Mutation in DNA Polymerase Beta Causes Spontaneous Chromosomal Instability and Inflammation-Associated Carcinogenesis in Mice

Abstract
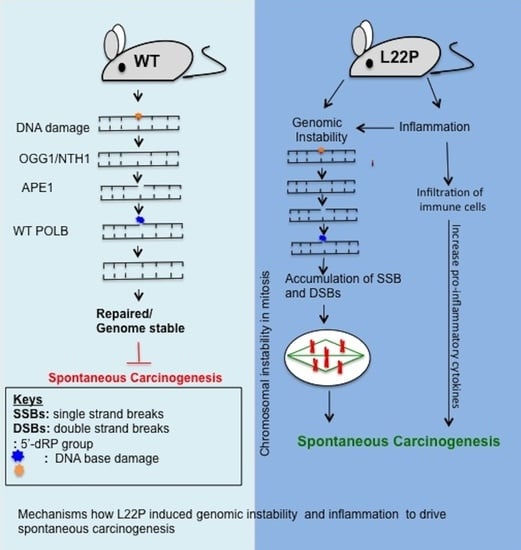
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
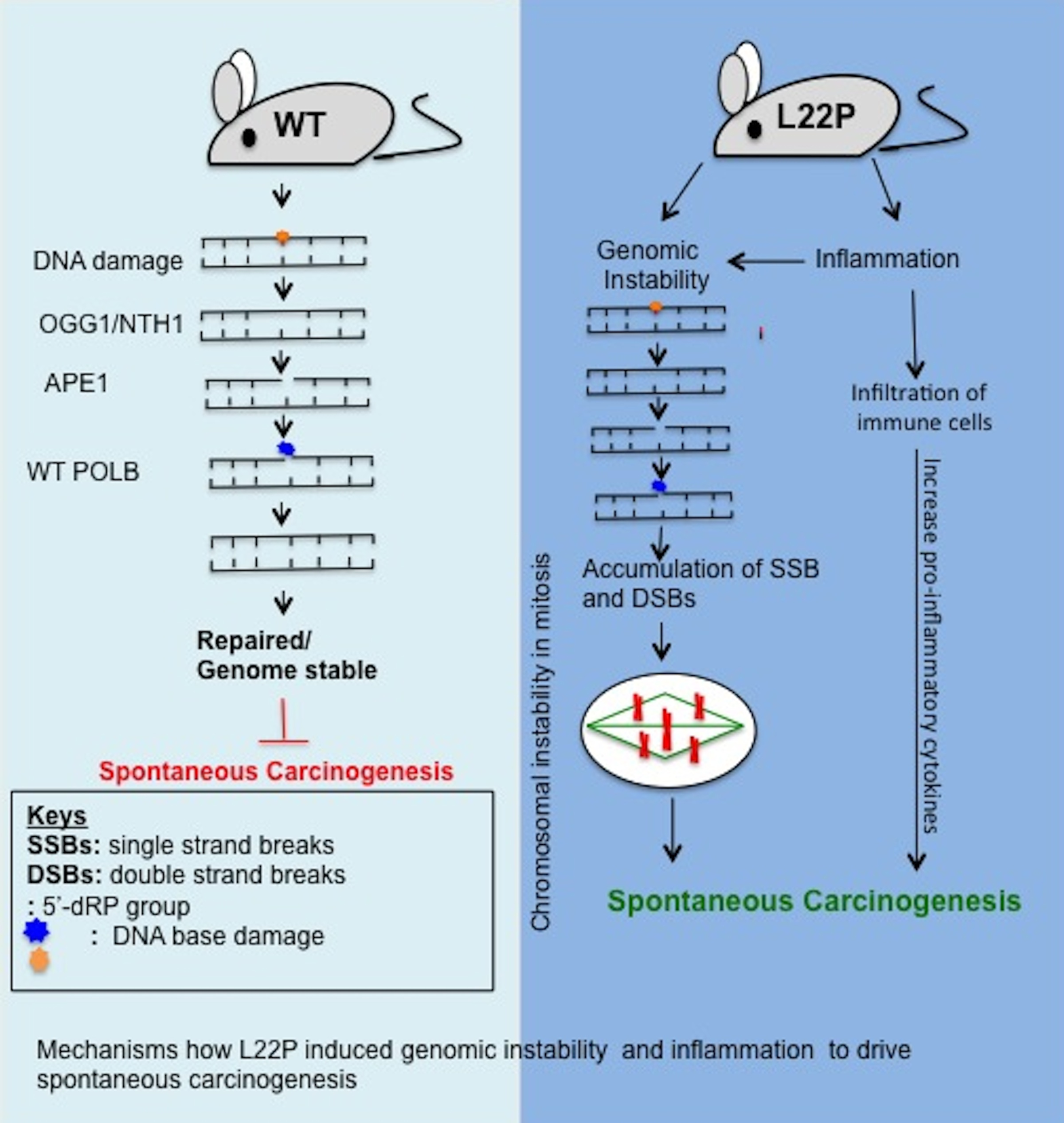
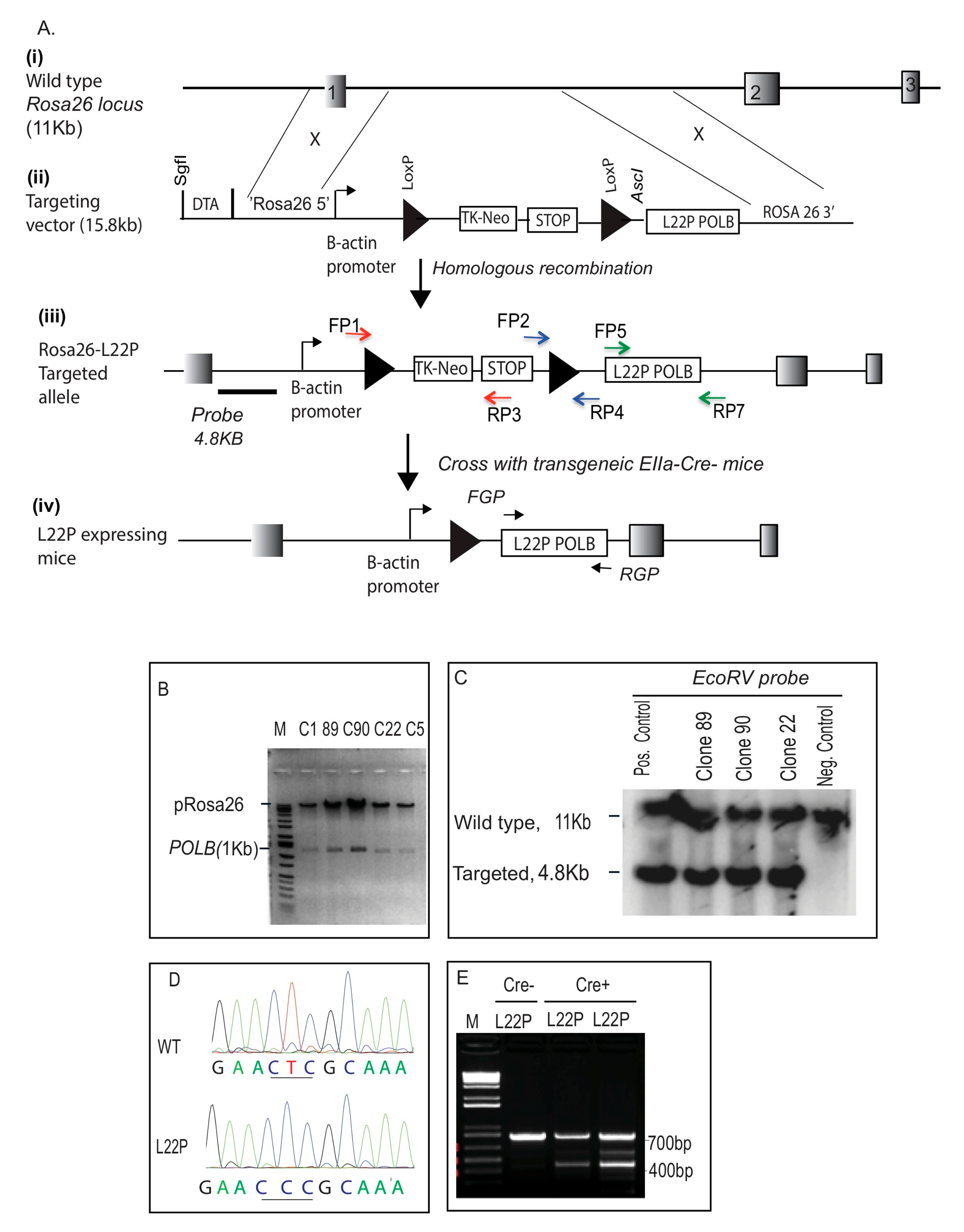
2.1. Generation of L22P Mutant Mice
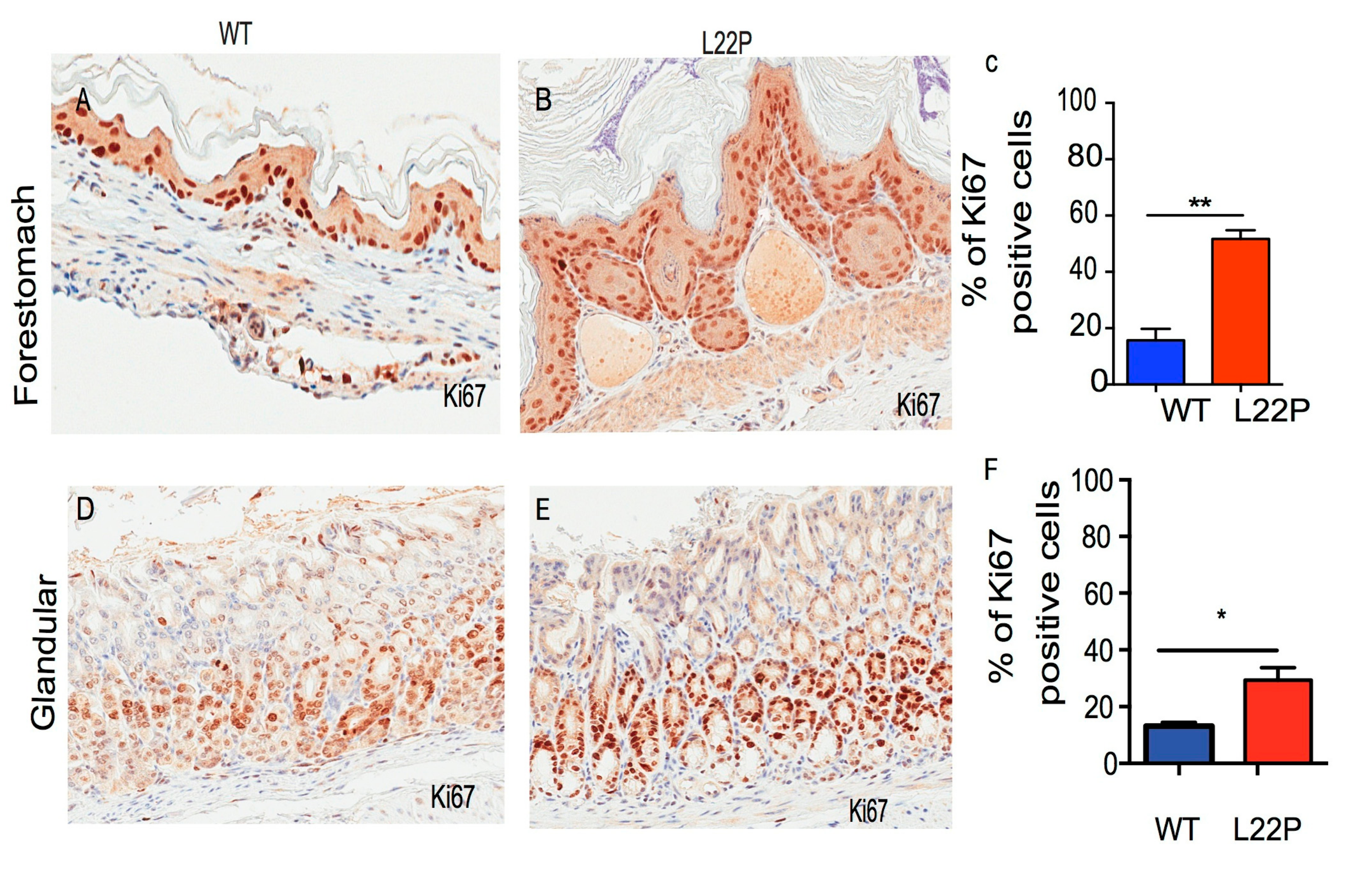
2.2. L22P Increases Cellular Proliferation in Stomach Cells
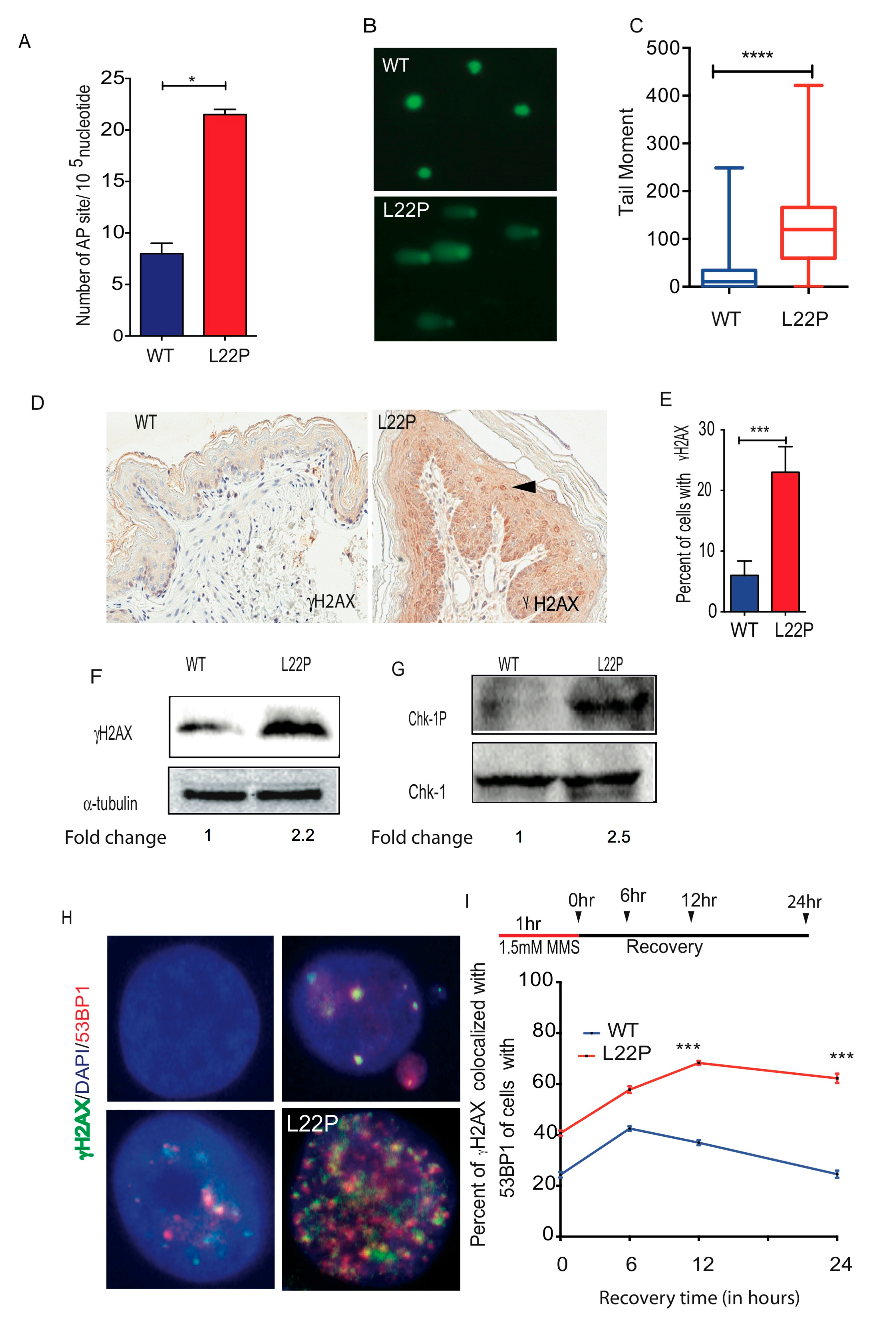
2.3. L22P Induces Significantly Increased BER Intermidates and DSBs
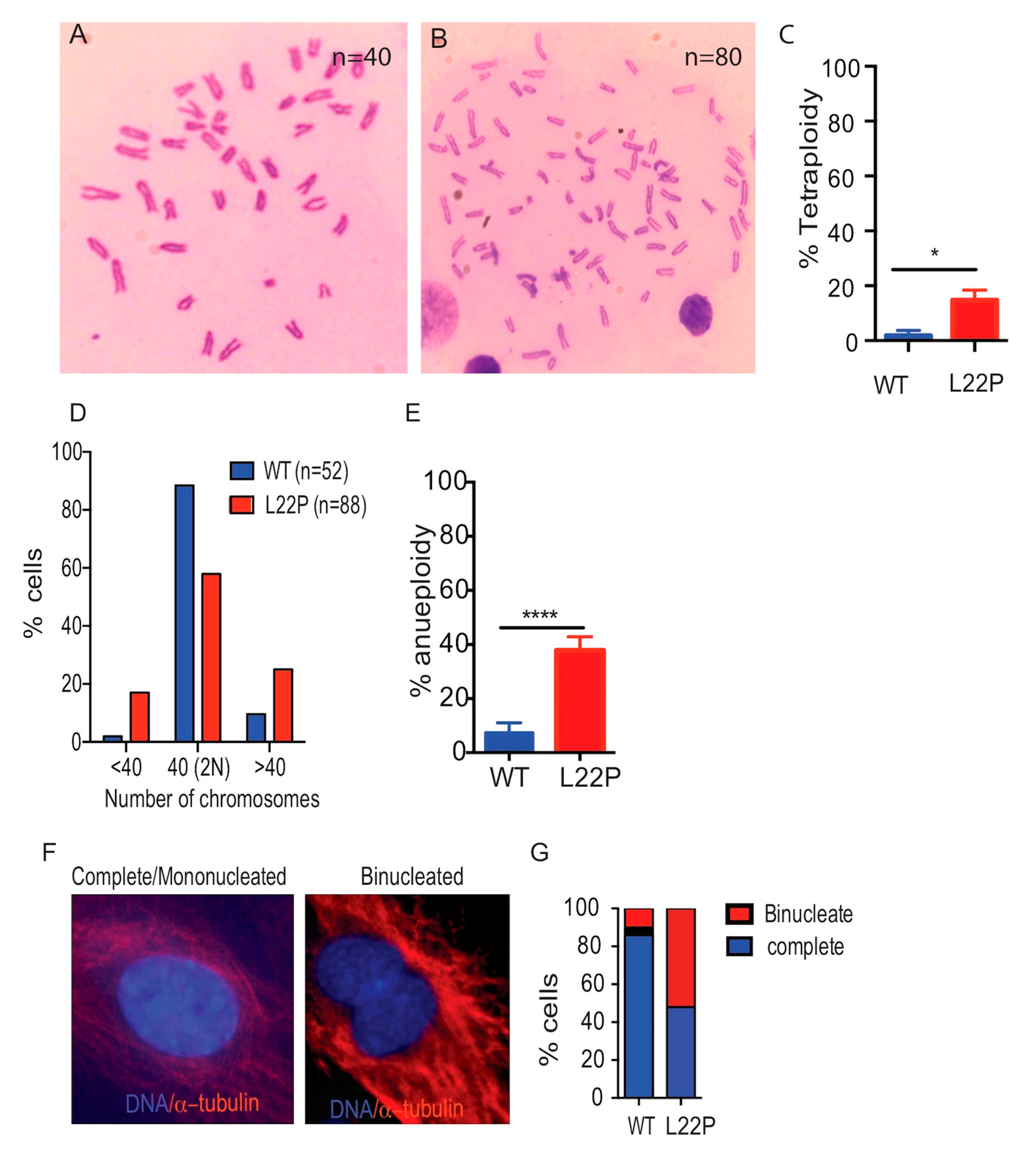
2.4. L22P Causes Tetraploidy and Aneuploidy
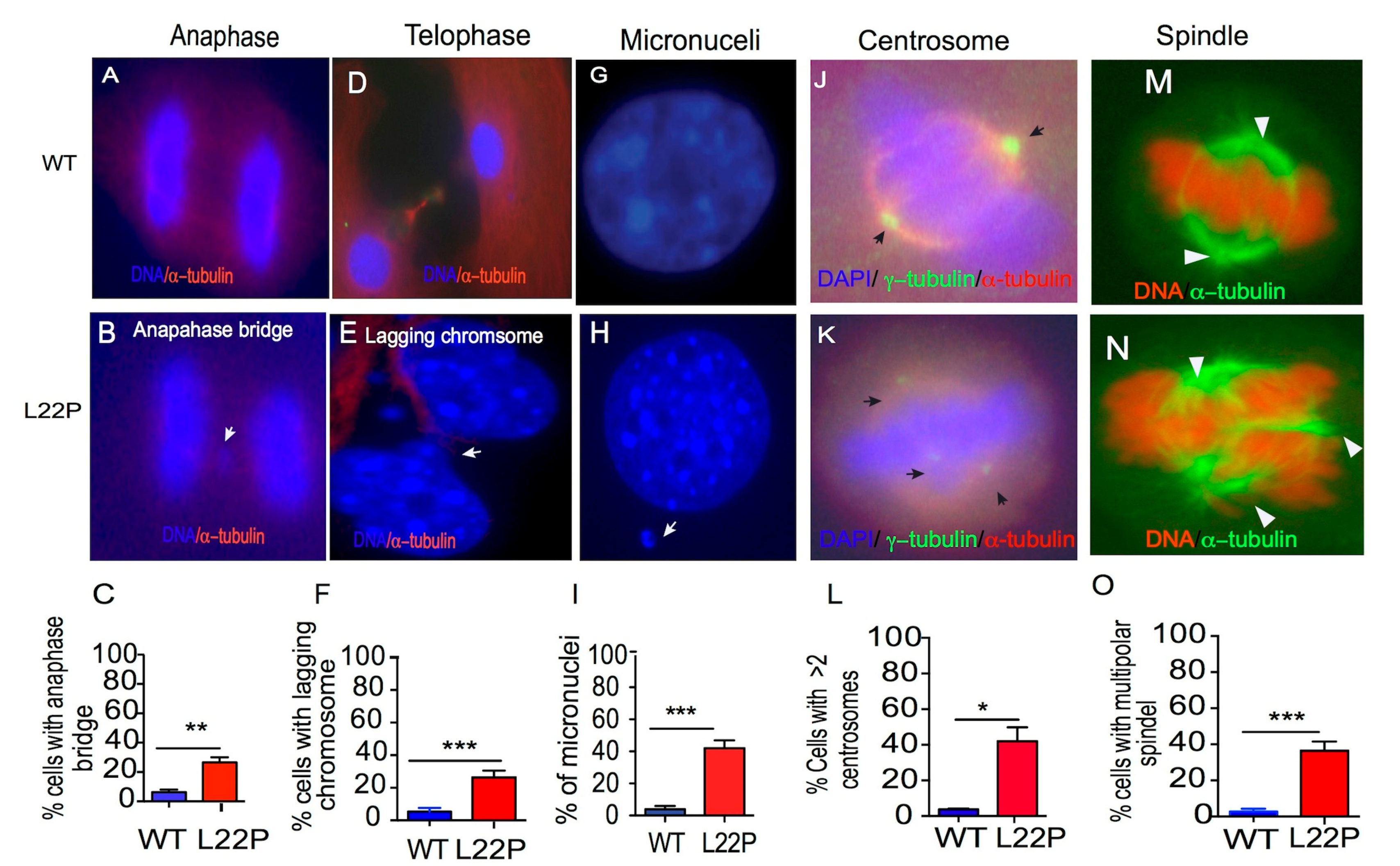
2.5. L22P Causes Mitotic Defects
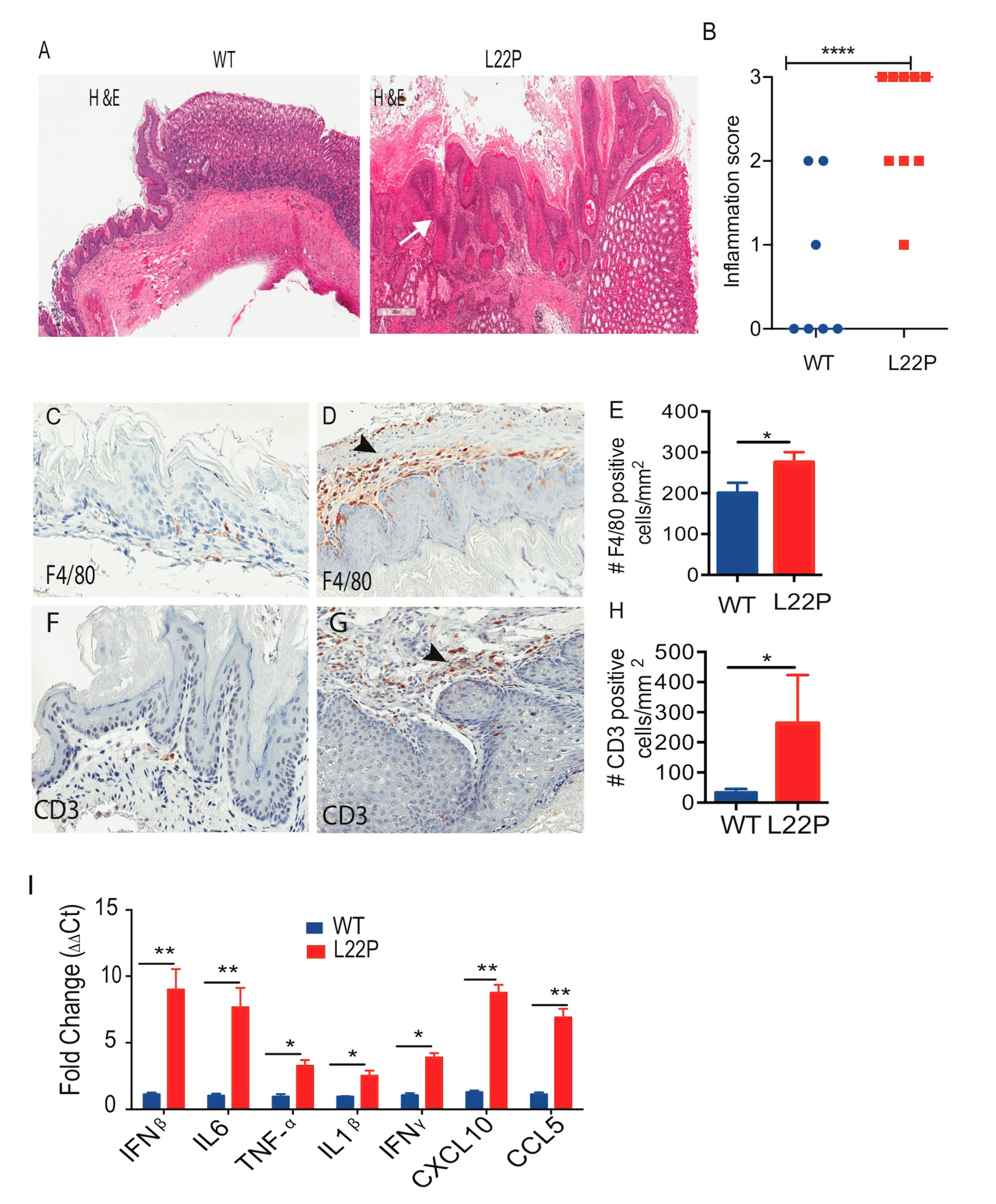
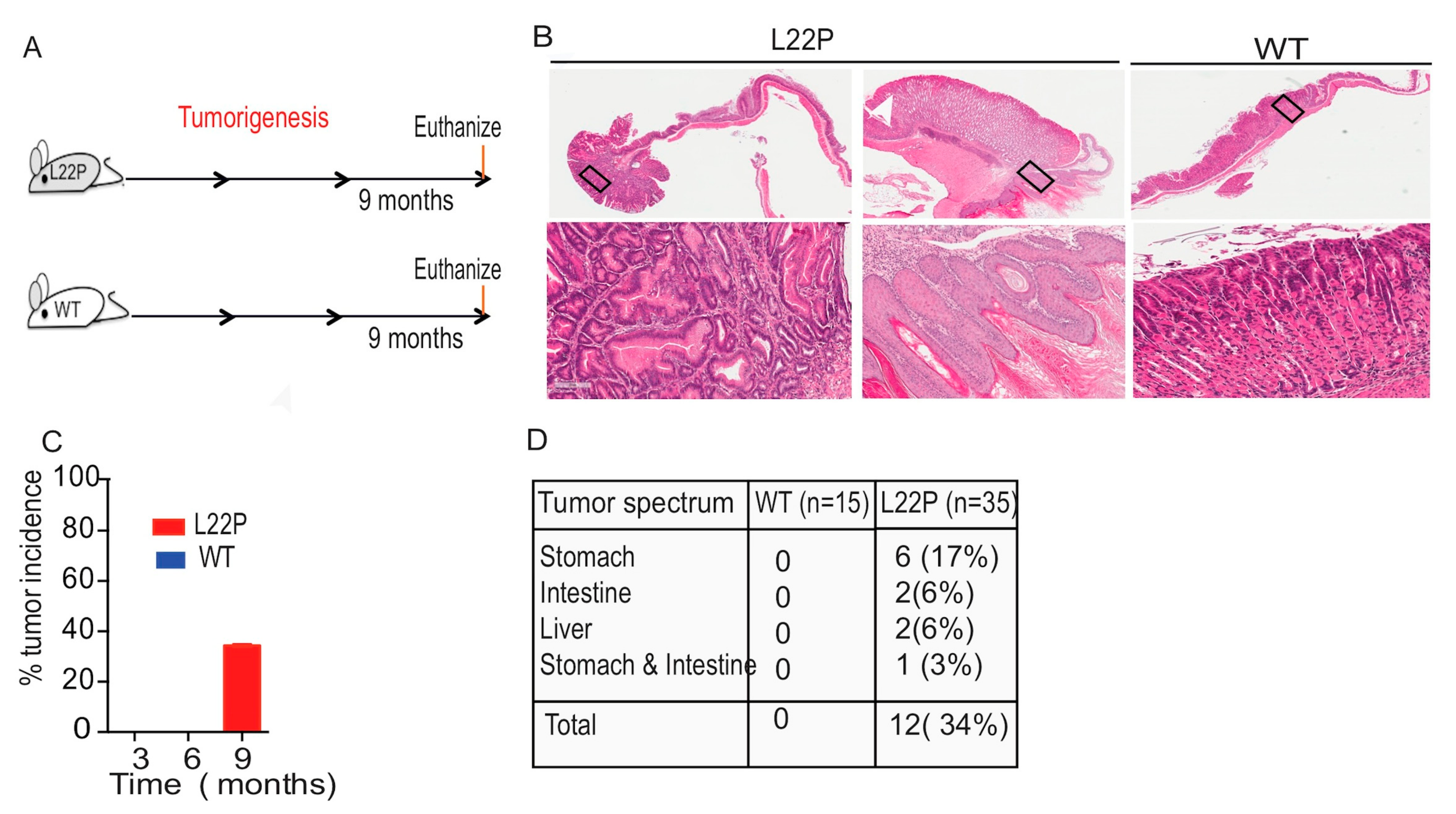
2.6. L22P Mice Exhibit an Inflammatory Response and Develop Tumors
3. Materials and Methods
3.1. Mouse Breeding and Expression of L22P
3.2. Immunohistochemical Analysis
3.3. Comet Assays
3.4. AP Site Assay
3.5. Immunofluorescence Localization
3.6. Quantitative Real-Time PCR
3.7. Immunoblotting
3.8. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N. Endogenous oxidative DNA damage, aging, and cancer. Free Radic. Res. Commun. 1989, 7, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Lindahl, T.; Sedgwick, B. DNA repair. Curr. Opin. Cell Biol. 1993, 5, 424–433. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., 3rd.; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Sobol, R.W.; Horton, J.K.; Kuhn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature 1996, 379, 183–186. [Google Scholar] [CrossRef]
- Sobol, R.W.; Prasad, R.; Evenski, A.; Baker, A.; Yang, X.P.; Horton, J.K.; Wilson, S.H. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature 2000, 405, 807–810. [Google Scholar] [CrossRef]
- Dianov, G.; Lindahl, T. Reconstitution of the DNA base excision-repair pathway. Curr. Biol. CB 1994, 4, 1069–1076. [Google Scholar] [CrossRef]
- Singhal, R.K.; Prasad, R.; Wilson, S.H. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J. Biol. Chem. 1995, 270, 949–957. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 1995, 269, 699–702. [Google Scholar] [CrossRef]
- Piersen, C.E.; Prasad, R.; Wilson, S.H.; Lloyd, R.S. Evidence for an imino intermediate in the DNA polymerase beta deoxyribose phosphate excision reaction. J. Biol. Chem. 1996, 271, 17811–17815. [Google Scholar] [CrossRef]
- Canitrot, Y.; Cazaux, C.; Frechet, M.; Bouayadi, K.; Lesca, C.; Salles, B.; Hoffmann, J.S. Overexpression of DNA polymerase beta in cell results in a mutator phenotype and a decreased sensitivity to anticancer drugs. Proc. Natl. Acad. Sci. USA 1998, 95, 12586–12590. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd.; McNeill, D.R. Base excision repair and the central nervous system. Neuroscience 2007, 145, 1187–1200. [Google Scholar] [CrossRef]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef]
- Sugo, N.; Aratani, Y.; Nagashima, Y.; Kubota, Y.; Koyama, H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000, 19, 1397–1404. [Google Scholar] [CrossRef]
- Sweasy, J.B.; Lang, T.; DiMaio, D. Is base excision repair a tumor suppressor mechanism? Cell Cycle 2006, 5, 250–259. [Google Scholar] [CrossRef][Green Version]
- Donigan, K.A.; Sun, K.W.; Nemec, A.A.; Murphy, D.L.; Cong, X.; Northrup, V.; Zelterman, D.; Sweasy, J.B. Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 2012, 287, 23830–23839. [Google Scholar] [CrossRef]
- Tan, X.H.; Zhao, M.; Pan, K.F.; Dong, Y.; Dong, B.; Feng, G.J.; Jia, G.; Lu, Y.Y. Frequent mutation related with overexpression of DNA polymerase beta in primary tumors and precancerous lesions of human stomach. Cancer Lett. 2005, 220, 101–114. [Google Scholar] [CrossRef]
- Nemec, A.A.; Donigan, K.A.; Murphy, D.L.; Jaeger, J.; Sweasy, J.B. Colon cancer-associated DNA polymerase beta variant induces genomic instability and cellular transformation. J. Biol. Chem. 2012, 287, 23840–23849. [Google Scholar] [CrossRef]
- Kidane, D.; Dalal, S.; Keh, A.; Liu, Y.; Zelterman, D.; Sweasy, J.B. DNA polymerase beta is critical for genomic stability of sperm cells. DNA Repair 2011, 10, 390–397. [Google Scholar] [CrossRef]
- Prasad, R.; Longley, M.J.; Sharief, F.S.; Hou, E.W.; Copeland, W.C.; Wilson, S.H. Human DNA polymerase theta possesses 5′-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res. 2009, 37, 1868–1877. [Google Scholar] [CrossRef]
- Bebenek, K.; Tissier, A.; Frank, E.G.; McDonald, J.P.; Prasad, R.; Wilson, S.H.; Woodgate, R.; Kunkel, T.A. 5′-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science 2001, 291, 2156–2159. [Google Scholar] [CrossRef]
- Iwanaga, A.; Ouchida, M.; Miyazaki, K.; Hori, K.; Mukai, T. Functional mutation of DNA polymerase beta found in human gastric cancer--inability of the base excision repair in vitro. Mutat. Res. 1999, 435, 121–128. [Google Scholar] [CrossRef]
- Dalal, S.; Chikova, A.; Jaeger, J.; Sweasy, J.B. The Leu22Pro tumor-associated variant of DNA polymerase beta is dRP lyase deficient. Nucleic Acids Res. 2008, 36, 411–422. [Google Scholar] [CrossRef][Green Version]
- Rozacky, J.; Nemec, A.A.; Sweasy, J.B.; Kidane, D. Gastric cancer associated variant of DNA polymerase beta (Leu22Pro) promotes DNA replication associated double strand breaks. Oncotarget 2015, 6, 24474–24487. [Google Scholar] [CrossRef]
- Salmon, E.D.; Cimini, D.; Cameron, L.A.; DeLuca, J.G. Merotelic kinetochores in mammalian tissue cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 553–568. [Google Scholar] [CrossRef]
- Thompson, S.L.; Compton, D.A. Chromosomes and cancer cells. Chromosome Res. 2011, 19, 433–444. [Google Scholar] [CrossRef]
- Goldsby, R.E.; Hays, L.E.; Chen, X.; Olmsted, E.A.; Slayton, W.B.; Spangrude, G.J.; Preston, B.D. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc. Natl. Acad. Sci. USA 2002, 99, 15560–15565. [Google Scholar] [CrossRef]
- Thiem, S.; Pierce, T.P.; Palmieri, M.; Putoczki, T.L.; Buchert, M.; Preaudet, A.; Farid, R.O.; Love, C.; Catimel, B.; Lei, Z.; et al. mTORC1 inhibition restricts inflammation-associated gastrointestinal tumorigenesis in mice. J. Clin. Investig. 2013, 123, 767–781. [Google Scholar] [CrossRef]
- Rogers, A.B.; Houghton, J. Helicobacter-based mouse models of digestive system carcinogenesis. Methods Mol. Biol. 2009, 511, 267–295. [Google Scholar]
- Rogers, A.B.; Taylor, N.S.; Whary, M.T.; Stefanich, E.D.; Wang, T.C.; Fox, J.G. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 2005, 65, 10709–10715. [Google Scholar] [CrossRef]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.V. OpenComet: An automated tool for comet assay image analysis. Redox. Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef]
- Sweasy, J.B.; Lang, T.; Starcevic, D.; Sun, K.W.; Lai, C.C.; Dimaio, D.; Dalal, S. Expression of DNA polymerase {beta} cancer-associated variants in mouse cells results in cellular transformation. Proc. Natl. Acad. Sci. USA 2005, 102, 14350–14355. [Google Scholar] [CrossRef]
- Yamtich, J.; Nemec, A.A.; Keh, A.; Sweasy, J.B. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012, 8, e1003052. [Google Scholar] [CrossRef]
- Kirby, T.W.; Derose, E.F.; Beard, W.A.; Shock, D.D.; Wilson, S.H.; London, R.E. Substrate rescue of DNA polymerase beta containing a catastrophic L22P mutation. Biochemistry 2014, 53, 2413–2422. [Google Scholar] [CrossRef]
- Huggins, C.F.; Chafin, D.R.; Aoyagi, S.; Henricksen, L.A.; Bambara, R.A.; Hayes, J.J. Flap endonuclease 1 efficiently cleaves base excision repair and DNA replication intermediates assembled into nucleosomes. Mol. Cell 2002, 10, 1201–1211. [Google Scholar] [CrossRef]
- Demple, B.; DeMott, M.S. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene 2002, 21, 8926–8934. [Google Scholar] [CrossRef][Green Version]
- Sung, J.S.; DeMott, M.S.; Demple, B. Long-patch base excision DNA repair of 2-deoxyribonolactone prevents the formation of DNA-protein cross-links with DNA polymerase beta. J. Biol. Chem. 2005, 280, 39095–39103. [Google Scholar] [CrossRef]
- Osborn, A.J.; Elledge, S.J.; Zou, L. Checking on the fork: The DNA-replication stress-response pathway. Trends. Cell Biol. 2002, 12, 509–516. [Google Scholar] [CrossRef]
- Hoffelder, D.R.; Luo, L.; Burke, N.A.; Watkins, S.C.; Gollin, S.M.; Saunders, W.S. Resolution of anaphase bridges in cancer cells. Chromosoma 2004, 112, 389–397. [Google Scholar] [CrossRef]
- Terradas, M.; Martin, M.; Tusell, L.; Genesca, A. DNA lesions sequestered in micronuclei induce a local defective-damage response. DNA Repair 2009, 8, 1225–1234. [Google Scholar] [CrossRef]
- Gisselsson, D. Classification of chromosome segregation errors in cancer. Chromosoma 2008, 117, 511–519. [Google Scholar] [CrossRef]
- Vig, B.K.; Paweletz, N. Kinetochores, centromeres, spindles and the induction of aneuploidy. Mutat. Res. 1988, 201, 259–269. [Google Scholar] [CrossRef]
- Doxsey, S. The centrosome—A tiny organelle with big potential. Nat. Genet. 1998, 20, 104–106. [Google Scholar] [CrossRef]
- Sluder, G.; Thompson, E.A.; Miller, F.J.; Hayes, J.; Rieder, C.L. The checkpoint control for anaphase onset does not monitor excess numbers of spindle poles or bipolar spindle symmetry. J. Cell Sci. 1997, 110, 421–429. [Google Scholar]
- Xu, X.; Weaver, Z.; Linke, S.P.; Li, C.; Gotay, J.; Wang, X.W.; Harris, C.C.; Ried, T.; Deng, C.X. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 1999, 3, 389–395. [Google Scholar] [CrossRef]
- Tutt, A.; Gabriel, A.; Bertwistle, D.; Connor, F.; Paterson, H.; Peacock, J.; Ross, G.; Ashworth, A. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr. Biol. CB 1999, 9, 1107–1110. [Google Scholar] [CrossRef]
- Dodson, H.; Bourke, E.; Jeffers, L.J.; Vagnarelli, P.; Sonoda, E.; Takeda, S.; Earnshaw, W.C.; Merdes, A.; Morrison, C. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 2004, 23, 3864–3873. [Google Scholar] [CrossRef]
- Brinkley, B.R. Managing the centrosome numbers game: From chaos to stability in cancer cell division. Trends. Cell Biol. 2001, 11, 18–21. [Google Scholar] [CrossRef]
- D’Assoro, A.B.; Lingle, W.L.; Salisbury, J.L. Centrosome amplification and the development of cancer. Oncogene 2002, 21, 6146–6153. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Balczon, R.; Bao, L.; Zimmer, W.E.; Brown, K.; Zinkowski, R.P.; Brinkley, B.R. Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J. Cell Biol. 1995, 130, 105–115. [Google Scholar] [CrossRef]
- Wong, C.; Stearns, T. Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat. Cell Biol. 2003, 5, 539–544. [Google Scholar] [CrossRef]
- Bergoglio, V.; Pillaire, M.J.; Lacroix-Triki, M.; Raynaud-Messina, B.; Canitrot, Y.; Bieth, A.; Gares, M.; Wright, M.; Delsol, G.; Loeb, L.A.; et al. Deregulated DNA polymerase beta induces chromosome instability and tumorigenesis. Cancer Res. 2002, 62, 3511–3514. [Google Scholar]
- Bockerstett, K.A.; DiPaolo, R.J. Regulation of Gastric Carcinogenesis by Inflammatory Cytokines. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 47–53. [Google Scholar] [CrossRef]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev. Cell 2017, 40, 313–322. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Klattenhoff, A.W.; Thakur, M.; Sebastian, M.; Kidane, D. Mutation in DNA Polymerase Beta Causes Spontaneous Chromosomal Instability and Inflammation-Associated Carcinogenesis in Mice. Cancers 2019, 11, 1160. https://doi.org/10.3390/cancers11081160
Zhao S, Klattenhoff AW, Thakur M, Sebastian M, Kidane D. Mutation in DNA Polymerase Beta Causes Spontaneous Chromosomal Instability and Inflammation-Associated Carcinogenesis in Mice. Cancers. 2019; 11(8):1160. https://doi.org/10.3390/cancers11081160
Chicago/Turabian StyleZhao, Shengyuan, Alex W. Klattenhoff, Megha Thakur, Manu Sebastian, and Dawit Kidane. 2019. "Mutation in DNA Polymerase Beta Causes Spontaneous Chromosomal Instability and Inflammation-Associated Carcinogenesis in Mice" Cancers 11, no. 8: 1160. https://doi.org/10.3390/cancers11081160
APA StyleZhao, S., Klattenhoff, A. W., Thakur, M., Sebastian, M., & Kidane, D. (2019). Mutation in DNA Polymerase Beta Causes Spontaneous Chromosomal Instability and Inflammation-Associated Carcinogenesis in Mice. Cancers, 11(8), 1160. https://doi.org/10.3390/cancers11081160