Risk Factors
2.1. Genetic
2.1.1. KRAS Mutations
2.1.2. EGFR Mutations
2.2. Cigarette Smoking
2.3. Inflammation
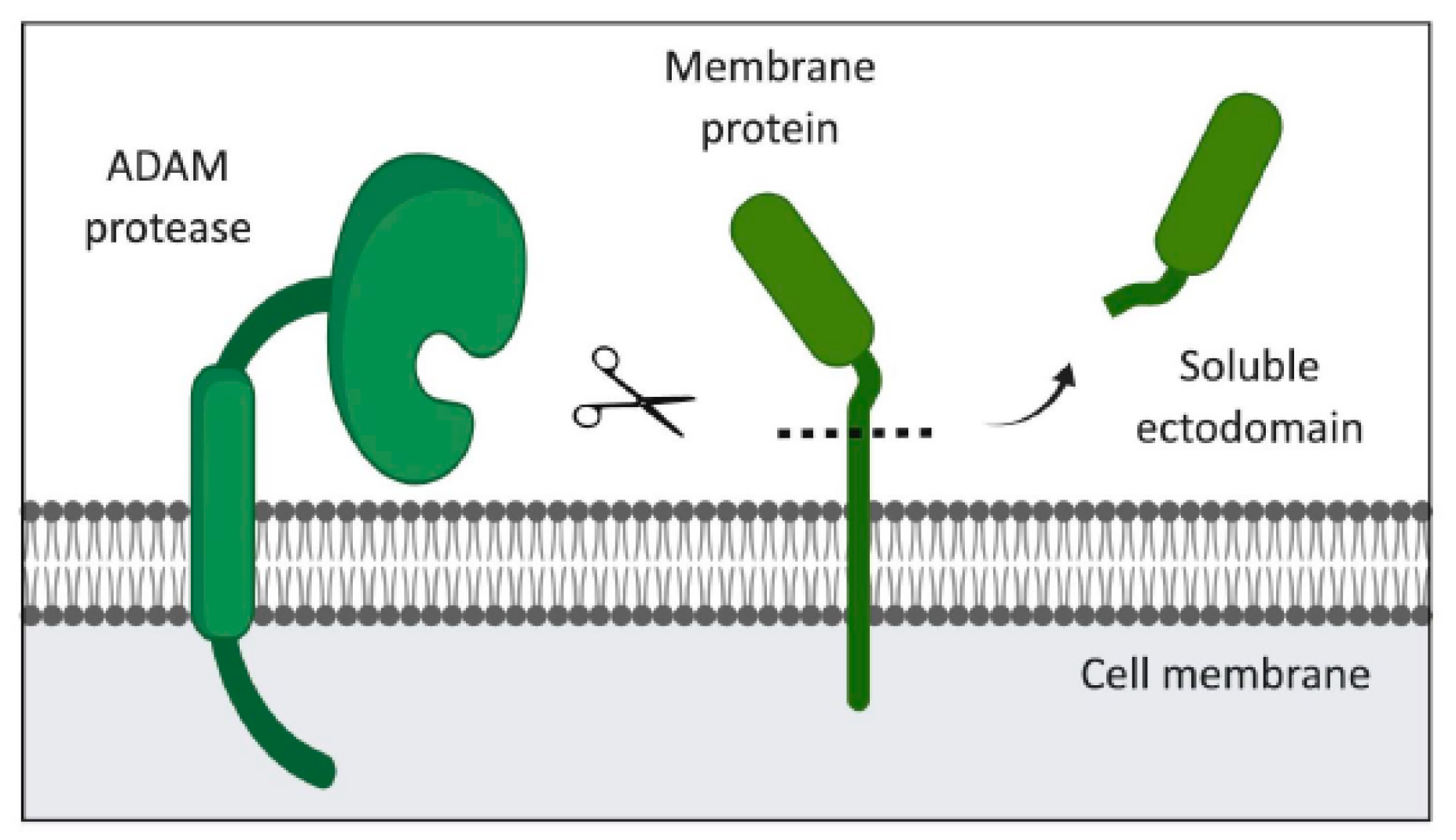
3. The ADAM Family of Proteases
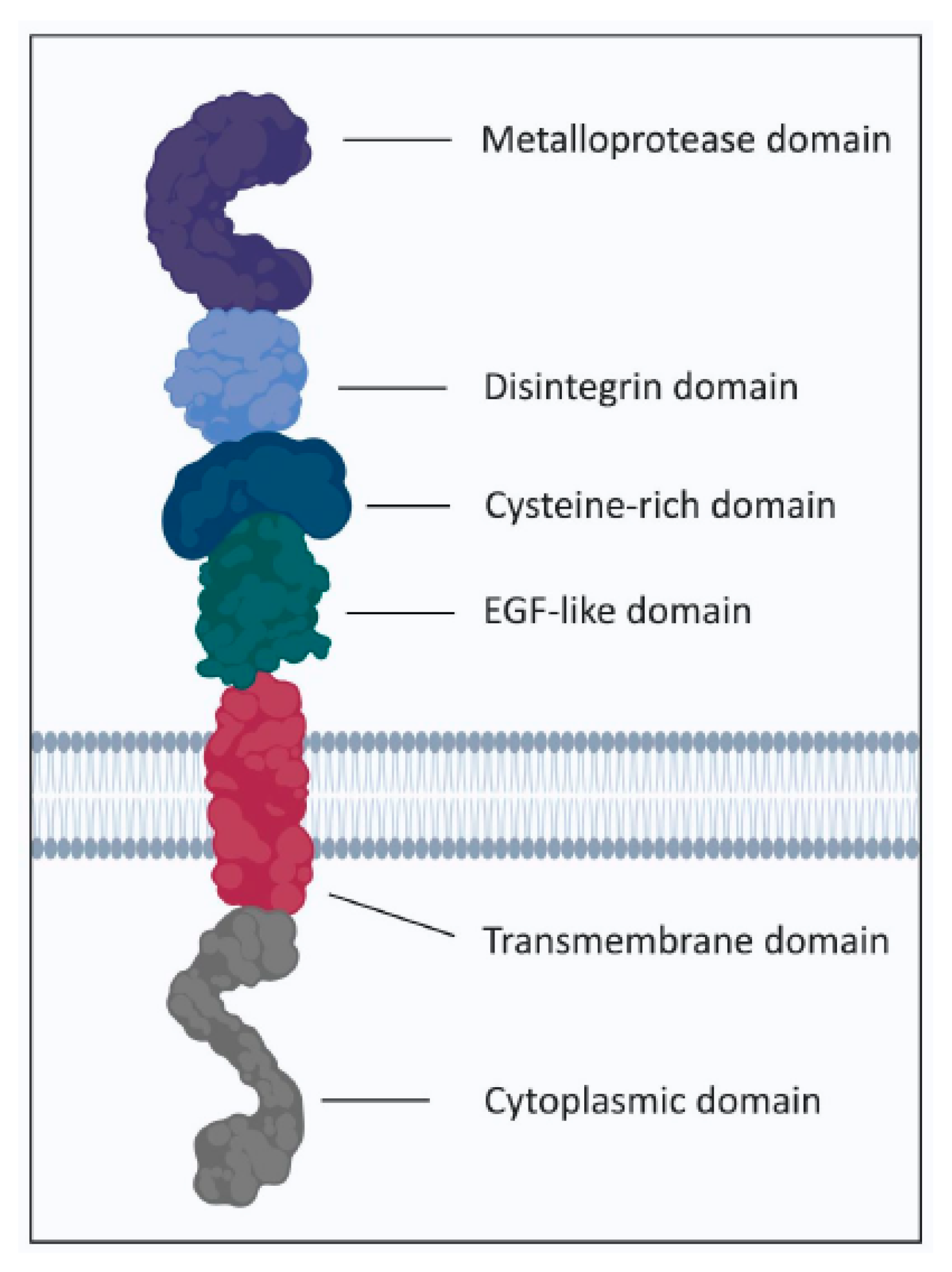
3.1. ADAM17
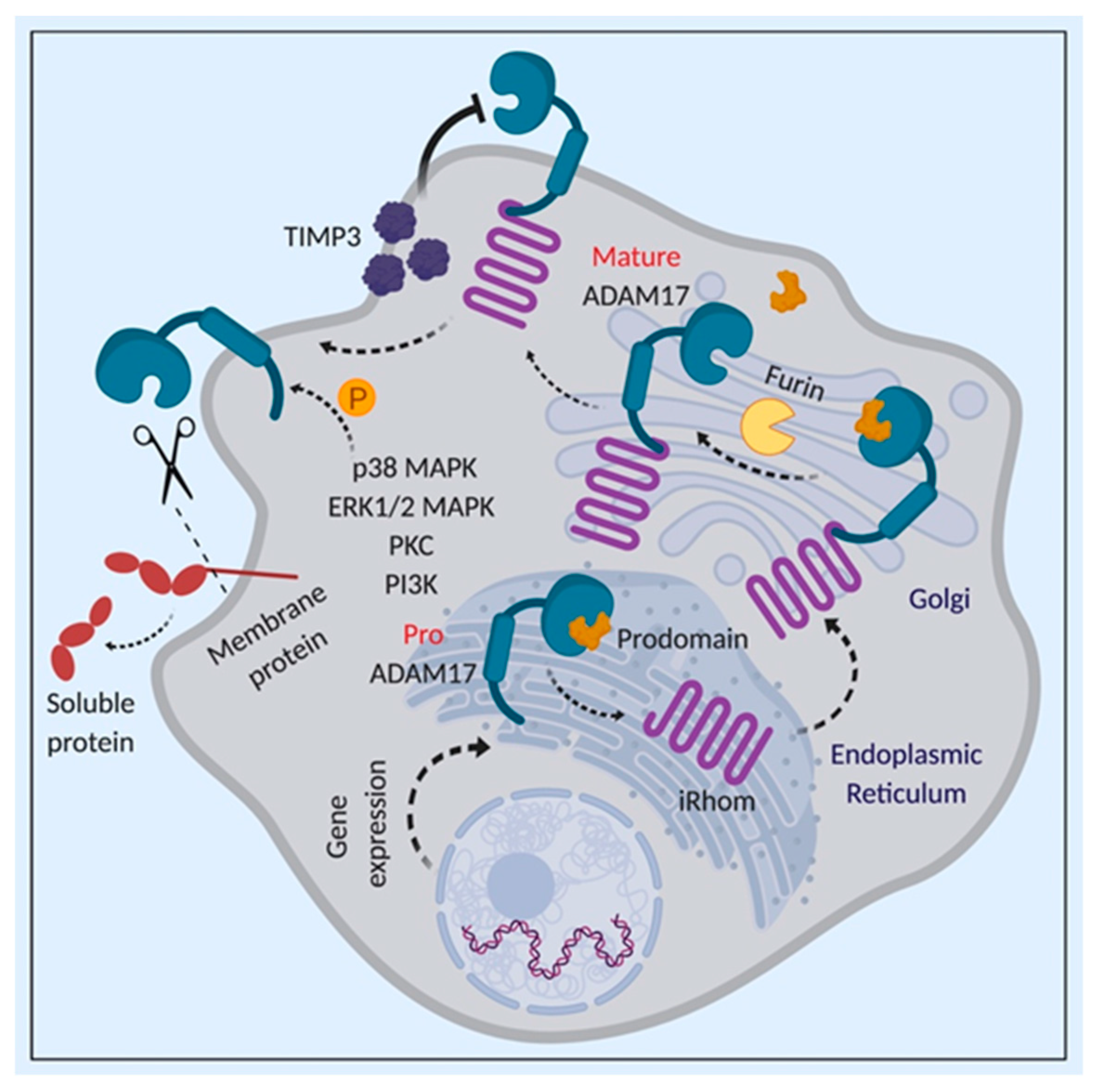
3.2. Regulation of ADAM17 Activity
4. ADAM17 and Its Role in Cancer
4.1. ADAM17 and Its Substrates in Lung Cancer
4.1.1. EGFR Ligands
4.1.2. Notch Signaling
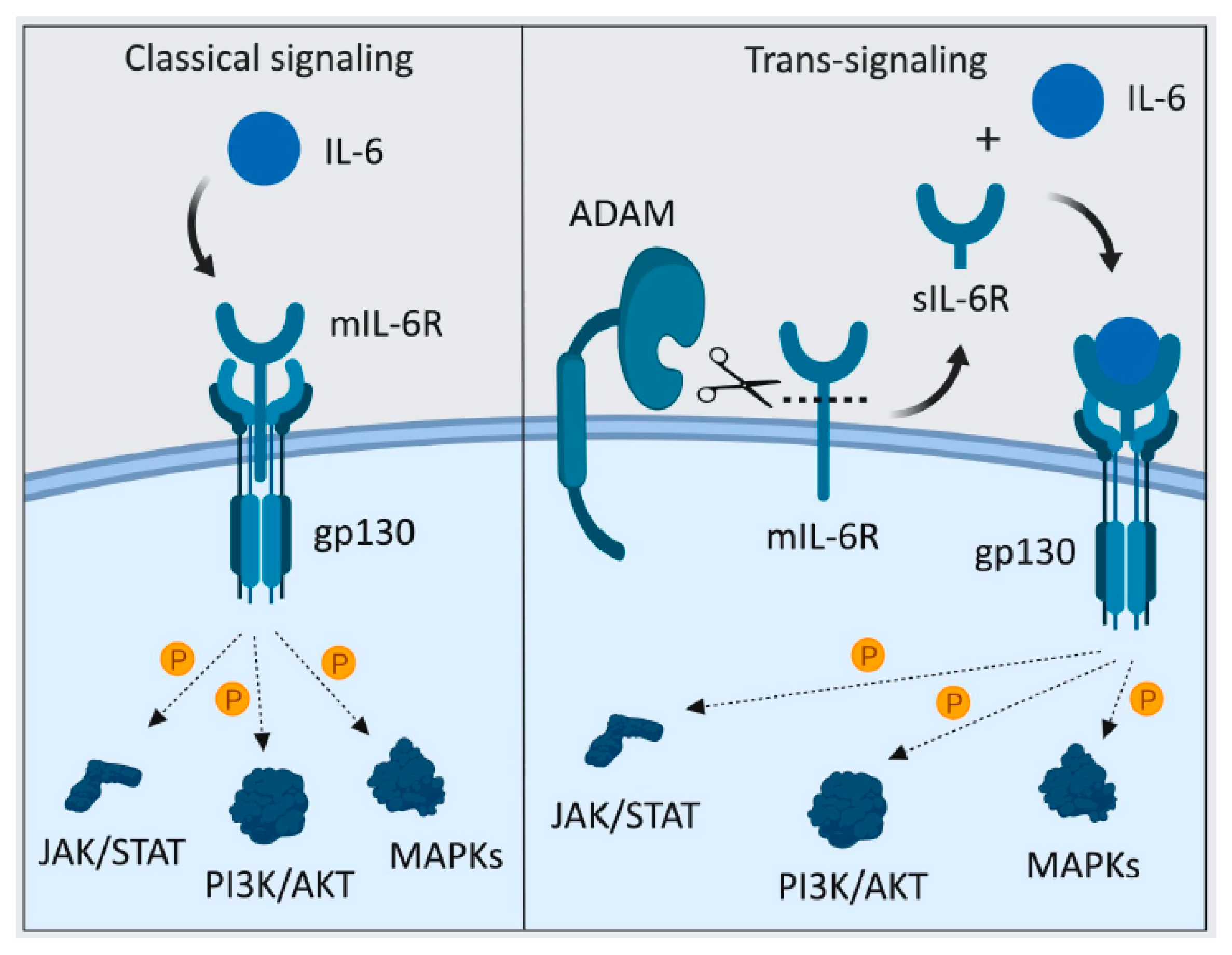
4.1.3. sIL-6R-Mediated IL-6 Trans-Signaling
5. Conclusions and Future Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. Adv Exp Med Biol. 2016, 893, 1–19. [Google Scholar] [PubMed]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.; Nguyen, D. Lineage factors and differentiation states in lung cancer progression. Oncogene 2015, 34, 5771. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.-L.; Jain, A.; Takano, A.; Newell, E.W.; Iyer, N.G.; Lim, W.-T.; Tan, E.-H.; Zhai, W.; Hillmer, A.M.; Tam, W.-L. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016, 17, e347–e362. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef]
- Santos, E.; Martin-Zanca, D.; Reddy, E.P.; Pierotti, M.A.; Della Porta, G.; Barbacid, M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 1984, 223, 661–664. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543. [Google Scholar]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Decker, P.A.; Alawi, E.A.; Zhu, Y.r.; Sanchez-Cespedes, M.; Yang, S.C.; Haasler, G.B.; Kajdacsy-Balla, A.; Demeure, M.J.; Sidransky, D. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer 2001, 92, 1525–1530. [Google Scholar] [CrossRef]
- Liao, J.; Shima, F.; Araki, M.; Ye, M.; Muraoka, S.; Sugimoto, T.; Kawamura, M.; Yamamoto, N.; Tamura, A.; Kataoka, T. Two conformational states of Ras GTPase exhibit differential GTP-binding kinetics. Biochem. Biophys. Res. Commun. 2008, 369, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Gysin, S.; Salt, M.; Young, A.; McCormick, F. Therapeutic strategies for targeting ras proteins. Genes Cancer 2011, 2, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Mayo, C.; Costa, C.; Magri, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Rosell, R. KRAS mutations in lung cancer. Clin. Lung Cancer 2013, 14, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell 2018, 33, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Amzallag, A.; Bagni, R.; Yi, M.; Afghani, S.; Burgan, W.; Fer, N.; Strathern, L.A.; Powell, K.; Smith, B. Differential effector engagement by oncogenic KRAS. Cell Rep. 2018, 22, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Köhler, J.; Zhou, Z.-W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q. KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 2018, 172, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S. Multivalent small-molecule pan-RAS inhibitors. Cell 2017, 168, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Wang, Z.; Zhang, X.; Cai, Z.; Zhang, J. Induction of CTLs by DCs pulsed with K-ras mutant peptide on the surface of nanoparticles in the treatment of pancreatic cancer. Oncol. Rep. 2011, 26, 215–221. [Google Scholar] [PubMed] [Green Version]
- Weng, T.; Yen, M.; Huang, C.; Hung, J.-J.; Chen, Y.; Chen, W.; Wang, C.; Chang, J.-Y.; Lai, M.-D. DNA vaccine elicits an efficient antitumor response by targeting the mutant Kras in a transgenic mouse lung cancer model. Gene Ther. 2014, 21, 888. [Google Scholar] [CrossRef] [PubMed]
- Wedén, S.; Klemp, M.; Gladhaug, I.P.; Møller, M.; Eriksen, J.A.; Gaudernack, G.; Buanes, T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int. J. Cancer 2011, 128, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.-i.; Restifo, N.P.; Yang, J.C. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef]
- Kohler, J.; Catalano, M.; Ambrogio, C. Back to the bench? MEK and ERK inhibitors for the treatment of KRAS mutant lung adenocarcinoma. Curr. Med. Chem. 2018, 25, 558–574. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; De Wit, N.; Gonçalves-Ribeiro, S. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961. [Google Scholar] [CrossRef]
- Lu, H.; Liu, C.; Velazquez, R.; Wang, H.; Dunkl, L.M.; Kazic-Legueux, M.; Haberkorn, A.; Billy, E.; Manchado, E.; Brachmann, S.M. SHP2 inhibition overcomes RTK-mediated pathway re-activation in KRAS mutant tumors treated with MEK inhibitors. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; Prodanovic, Z.; et al. ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 2019, 11, 4. [Google Scholar] [CrossRef]
- Burgess, A.W. EGFR family: Structure physiology signalling and therapeutic targets. Growth Factors 2008, 26, 263–274. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney Jr, D.W.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Leahy, D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef]
- Carpenter, G. ErbB-4: Mechanism of action and biology. Exp Cell Res. 2003, 284, 66–77. [Google Scholar] [CrossRef]
- Grandis, J.R.; Melhem, M.F.; Gooding, W.E.; Day, R.; Holst, V.A.; Wagener, M.M.; Drenning, S.D.; Tweardy, D.J. Levels of TGF-α and EGFR protein in head and neck squamous cell carcinoma and patient survival. J. Natl. Cancer Inst. 1998, 90, 824–832. [Google Scholar] [CrossRef]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Nam, H.K.; Park, W.S.; Nam, S.W.; Kim, M.S.; Sun, D.I.; Lee, Y.S.; Jang, J.J. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2005, 11, 2879–2882. [Google Scholar] [CrossRef]
- Rusch, V.; Baselga, J.; Cordon-Cardo, C.; Orazem, J.; Zaman, M.; Hoda, S.; McIntosh, J.; Kurie, J.; Dmitrovsky, E. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1993, 53, 2379–2385. [Google Scholar]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Spano, J.-P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.-F. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Barber, T.D.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N. Engl. J. Med. 2004, 351, 2883. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Gerald, W.L.; Li, A.R.; Pan, Q.; Lal, P.; Ladanyi, M.; Chen, B. EGFR gene amplification in breast cancer: Correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod. Pathol. 2005, 18, 1027. [Google Scholar] [CrossRef] [PubMed]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; DelGiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef]
- Sheng, Q.; Liu, J. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br. J. Cancer 2011, 104, 1241. [Google Scholar] [CrossRef] [PubMed]
- Terashima, M.; Kitada, K.; Ochiai, A.; Ichikawa, W.; Kurahashi, I.; Sakuramoto, S.; Katai, H.; Sano, T.; Imamura, H.; Sasako, M. Impact of expression of human epidermal growth factor receptors EGFR and ERBB2 on survival in stage II/III gastric cancer. Clin. Cancer Res. 2012, 18, 5992–6000. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Learn, C.A.; Archer, G.E.; McLendon, R.E.; Chewning, T.A.; Tuck, F.L.; Pracyk, J.B.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D. Brain tumors in mice are susceptible to blockade of epidermal growth factor receptor (EGFR) with the oral, specific, EGFR-tyrosine kinase inhibitor ZD1839 (iressa). Clin. Cancer Res. 2002, 8, 3496–3502. [Google Scholar] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef]
- Husgafvel-Pursiainen, K.; Hackman, P.; Ridanpää, M.; Anttila, S.; Karjalainen, A.; Partanen, T.; Taikina-Aho, O.; Heikkilä, L.; Vainio, H. K-ras mutations in human adenocarcinoma of the lung: Association with smoking and occupational exposure to asbestos. Int. J. Cancer 1993, 53, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Ward, W.H.; Cook, P.N.; Slater, A.M.; Davies, D.H.; Holdgate, G.A. Epidermal growth factor receptor tyrosine kinase: Investigation of catalytic mechanism, structure-based searching and discovery of a potent inhibitor. Biochem. Pharmacol. 1994, 48, 659–666. [Google Scholar] [CrossRef]
- Wakeling, A.E.; Guy, S.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Barker, A.J.; Gibson, K.H. ZD1839 (Iressa): An orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002, 62, 5749–5754. [Google Scholar]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar] [PubMed]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’haens, G.; Pintér, T.; Lim, R.; Bodoky, G. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-D.; Jia, X.-C.; Corvalan, J.R.; Wang, P.; Davis, C.G. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit. Rev. Oncol. Hematol. 2001, 38, 17–23. [Google Scholar] [CrossRef]
- Giusti, R.M.; Shastri, K.; Pilaro, A.M.; Fuchs, C.; Cordoba-Rodriguez, R.; Koti, K.; Rothmann, M.; Men, A.Y.; Zhao, H.; Hughes, M. US Food and Drug Administration approval: Panitumumab for epidermal growth factor receptor–expressing metastatic colorectal carcinoma with progression following fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens. Clin. Cancer Res. 2008, 14, 1296–1302. [Google Scholar] [CrossRef]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch Jr, T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non–small cell lung cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef]
- Campiglio, M.; Locatelli, A.; Olgiati, C.; Normanno, N.; Somenzi, G.; Viganò, L.; Fumagalli, M.; Ménard, S.; Gianni, L. Inhibition of proliferation and induction of apoptosis in breast cancer cells by the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor ZD1839 (‘Iressa’) is independent of EGFR expression level. J. Cell. Physiol. 2004, 198, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, L.; Fenchel, K.; Dempke, W.C. Improved overall survival following tyrosine kinase inhibitor treatment in advanced or metastatic non-small-cell lung cancer—the Holy Grail in cancer treatment? Transl. Lung Cancer Res. 2015, 4, 223. [Google Scholar] [PubMed]
- Giaccone, G.; Herbst, R.S.; Manegold, C.; Scagliotti, G.; Rosell, R.; Miller, V.; Natale, R.B.; Schiller, J.H.; von Pawel, J.; Pluzanska, A. Gefitinib in combination with gemcitabine and cisplatin in advanced non–small-cell lung cancer: A phase III trial—INTACT 1. J. Clin. Oncol. 2004, 22, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R. Chronic obstructive pulmonary disease and lung cancer: Common pathogenesis, shared clinical challenges. Proc. Am. Thorac. Soc. 2012, 9, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Cigarette smoking: Cancer risks, carcinogens, and mechanisms. Langenbecks Arch. Surg. 2006, 391, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Akopyan, G.; Bonavida, B. Understanding tobacco smoke carcinogen NNK and lung tumorigenesis. Int. J. Oncol. 2006, 29, 745–752. [Google Scholar] [CrossRef]
- Hecht, S.; Hoffmann, D. N-nitroso compounds and tobacco-induced cancers in man. IARC Sci. Publ. 1990, 105, 54–61. [Google Scholar]
- Hecht, S.S. Research opportunities related to establishing standards for tobacco products under the Family Smoking Prevention and Tobacco Control Act. Nicotine Tob. Res. 2011, 14, 18–28. [Google Scholar] [CrossRef]
- Thomas, J.L.; Hecht, S.S.; Luo, X.; Ming, X.; Ahluwalia, J.S.; Carmella, S.G. Thirdhand tobacco smoke: A tobacco-specific lung carcinogen on surfaces in smokers’ homes. Nicotine Tob. Res. 2013, 16, 26–32. [Google Scholar] [CrossRef]
- Rivenson, A.; Hoffmann, D.; Prokopczyk, B.; Amin, S.; Hecht, S.S. Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and Areca-derived N-nitrosamines. Cancer Res. 1988, 48, 6912–6917. [Google Scholar]
- Hecht, S.S.; Adams, J.D.; Numoto, S.; Hoffmann, D. Induction of respiratory tract tumors in Syrian golden hamsters by a single dose of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and the effect of smoke inhalation. Carcinogenesis 1983, 4, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, A.; Lin, D.; Stoner, G.D.; Radok, P.; Furuya, K.; Hecht, S.S.; Schut, H.A.; Klaunig, J.E. Comparative carcinogenicity in A/J mice and metabolism by cultured mouse peripheral lung of N′-nitrosonornicotine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, and their analogues. Cancer Res. 1983, 43, 1223–1229. [Google Scholar] [PubMed]
- Belinsky, S.A.; Devereux, T.R.; Foley, J.F.; Maronpot, R.R.; Anderson, M.W. Role of the Alveor Type II Cell in the Development and Progression of Pulmonary Tumors Induced bt 4-(Methylnitrosamino)-(3-pyridyl)-1-butanone in the A/J Mouse. Cancer Res. 1992, 52, 3164–3173. [Google Scholar] [PubMed]
- Fujimoto, J.; Kadara, H.; Men, T.; van Pelt, C.; Lotan, D.; Lotan, R. Comparative functional genomics analysis of NNK tobacco-carcinogen induced lung adenocarcinoma development in Gprc5a-knockout mice. PLoS ONE 2010, 5, e11847. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, A.; Bartels, T.; Klocke, R.; Paul, D.; Halter, R. Increased susceptibility to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in transgenic mice overexpressing c-myc and epidermal growth factor in alveolar type II cells. J. Cancer Res. Clin. Oncol. 2003, 129, 71–75. [Google Scholar] [PubMed]
- Yang, Y.; Zhang, Z.; Mukherjee, A.B.; Linnoila, R.I. Increased susceptibility of mice lacking Clara cell 10-kDa protein to lung tumorigenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a potent carcinogen in cigarette smoke. J. Biol. Chem. 2004, 279, 29336–29340. [Google Scholar] [CrossRef]
- Hecht, S.S. Recent studies on mechanisms of bioactivation and detoxification of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific lung carcinogen. Crit. Rev. Toxicol. 1996, 26, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Demizu, Y.; Sasaki, R.; Trachootham, D.; Pelicano, H.; Colacino, J.A.; Liu, J.; Huang, P. Alterations of cellular redox state during NNK-induced malignant transformation and resistance to radiation. Antioxid. Redox Signal. 2008, 10, 951–961. [Google Scholar] [CrossRef]
- Ho, Y.-S.; Chen, C.-H.; Wang, Y.-J.; Pestell, R.G.; Albanese, C.; Chen, R.-J.; Chang, M.-C.; Jeng, J.-H.; Lin, S.-Y.; Liang, Y.-C. Tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFκB activation and cyclin D1 up-regulation. Toxicol. Appl. Pharmacol. 2005, 205, 133–148. [Google Scholar] [CrossRef]
- Jin, Z.; Gao, F.; Flagg, T.; Deng, X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J. Biol. Chem. 2004, 279, 40209–40219. [Google Scholar] [CrossRef]
- Jin, Z.; May, W.S.; Gao, F.; Flagg, T.; Deng, X. Bcl2 suppresses DNA repair by enhancing c-Myc transcriptional activity. J. Biol. Chem. 2006, 281, 14446–14456. [Google Scholar] [CrossRef] [PubMed]
- Jull, B.; Plummer, H.; Schuller, H. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. Clin. Oncol. 2001, 127, 707–717. [Google Scholar] [PubMed]
- Miller, A.; Brooks, G.D.; Mcleod, L.; Ruwanpura, S.; Jenkins, B.J. Differential involvement of gp130 signalling pathways in modulating tobacco carcinogen-induced lung tumourigenesis. Oncogene 2015, 34, 1510. [Google Scholar] [CrossRef] [PubMed]
- El-Bayoumy, K.; Iatropoulos, M.; Amin, S.; Hoffmann, D.; Wynder, E.L. Increased expression of cyclooxygenase-2 in rat lung tumors induced by the tobacco-specific nitrosamine 4-(methylnitrosamino)-4-(3-pyridyl)-1-butanone. Cancer Res. 1999, 59, 1400–1403. [Google Scholar] [PubMed]
- Houghton, A.M.; Mouded, M.; Shapiro, S.D. Common origins of lung cancer and COPD. Nat. Med. 2008, 14, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Cheung, A.F.; Mazumdar, C.; Winslow, M.M.; Bronson, R.; Schmidt, L.M.; Crowley, D.; Chen, J.; Jacks, T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 2011, 19, 72–85. [Google Scholar] [CrossRef]
- Bellocq, A.; Antoine, M.; Flahault, A.; Philippe, C.; Crestani, B.; Bernaudin, J.F.; Mayaud, C.; Milleron, B.; Baud, L.; Cadranel, J. Neutrophil alveolitis in bronchioloalveolar carcinoma: Induction by tumor-derived interleukin-8 and relation to clinical outcome. Am. J. Pathol. 1998, 152, 83–92. [Google Scholar]
- Zaynagetdinov, R.; Sherrill, T.P.; Polosukhin, V.V.; Han, W.; Ausborn, J.A.; McLoed, A.G.; McMahon, F.B.; Gleaves, L.A.; Degryse, A.L.; Stathopoulos, G.T.; et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J. Immunol. 2011, 187, 5703–5711. [Google Scholar] [CrossRef]
- Vignaud, J.-M.; Marie, B.; Klein, N.; Plénat, F.; Pech, M.; Borrelly, J.; Martinet, N.; Duprez, A.; Martinet, Y. The role of platelet-derived growth factor production by tumor-associated macrophages in tumor stroma formation in lung cancer. Cancer Res. 1994, 54, 5455–5463. [Google Scholar]
- Ji, H.; Houghton, A.; Mariani, T.; Perera, S.; Kim, C.; Padera, R.; Tonon, G.; McNamara, K.; Marconcini, L.; Hezel, A. K-ras activation generates an inflammatory response in lung tumors. Oncogene 2006, 25, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 5, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Numasaki, M.; Watanabe, M.; Suzuki, T.; Takahashi, H.; Nakamura, A.; McAllister, F.; Hishinuma, T.; Goto, J.; Lotze, M.T.; Kolls, J.K. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J. Immunol. 2005, 175, 6177–6189. [Google Scholar] [CrossRef] [PubMed]
- Ancrile, B.; Lim, K.-H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Invest. 2007, 117, 3846–3856. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.; Jenkins, B.J. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef]
- Chen, J.J.; Yao, P.-L.; Yuan, A.; Hong, T.-M.; Shun, C.-T.; Kuo, M.-L.; Lee, Y.-C.; Yang, P.-C. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: Its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin. Cancer Res. 2003, 9, 729–737. [Google Scholar]
- White, E.S.; Flaherty, K.R.; Carskadon, S.; Brant, A.; Iannettoni, M.D.; Yee, J.; Orringer, M.B.; Arenberg, D.A. Macrophage migration inhibitory factor and CXC chemokine expression in non-small cell lung cancer: Role in angiogenesis and prognosis. Clin. Cancer Res. 2003, 9, 853–860. [Google Scholar]
- Yatsunami, J.; Tsuruta, N.; Ogata, K.; Wakamatsu, K.; Takayama, K.; Kawasaki, M.; Nakanishi, Y.; Hara, N.; Hayashi, S.-i. Interleukin-8 participates in angiogenesis in non-small cell, but not small cell carcinoma of the lung. Cancer Lett. 1997, 120, 101–108. [Google Scholar] [CrossRef]
- Arenberg, D.A.; Kunkel, S.L.; Polverini, P.J.; Glass, M.; Burdick, M.D.; Strieter, R.M. Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. J. Clin. Invest. 1996, 97, 2792–2802. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Jenkins, B.J. Potential efficacy of interleukin-1β inhibition in lung cancer. Lancet 2017, 390, 1813–1814. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Feng, W.; Kang, S.; Zhang, Y.; Shen, J.; He, J.; Jiang, L.; Wang, W.; Guo, Z.; Peng, G. Evaluation of the efficacy and safety of anti-PD-1 and anti-PD-L1 antibody in the treatment of non-small cell lung cancer (NSCLC): A meta-analysis. J. Thorac. Dis. 2015, 7, 455. [Google Scholar] [PubMed]
- He, J.; Hu, Y.; Hu, M.; Li, B. Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci. Rep. 2015, 5, 13110. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef]
- Caescu, C.I.; Jeschke, G.R.; Turk, B.E. Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 2009, 424, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.; Niu, X.-D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, K.W.; Carpenter, G. ErbB-4 and TNF-alpha converting enzyme localization to membrane microdomains. Biochem. Biophys. Res. Commun. 2006, 350, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Zimina, E.P.; Bruckner-Tuderman, L.; Franzke, C.W. Shedding of collagen XVII ectodomain depends on plasma membrane microenvironment. J. Biol. Chem. 2005, 280, 34019–34024. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.-L.C.; Milla, M.E.; Burkhart, W.; Carter, H.L.; Chen, W.-J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; Hoffman, C.R. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Grötzinger, J.; Lorenzen, I.; Düsterhöft, S. Molecular insights into the multilayered regulation of ADAM17: The role of the extracellular region. Biochim. Biophys. Acta Mol. Cell Res. 2017, 11, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.F.; Qiu, T.H.; Sunnarborg, S.W.; Chang, A.; Zhang, C.; Patterson, C.; Lee, D.C. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 2003, 22, 2704–2716. [Google Scholar] [CrossRef] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternlicht, M.D.; Sunnarborg, S.W.; Kouros-Mehr, H.; Yu, Y.; Lee, D.C.; Werb, Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development 2005, 132, 3923–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkstra, A.; Postma, D.S.; Noordhoek, J.A.; Lodewijk, M.E.; Kauffman, H.F.; ten Hacken, N.H.; Timens, W. Expression of ADAMs (“a disintegrin and metalloprotease”) in the human lung. Virchows Arch. 2009, 454, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Paulissen, G.; Rocks, N.; Gueders, M.M.; Crahay, C.; Quesada-Calvo, F.; Bekaert, S.; Hacha, J.; El Hour, M.; Foidart, J.M.; Noel, A.; et al. Role of ADAM and ADAMTS metalloproteinases in airway diseases. Respir. Res. 2009, 10, 127. [Google Scholar] [CrossRef]
- Dreymueller, D.; Martin, C.; Kogel, T.; Pruessmeyer, J.; Hess, F.M.; Horiuchi, K.; Uhlig, S.; Ludwig, A. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol. Med. 2012, 4, 412–423. [Google Scholar] [CrossRef]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef]
- Weskamp, G.; Mendelson, K.; Swendeman, S.; Le Gall, S.; Ma, Y.; Lyman, S.; Hinoki, A.; Eguchi, S.; Guaiquil, V.; Horiuchi, K. Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ. Res. 2010, 106, 932–940. [Google Scholar] [CrossRef]
- Arndt, P.G.; Strahan, B.; Wang, Y.; Long, C.; Horiuchi, K.; Walcheck, B. Leukocyte ADAM17 regulates acute pulmonary inflammation. PLoS ONE 2011, 6, e19938. [Google Scholar] [CrossRef]
- Bergmeier, W.; Piffath, C.L.; Cheng, G.; Dole, V.S.; Zhang, Y.; von Andrian, U.H.; Wagner, D.D. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ. Res. 2004, 95, 677–683. [Google Scholar] [CrossRef]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.I.; McLeod, L.; Yu, L.; Ebi, H.; Ruwanpura, S.; Sagi, I.; Rose-John, S.; Jenkins, B.J. The ADAM17 protease promotes tobacco smoke carcinogen-induced lung tumourigenesis. Carcinogenesis 2019. in-press. [Google Scholar]
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knösel, T.; et al. ADAM17 is required for EGF-R–induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225. [Google Scholar] [CrossRef] [PubMed]
- Arribas, J.; Coodly, L.; Vollmer, P.; Kishimoto, T.K.; Rose-John, S.; Massague, J. Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J. Biol. Chem. 1996, 271, 11376–11382. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.C.; Blobel, C.P. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PLoS ONE 2012, 7, e31600. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Godinho, S.I.; Uppington, K.M.; Whittington, H.A.; Millar, A.B. Contribution of TNF-alpha converting enzyme and proteinase-3 to TNF-alpha processing in human alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2006, 34, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Sun, L.; Neppl, C.; Siggs, O.M.; Le Gall, S.M.; Tomisato, W.; Li, X.; Du, X.; Maennel, D.N.; Blobel, C.P.; et al. MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 19967–19972. [Google Scholar] [CrossRef] [Green Version]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Garbers, C.; Rose-John, S. ADAM17: A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011, 32, 380–387. [Google Scholar] [CrossRef]
- Rose-John, S. ADAM17, shedding, TACE as therapeutic targets. Pharmacol. Res. 2013, 71, 19–22. [Google Scholar] [CrossRef]
- Schlondorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem. J. 2000, 347, 131–138. [Google Scholar] [CrossRef]
- Milla, M.E.; Leesnitzer, M.A.; Moss, M.L.; Clay, W.C.; Carter, H.L.; Miller, A.B.; Su, J.L.; Lambert, M.H.; Willard, D.H.; Sheeley, D.M.; et al. Specific sequence elements are required for the expression of functional tumor necrosis factor-alpha-converting enzyme (TACE). J. Biol. Chem. 1999, 274, 30563–30570. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, P.E.; Solomon, A.; Miller, A.B.; Leesnitzer, M.A.; Sagi, I.; Milla, M.E. Inhibition of the tumor necrosis factor-α-converting enzyme by its pro domain. J. Biol. Chem. 2004, 279, 31638–31645. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cohen, T.; Romi, E.; Levin, M.; Peleg, Y.; Arad, U.; Yaron, A.; Milla, M.E.; Sagi, I. Harnessing the natural inhibitory domain to control TNFalpha Converting Enzyme (TACE) activity in vivo. Sci. Rep. 2016, 6, 35598. [Google Scholar] [CrossRef] [PubMed]
- Cavadas, M.; Oikonomidi, I.; Gaspar, C.J.; Burbridge, E.; Badenes, M.; Felix, I.; Bolado, A.; Hu, T.; Bileck, A.; Gerner, C.; et al. Phosphorylation of iRhom2 Controls Stimulated Proteolytic Shedding by the Metalloprotease ADAM17/TACE. Cell Rep. 2017, 21, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Christova, Y.; Adrain, C.; Bambrough, P.; Ibrahim, A.; Freeman, M. Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 2013, 14, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Dulloo, I.; Muliyil, S.; Freeman, M. The molecular, cellular and pathophysiological roles of iRhom pseudoproteases. Open Biol. 2019, 9, 190003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemberg, M.K.; Freeman, M. Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 2007, 17, 1634–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieve, A.G.; Xu, H.; Künzel, U.; Bambrough, P.; Sieber, B.; Freeman, M. Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. Elife 2017, 6, e23968. [Google Scholar] [CrossRef]
- Groth, E.; Pruessmeyer, J.; Babendreyer, A.; Schumacher, J.; Pasqualon, T.; Dreymueller, D.; Higashiyama, S.; Lorenzen, I.; Grötzinger, J.; Cataldo, D. Stimulated release and functional activity of surface expressed metalloproteinase ADAM17 in exosomes. Biochim. Biophys. Acta Mol. Cell. Res. 2016, 1863, 2795–2808. [Google Scholar] [CrossRef]
- Diaz-Rodriguez, E.; Montero, J.C.; Esparís-Ogando, A.; Yuste, L.; Pandiella, A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: A potential role in regulated shedding. Mol. Biol. Cell 2002, 13, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Derynck, R. Ectodomain shedding of TGF-α and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. 1999, 18, 6962–6972. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Derynck, R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 2010, 37, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Chauhan, A.K.; Canault, M.; Walsh, M.T.; Bergmeier, W.; Wagner, D.D. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc. Res. 2009, 84, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soond, S.M.; Everson, B.; Riches, D.W.; Murphy, G. ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 2005, 118, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, T.; von der Heyde, J.; Bartsch, K.; Garbers, C.; Schulze-Osthoff, K.; Chalaris, A.; Murphy, G.; Rose-John, S.; Rabe, B. Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J. 2014, 28, 4847–4856. [Google Scholar] [CrossRef] [PubMed]
- Amour, A.; Slocombe, P.M.; Webster, A.; Butler, M.; Knight, C.G.; Smith, B.J.; Stephens, P.E.; Shelley, C.; Hutton, M.; Knäuper, V. TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998, 435, 39–44. [Google Scholar] [CrossRef]
- Wisniewska, M.; Goettig, P.; Maskos, K.; Belouski, E.; Winters, D.; Hecht, R.; Black, R.; Bode, W. Structural determinants of the ADAM inhibition by TIMP-3: Crystal structure of the TACE-N-TIMP-3 complex. J. Mol. Biol. 2008, 381, 1307–1319. [Google Scholar] [CrossRef]
- Mohammed, F.F.; Smookler, D.S.; Taylor, S.E.; Fingleton, B.; Kassiri, Z.; Sanchez, O.H.; English, J.L.; Matrisian, L.M.; Au, B.; Yeh, W.-C. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat. Genet. 2004, 36, 969–977. [Google Scholar] [CrossRef]
- Chapnick, D.A.; Bunker, E.; Liu, X. A biosensor for the activity of the “sheddase” TACE (ADAM17) reveals novel and cell type-specific mechanisms of TACE activation. Sci. Signal. 2015, 8, rs1. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Park, D.; Kim, C. Disruption of TACE-filamin interaction can inhibit TACE-mediated ectodomain shedding. Biochem. Biophys. Res. Commun. 2017, 490, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-López, M.D.; Gilsanz, A.; Yáñez-Mó, M.; Ovalle, S.; Lafuente, E.M.; Domínguez, C.; Monk, P.N.; González-Alvaro, I.; Sánchez-Madrid, F.; Cabañas, C. The sheddase activity of ADAM17/TACE is regulated by the tetraspanin CD9. Cell. Mol. Life Sci. 2011, 68, 3275–3292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Herrera, A.H.; Li, Y.; Belani, K.K.; Walcheck, B. Regulation of mature ADAM17 by redox agents for L-selectin shedding. J. Immunol. 2009, 182, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Sidhu, S.S.; Mengistab, A.; Gallup, M.; Basbaum, C. TACE/ADAM-17 phosphorylation by PKC-epsilon mediates premalignant changes in tobacco smoke-exposed lung cells. PLoS ONE 2011, 6, e17489. [Google Scholar] [CrossRef]
- Tellier, E.; Canault, M.; Rebsomen, L.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell Res. 2006, 312, 3969–3980. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Armbruster, N.; Miller, M.A.; Cermeno, E.; Hartmann, M.; Bell, G.W.; Root, D.E.; Lauffenburger, D.A.; Lodish, H.F.; Herrlich, A. Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 9776–9781. [Google Scholar] [CrossRef] [Green Version]
- Düsterhöft, S.; Höbel, K.; Oldefest, M.; Lokau, J.; Waetzig, G.H.; Chalaris, A.; Garbers, C.; Scheller, J.; Rose-John, S.; Lorenzen, I. A disintegrin and metalloprotease 17 dynamic interaction sequence, the sweet tooth for the human interleukin 6 receptor. J. Biol. Chem. 2014, 289, 16336–16348. [Google Scholar] [CrossRef]
- Blanchot-Jossic, F.; Jarry, A.; Masson, D.; Bach-Ngohou, K.; Paineau, J.; Denis, M.G.; Laboisse, C.L.; Mosnier, J.F. Up-regulated expression of ADAM17 in human colon carcinoma: Co-expression with EGFR in neoplastic and endothelial cells. J. Pathol. 2005, 207, 156–163. [Google Scholar] [CrossRef]
- Kornfeld, J.; Meder, S.; Wohlberg, M.; Friedrich, R.; Rau, T.; Riethdorf, L.; Löning, T.; Pantel, K.; Riethdorf, S. Overexpression of TACE and TIMP3 mRNA in head and neck cancer: Association with tumour development and progression. Br. J. Cancer 2011, 104, 138–145. [Google Scholar] [CrossRef]
- Cai, M.; Wang, Z.; Zhang, J.; Zhou, H.; Jin, L.; Bai, R.; Weng, Y. Adam17, a target of Mir-326, promotes Emt-induced cells invasion in lung adenocarcinoma. Cell. Physiol. Biochem. 2015, 36, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.-S.; Zhang, J.; Zhao, W.-L.; Dong, X.-C.; Wang, J.-L. ADAM17 is overexpressed in non-small cell lung cancer and its expression correlates with poor patient survival. Tumor Biol. 2013, 34, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.M.; Ryan, B.M.; Hill, A.D.; McDermott, E.; O’Higgins, N.; Duffy, M.J. ADAM-17 expression in breast cancer correlates with variables of tumor progression. Clin. Cancer Res. 2007, 13, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; McGowan, P.M.; Mullooly, M.; Murray, A.; Synnott, N.; O’Donovan, N.; Flanagan, L.; Tape, C.J.; Murphy, G.; Crown, J. Targeting ADAM-17 with an inhibitory monoclonal antibody has antitumour effects in triple-negative breast cancer cells. Br. J. Cancer 2015, 112, 1895. [Google Scholar] [CrossRef]
- Capone, C.; Dabertrand, F.; Baron-Menguy, C.; Chalaris, A.; Ghezali, L.; Domenga-Denier, V.; Schmidt, S.; Huneau, C.; Rose-John, S.; Nelson, M.T. Mechanistic insights into a TIMP3-sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics. Elife 2016, 5, e17536. [Google Scholar] [CrossRef]
- Fang, W.; Qian, J.; Wu, Q.; Chen, Y.; Yu, G. ADAM-17 expression is enhanced by FoxM1 and is a poor prognostic sign in gastric carcinoma. J. Surg. Res. 2017, 220, 223–233. [Google Scholar] [CrossRef]
- Kenny, P.A.; Bissell, M.J. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J. Clin. Invest. 2007, 117, 337–345. [Google Scholar] [CrossRef]
- Zheng, X.; Jiang, F.; Katakowski, M.; Zhang, Z.G.; Lu, Q.-e.; Chopp, M. ADAM17 promotes breast cancer cell malignant phenotype through EGFR-PI3K-AKT activation. Cancer Biol. Ther. 2009, 8, 1045–1054. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Sun, X.; Feng, T.; Zou, H.; Jiang, Y.; Liu, Z.; Zhao, D.; Yu, X. ADAM17 regulates prostate cancer cell proliferation through mediating cell cycle progression by EGFR/PI3K/AKT pathway. Mol. Cell. Biochem. 2012, 359, 235–243. [Google Scholar] [CrossRef]
- Ye, J.; Yuen, S.M.; Murphy, G.; Xie, R.; Kwok, H.F. Anti-tumor effects of a ‘human & mouse cross-reactive’anti-ADAM17 antibody in a pancreatic cancer model in vivo. Eur. J. Pharm. Sci. 2017, 110, 62–69. [Google Scholar]
- Richards, F.M.; Tape, C.J.; Jodrell, D.I.; Murphy, G. Anti-tumour effects of a specific anti-ADAM17 antibody in an ovarian cancer model in vivo. PLoS ONE 2012, 7, e40597. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-B.S.; Peyton, M.; He, B.; Liu, C.; Girard, L.; Caudler, E.; Lo, Y.; Baribaud, F.; Mikami, I.; Reguart, N. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 2006, 10, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witters, L.; Scherle, P.; Friedman, S.; Fridman, J.; Caulder, E.; Newton, R.; Lipton, A. Synergistic inhibition with a dual epidermal growth factor receptor/HER-2/neu tyrosine kinase inhibitor and a disintegrin and metalloprotease inhibitor. Cancer Res. 2008, 68, 7083–7089. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Bradley, E.; Levy, R.; Doval, D.; Bondarde, S.; Sahoo, T.; Lokanatha, D.; Julka, P.; Nagarkar, R.; Friedman, S. Clinical benefit of INCB7839, a potent and selective ADAM inhibitor, in combination with trastuzumab in patients with metastatic HER2+ breast cancer. J. Clin. Oncol. 2010, 28, 3025. [Google Scholar] [CrossRef]
- Hirata, S.; Murata, T.; Suzuki, D.; Nakamura, S.; Jono-Ohnishi, R.; Hirose, H.; Sawaguchi, A.; Nishimura, S.; Sugimoto, N.; Eto, K. Selective Inhibition of ADAM17 Efficiently Mediates Glycoprotein Ibα Retention During Ex Vivo Generation of Human Induced Pluripotent Stem Cell-Derived Platelets. Stem Cells Transl. Med. 2017, 6, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Benaich, N.; Tape, C.; Kwok, H.F.; Murphy, G. Targeting the sheddase activity of ADAM17 by an anti-ADAM17 antibody D1 (A12) inhibits head and neck squamous cell carcinoma cell proliferation and motility via blockage of bradykinin induced HERs transactivation. Int. J. Biol. Sci. 2014, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, A.; Seidl, S.; Vlachou, P.; Michel, L.; Mitova, N.; Schatz, N.; Specht, K.; Koch, I.; Schuster, T.; Grundler, R. ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non–small cell lung cancer. Cancer Res. 2010, 70, 5368–5378. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bender, S.; Zimmermann, M.; Riesterer, O.; Broggini-Tenzer, A.; Pruschy, M.N. Secretome Signature Identifies ADAM17 as Novel Target for Radiosensitization of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2016, 22, 4428–4439. [Google Scholar] [CrossRef]
- Friedman, S.; Levy, R.; Garrett, W.; Doval, D.; Bondarde, S.; Sahoo, T.; Lokanatha, D.; Julka, P.; Shenoy, K.; Nagarkar, R. Clinical Benefit of INCB7839, a Potent and Selective Inhibitor of ADAM10 and ADAM17, in Combination with Trastuzumab in Metastatic HER2 Positive Breast Cancer Patients. Cancer Res. 2009, 69 (Suppl. 24). [Google Scholar] [CrossRef]
- Infante, J.; Burris, H.; Lewis, N.; Donehower, R.; Redman, J.; Friedman, S.; Scherle, P.; Fridman, J.; Li, J.; Emm, T. A multicenter phase Ib study of the safety, pharmacokinetics, biological activity and clinical efficacy of INCB7839, a potent and selective inhibitor of ADAM10 and ADAM17. Breast Cancer Res. Treat. 2007, 106 (Suppl. 1), 1. [Google Scholar] [CrossRef]
- Pecot, C.V.; Calin, G.A.; Coleman, R.L.; Lopez-Berestein, G.; Sood, A.K. RNA interference in the clinic: Challenges and future directions. Nat. Rev. Cancer 2011, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, Y.; Qian, M.; Ma, C.; Jing, H.; Wen, Z.; Qian, D. ADAM17 silencing suppresses the migration and invasion of non-small cell lung cancer. Mol. Med. Report. 2014, 9, 1935–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoda, M.; Kimura, T.; Tohmonda, T.; Morioka, H.; Matsumoto, M.; Okada, Y.; Toyama, Y.; Horiuchi, K. Systemic overexpression of TNFα-converting enzyme does not lead to enhanced shedding activity in vivo. PLoS ONE 2013, 8, e54412. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-S.; Gilligan, D.; Pacey, S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Borrell-Pagès, M.; Rojo, F.; Albanell, J.; Baselga, J.; Arribas, J. TACE is required for the activation of the EGFR by TGF-α in tumors. EMBO J. 2003, 22, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; McLeod, L.; Alhayyani, S.; Szczepny, A.; Watkins, D.; Chen, W.; Enriori, P.; Ferlin, W.; Ruwanpura, S.; Jenkins, B. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene 2017, 36, 3059. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Tomoshige, K.; Guo, M.; Tsuchiya, T.; Fukazawa, T.; Fink-Baldauf, I.M.; Stuart, W.D.; Naomoto, Y.; Nagayasu, T.; Maeda, Y. An EGFR ligand promotes EGFR-mutant but not KRAS-mutant lung cancer in vivo. Oncogene 2018, 1, 3894. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. 1), S24–S31. [Google Scholar] [CrossRef]
- Brachmann, R.; Lindquist, P.B.; Nagashima, M.; Kohr, W.; Lipari, T.; Napier, M.; Derynck, R. Transmembrane TGF-alpha precursors activate EGF/TGF-alpha receptors. Cell 1989, 56, 691–700. [Google Scholar] [CrossRef]
- Higashiyama, S.; Iwamoto, R.; Goishi, K.; Raab, G.; Taniguchi, N.; Klagsbrun, M.; Mekada, E. The membrane protein CD9/DRAP 27 potentiates the juxtacrine growth factor activity of the membrane-anchored heparin-binding EGF-like growth factor. J. Cell Biol. 1995, 128, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.T.; Winchell, L.F.; McCune, B.K.; Earp, H.S.; Teixido, J.; Massague, J.; Herman, B.; Lee, D.C. The TGF-alpha precursor expressed on the cell surface binds to the EGF receptor on adjacent cells, leading to signal transduction. Cell 1989, 56, 495–506. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, D.; Li, W.; Liang, J.; Gentry, L.E.; Brattain, M.G. Defective cleavage of membrane bound TGF [alpha] leads to enhanced activation of the EGF receptor in malignant cells. Oncogene 2000, 19, 1901. [Google Scholar] [CrossRef] [PubMed]
- Hurbin, A.; Dubrez, L.; Coll, J.-L.; Favrot, M.-C. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. J. Biol. Chem. 2002, 277, 49127–49133. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Daigo, Y.; Takano, A.; Taniwaki, M.; Kato, T.; Hayama, S.; Murakami, H.; Takeshima, Y.; Inai, K.; Nishimura, H. Increases of amphiregulin and transforming growth factor-α in serum as predictors of poor response to gefitinib among patients with advanced non–small cell lung cancers. Cancer Res. 2005, 65, 9176–9184. [Google Scholar] [CrossRef] [PubMed]
- Logeat, F.; Bessia, C.; Brou, C.; LeBail, O.; Jarriault, S.; Seidah, N.G.; Israël, A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. USA 1998, 95, 8108–8112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, W.R.; Arnett, K.L.; Blacklow, S.C. The molecular logic of Notch signaling--a structural and biochemical perspective. J. Cell Sci. 2008, 121, 3109–3119. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Bozkulak, E.C.; Weinmaster, G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [PubMed]
- Eliasz, S.; Liang, S.; Chen, Y.; De Marco, M.A.; Machek, O.; Skucha, S.; Miele, L.; Bocchetta, M. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene 2010, 29, 2488. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, A.; Mazur, P.; Anton, M.; Rudelius, M.; Schwamborn, K.; Feuchtinger, A.; Behnke, K.; Walch, A.; Braren, R.; Peschel, C. Opposing role of Notch1 and Notch2 in a Kras G12D-driven murine non-small cell lung cancer model. Oncogene 2015, 34, 578. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 1, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Garbers, C.; Rose-John, S. Interleukin-6: From basic biology to selective blockade of pro-inflammatory activities. Semin. Immunol. 2014, 26, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.A.; Revez, J.A.; Loh, Z.; Simpson, J.; Zhang, V.; Bain, L.; Varelias, A.; Rose-John, S.; Blumenthal, A.; Smyth, M.J.; et al. Allergen-induced IL-6 trans-signaling activates gammadelta T cells to promote type 2 and type 17 airway inflammation. J. Allergy Clin. Immunol. 2015, 136, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; McLeod, L.; Dousha, L.F.; Seow, H.J.; Alhayyani, S.; Tate, M.D.; Deswaerte, V.; Brooks, G.D.; Bozinovski, S.; MacDonald, M.; et al. Therapeutic Targeting of the IL-6 Trans-Signaling/Mechanistic Target of Rapamycin Complex 1 Axis in Pulmonary Emphysema. Am. J. Respir. Crit. Care Med. 2016, 194, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Rousseau, F.; Guilhot, F.; Malinge, P.; Magistrelli, G.; Herren, S.; Jones, S.A.; Jones, G.W.; Scheller, J.; Lissilaa, R. Novel insights into interleukin 6 (IL-6) cis-and trans-signaling pathways by differentially manipulating the assembly of the IL-6 signaling complex. J. Biol. Chem. 2015, 290, 26943–26953. [Google Scholar] [CrossRef]
- Schumacher, N.; Meyer, D.; Mauermann, A.; von der Heyde, J.; Wolf, J.; Schwarz, J.; Knittler, K.; Murphy, G.; Michalek, M.; Garbers, C. Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by a disintegrin and metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. J. Biol. Chem. 2015, 290, 26059–26071. [Google Scholar] [CrossRef]
- Rose-John, S.; Winthrop, K.; Calabrese, L. The role of IL-6 in host defence against infections: Immunobiology and clinical implications. Nat. Rev. Rheumatol. 2017, 13, 399. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Main Effect | Model | Reference |
|---|---|---|---|
| Neutrophils | Poor prognosis | Human patients | [88] |
| Macrophages | Promoting lung carcinogenesis | Urethane-induced lung cancer mouse model | [89] |
| IL-17+ T cells | Enhancing tumour proliferation and angiogenesis | Kras mutant mouse model | [92] |
| Commensal microbiota | induce tumour cell proliferation via activating lung-resident γδ T cells | Mouse model harbouring Kras mutation and Trp53 deficiency | [93] |
| IL-17 | Promoting tumour angiogenesis | NSCLC xenografts | [94] |
| IL-6 | Activation of IL-6 trans-signaling | Kras mutant mouse model | [95,96,97] |
| IL-8 | Promoting tumour angiogenesis | Human patients | [98,99,100,101] |
| IL-1β | Enhance lung cancer incidence and mortality | Human patients (CANTOS) | [103] |
| Agent | Type of Agent | Disease Setting | Substrate Inhibited | Reference |
|---|---|---|---|---|
| INCB3619 | Small molecule inhibitor | Breast cancer and NSCLC | Heregulin | [182] |
| INCB7839 | Small molecule inhibitor | Breast cancer | HER2 | [183,184] |
| KP-457 | Small molecule inhibitor | Thrombus formation | Glycoprotein Ibα (GPIbα) | [185] |
| Prodomain | Peptide | LAC and inflammatory diseases | TNFα and IL-6R | [29,131,143] |
| D1(A12) | Antibody | Triple-negative breast cancer and head and neck squamous cell carcinoma | TNFα, TGFα, amphiregulin and TNFR1 | [174,186] |
| A9(B8) | Antibody | Pancreatic ductal adenoma | Amphiregulin | [180] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saad, M.I.; Rose-John, S.; Jenkins, B.J. ADAM17: An Emerging Therapeutic Target for Lung Cancer. Cancers 2019, 11, 1218. https://doi.org/10.3390/cancers11091218
Saad MI, Rose-John S, Jenkins BJ. ADAM17: An Emerging Therapeutic Target for Lung Cancer. Cancers. 2019; 11(9):1218. https://doi.org/10.3390/cancers11091218
Chicago/Turabian StyleSaad, Mohamed I., Stefan Rose-John, and Brendan J. Jenkins. 2019. "ADAM17: An Emerging Therapeutic Target for Lung Cancer" Cancers 11, no. 9: 1218. https://doi.org/10.3390/cancers11091218