Interphase Cytogenetic Analysis of Micronucleated and Multinucleated Cells Supports the Premature Chromosome Condensation Hypothesis as the Mechanistic Origin of Chromothripsis

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
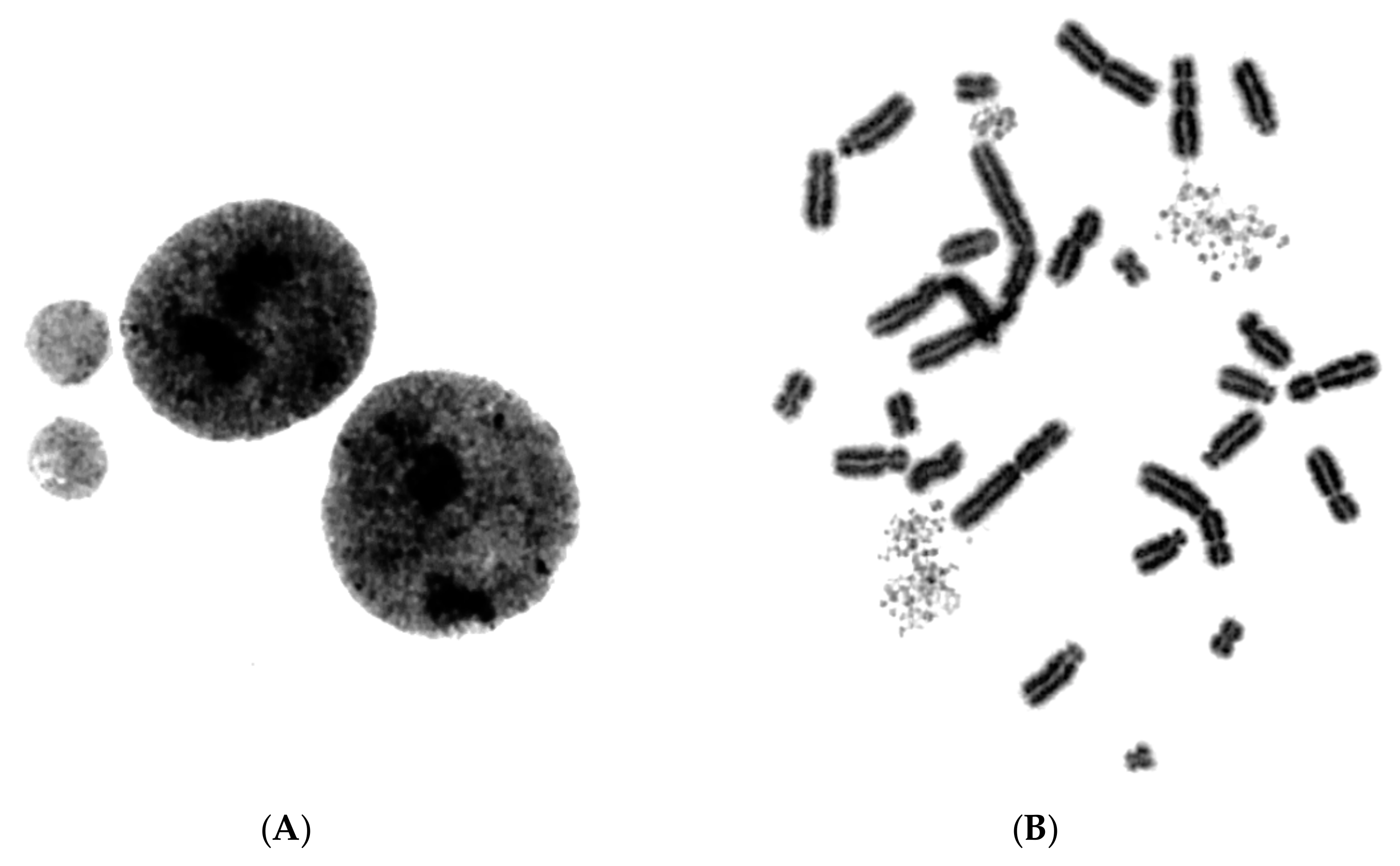{kind=link}
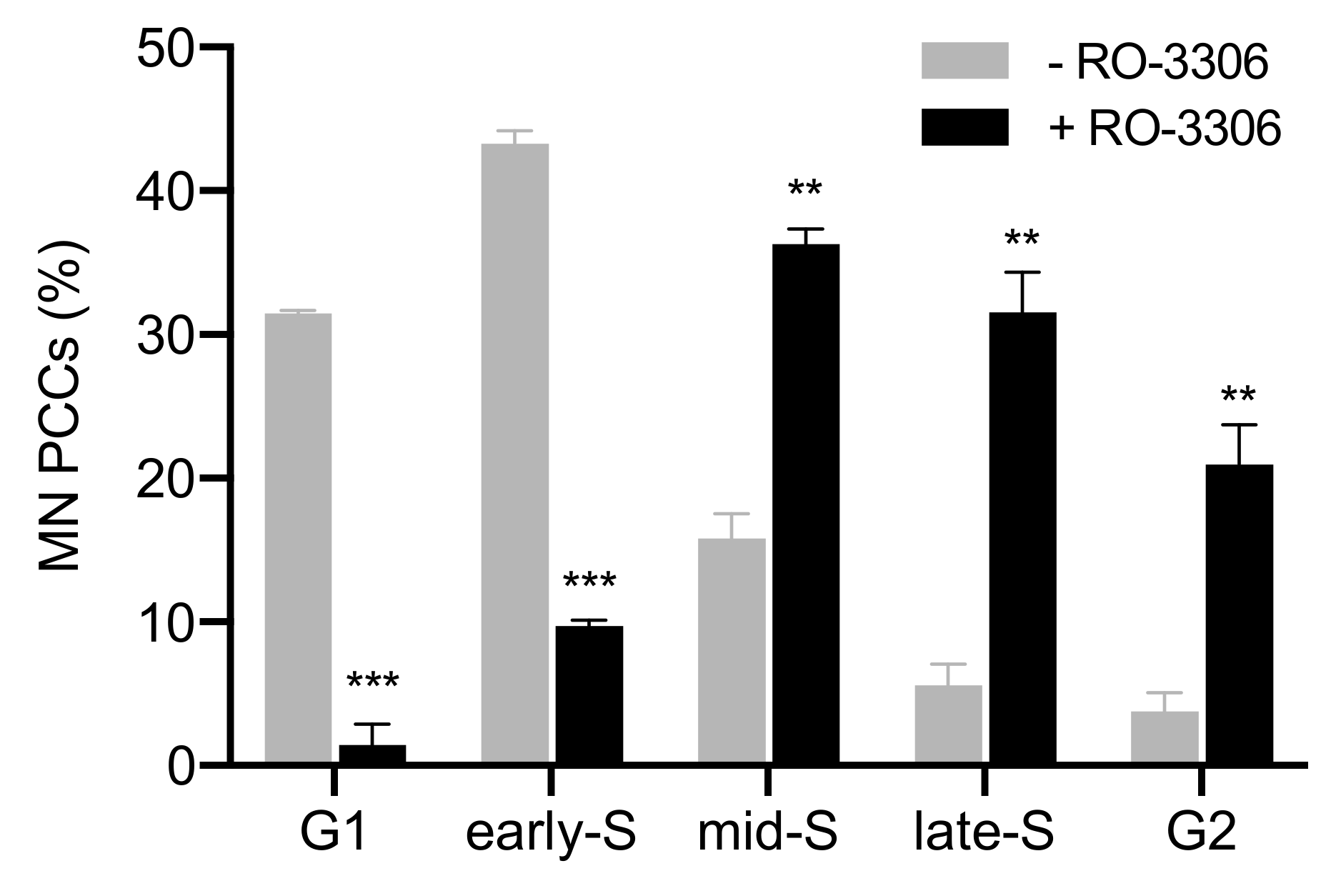{kind=link}
{kind=link}
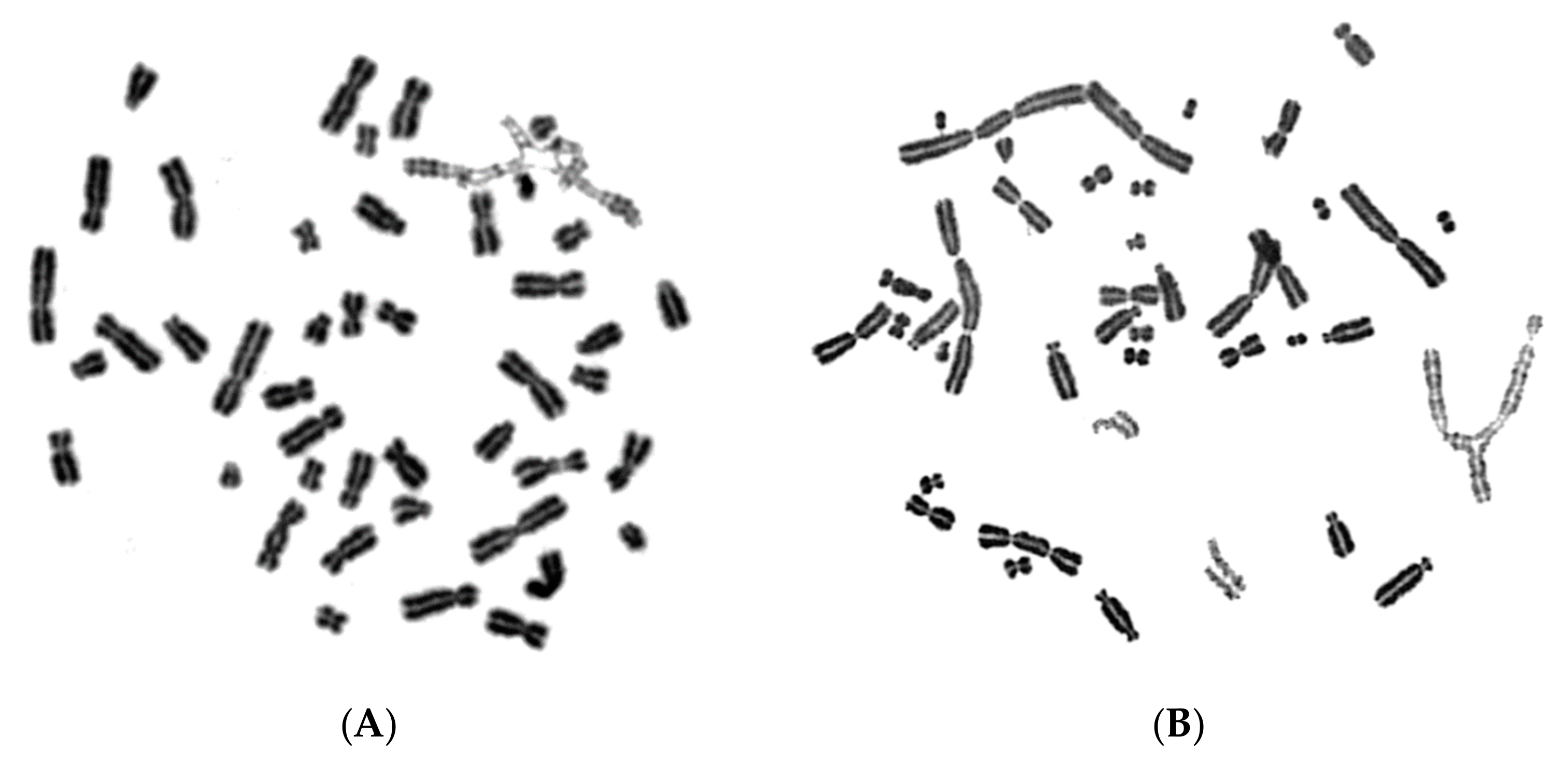{kind=link}
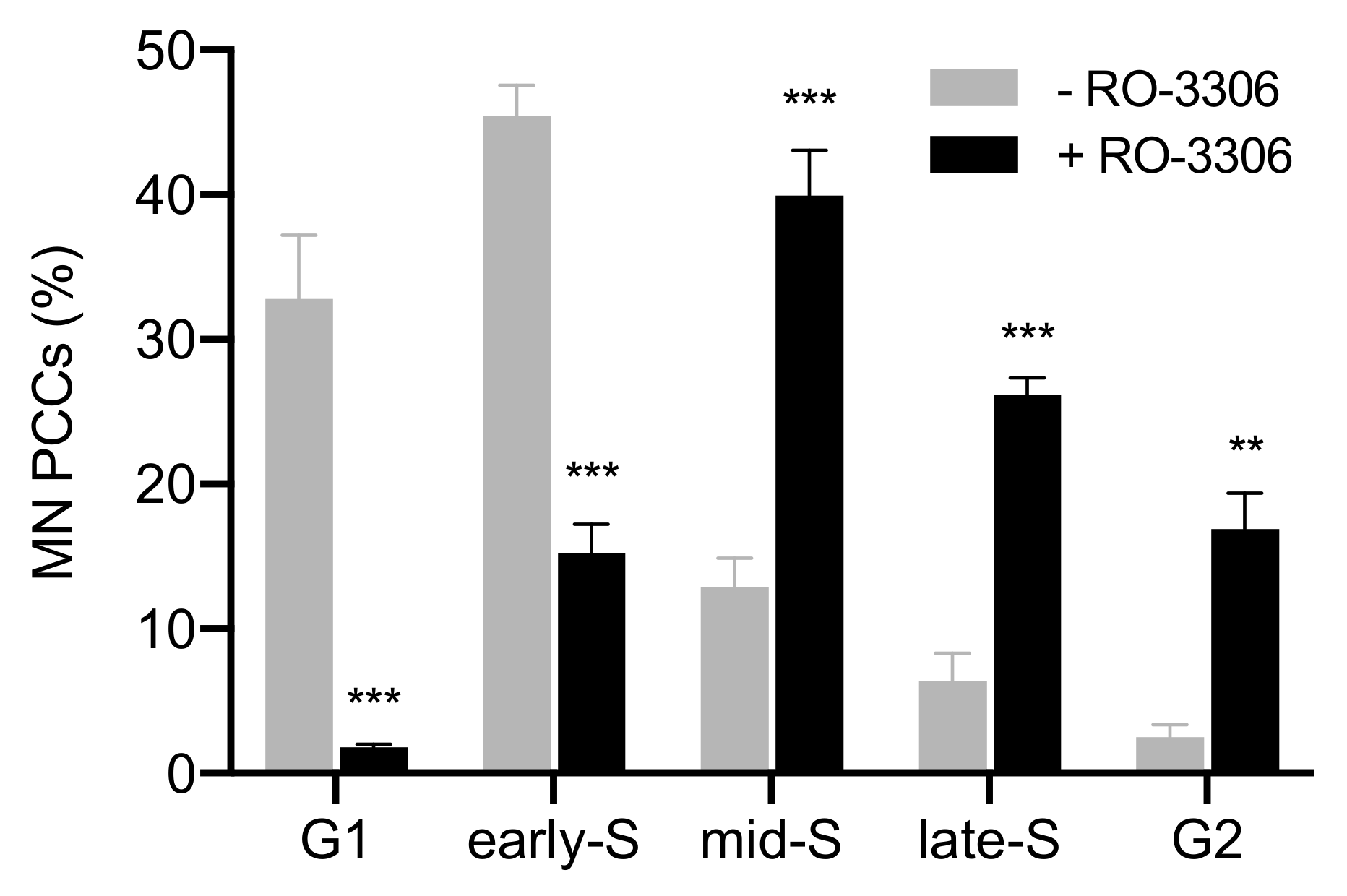{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
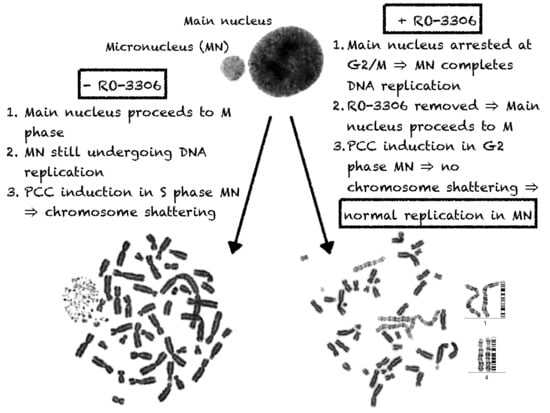
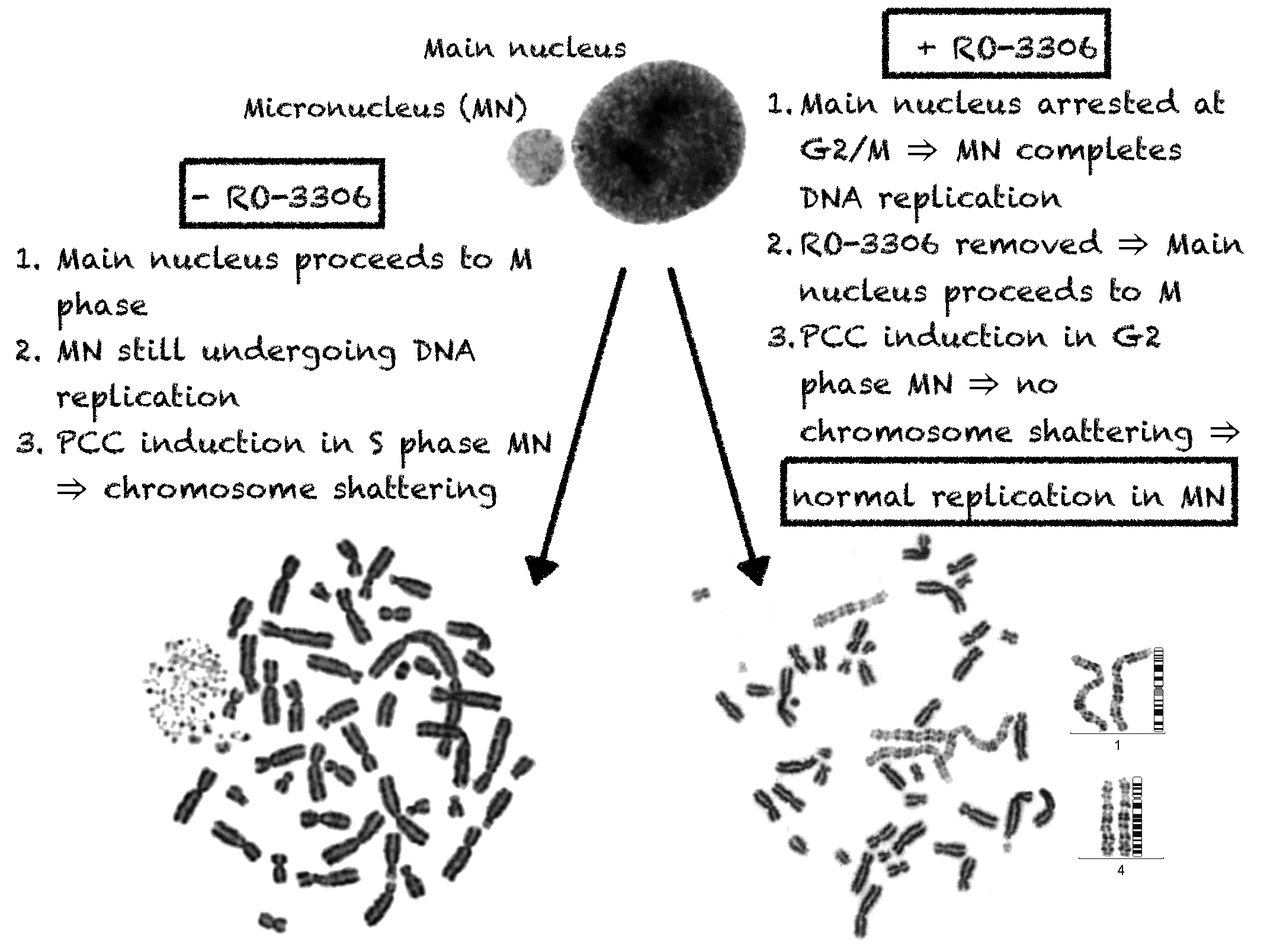
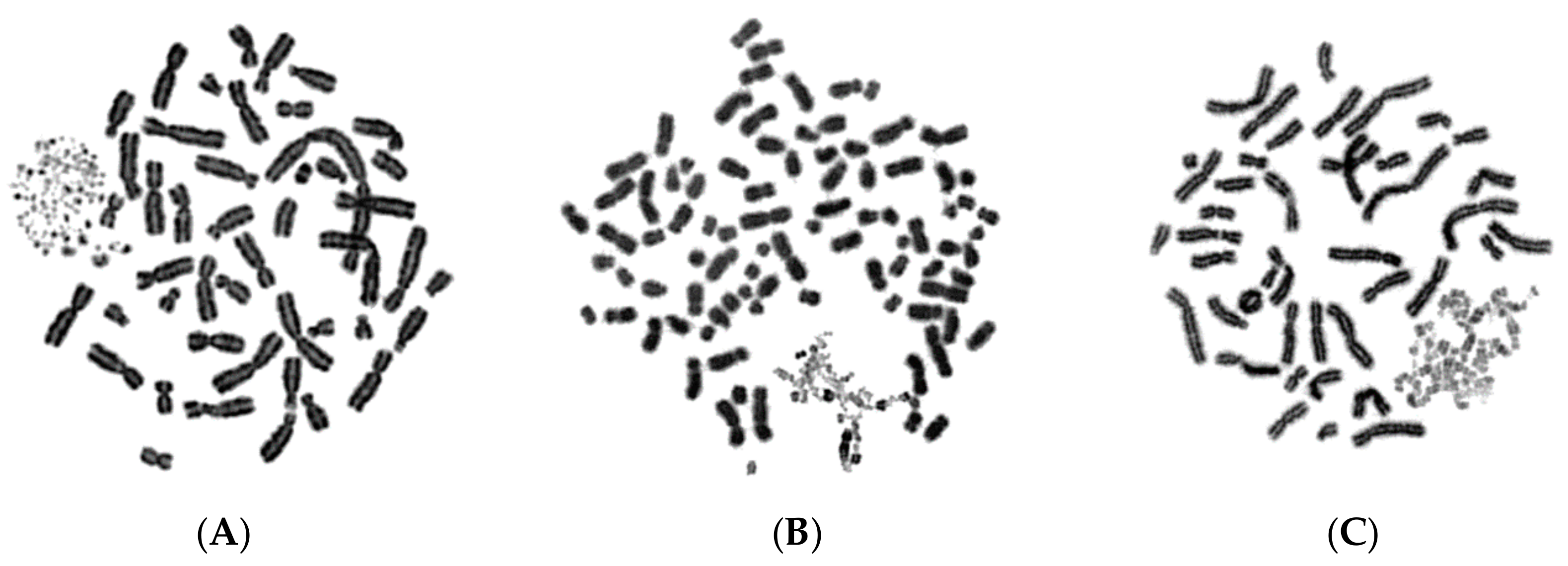
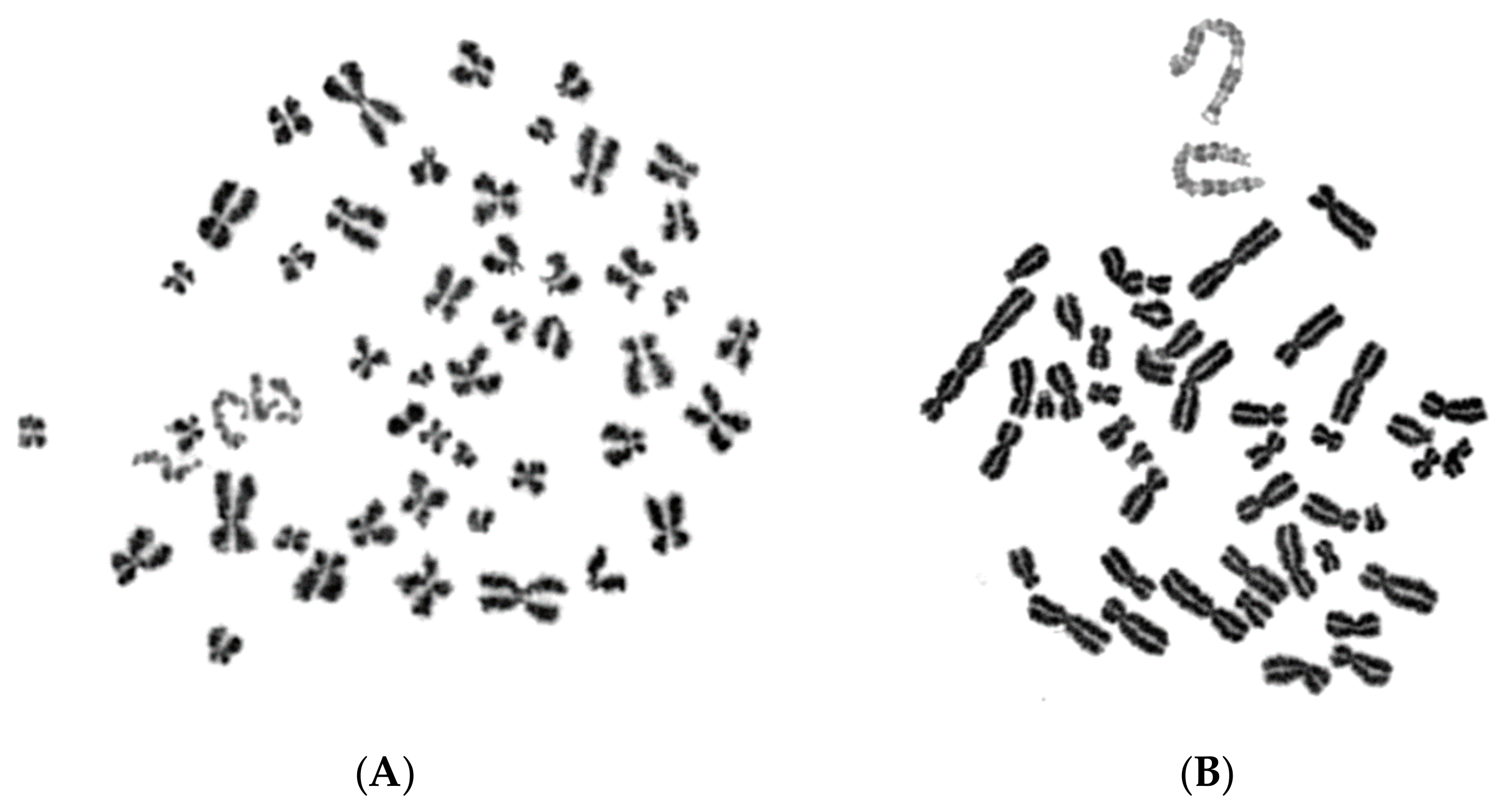
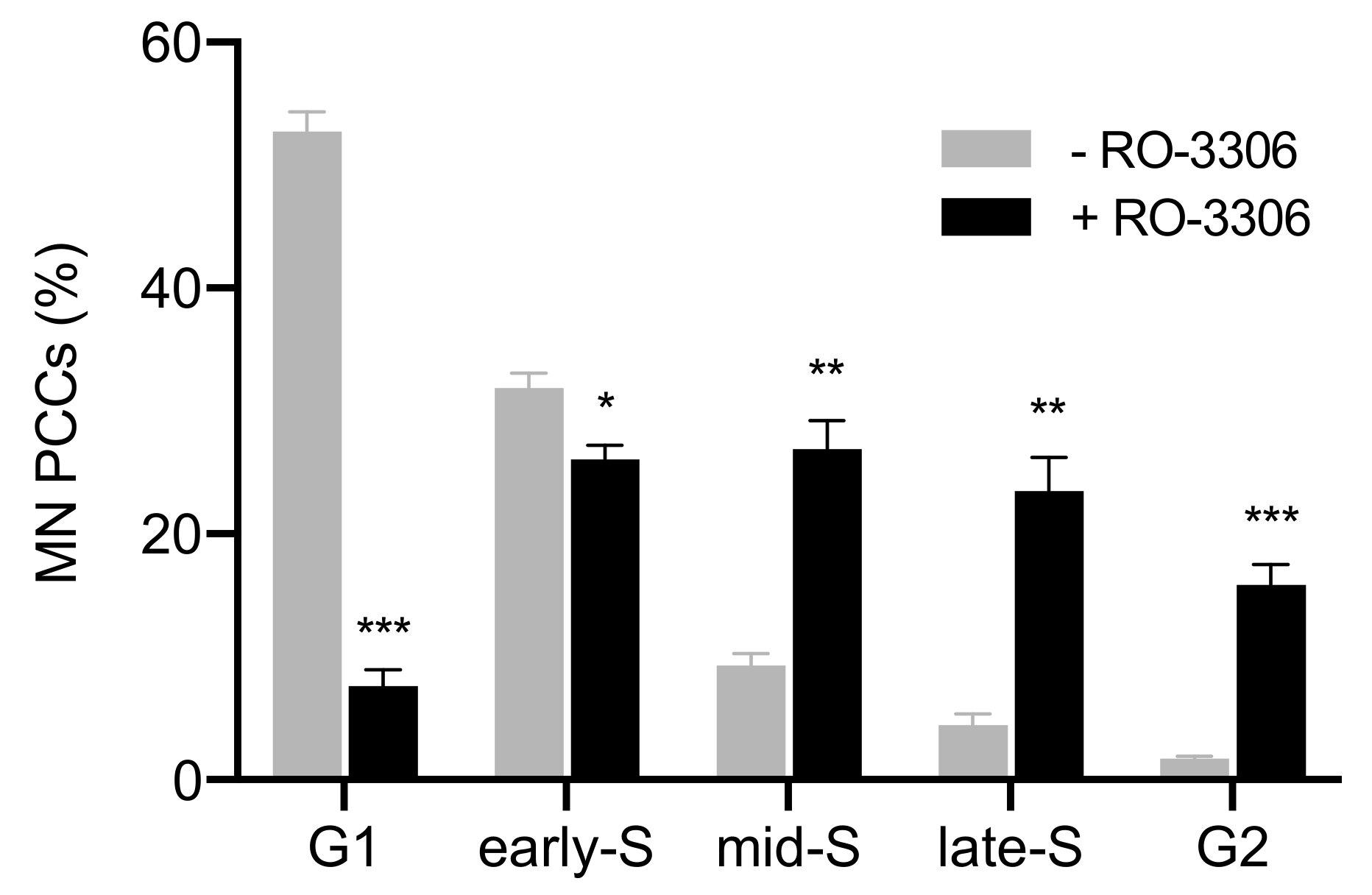
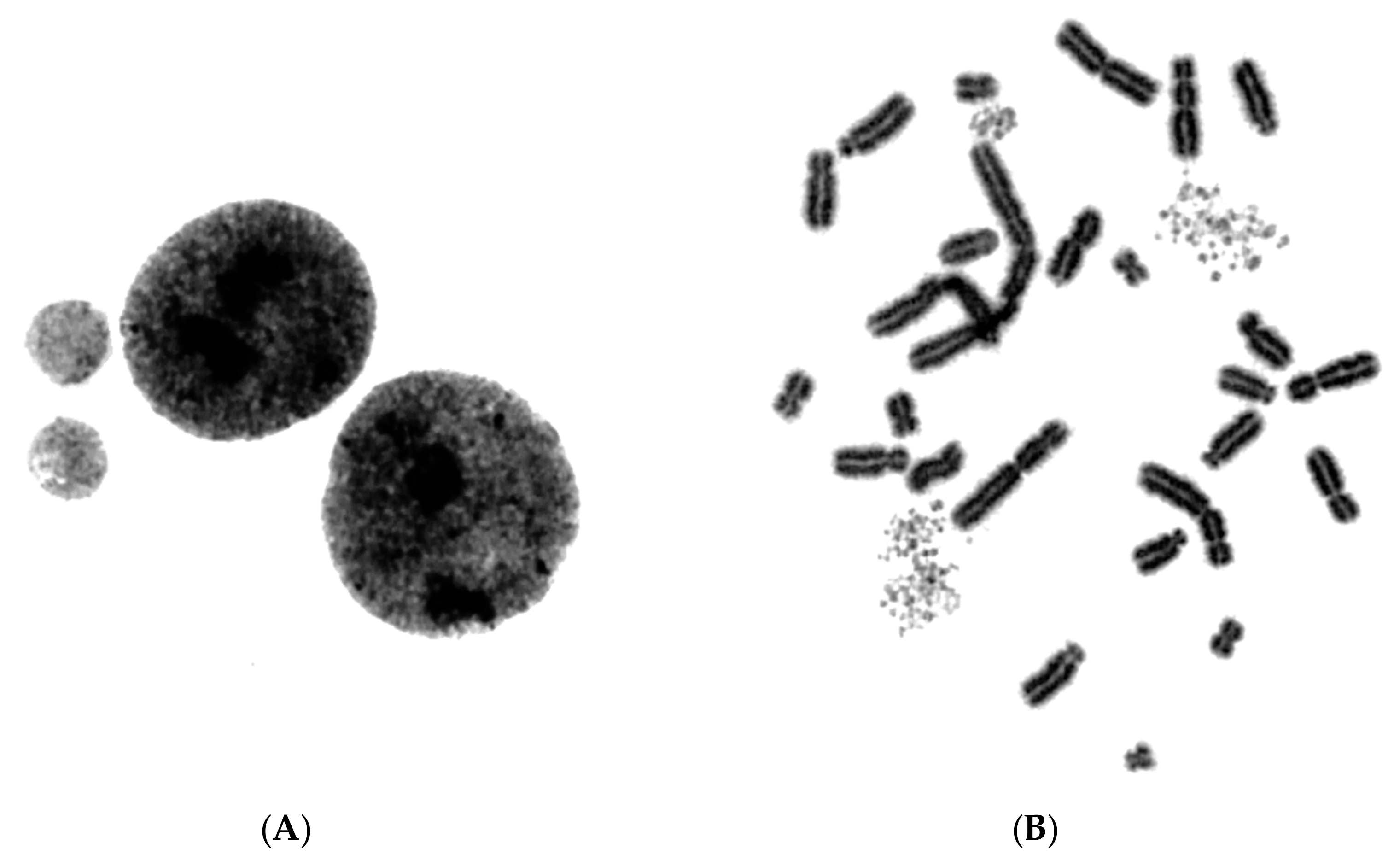
2.1. In Asynchronous Micronucleated Cells Generated by γ-Irradiation of G0-lymphocytes, the PCC Process Triggers Shattering of Chromosomes in MN Still Undergoing DNA Replication When Main Nuclei Enter Mitosis
2.2. The PCC Process Underlies the Mechanistic Origin of Chromosome Shattering by a One-Step Cellular Catastrophic Event in Asynchronous Micronucleated Cells Generated by γ-Irradiation of Lymphocytes in the G1/S Phase Border
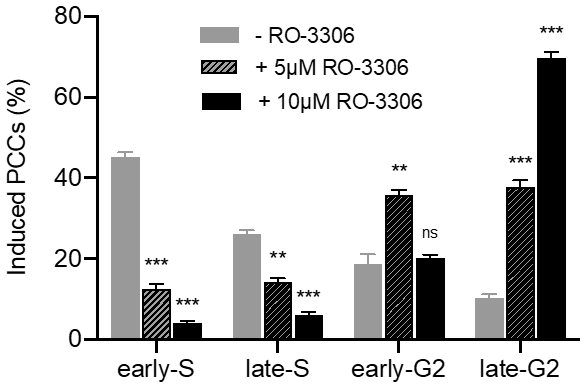
2.3. The PCC Process Induces Shattering of Chromosomes in MN Still Replicating DNA When Main Nuclei Enter Mitosis in Asynchronous Micronucleated Cells Generated by γ-Irradiation of Mitotic CHO Cells
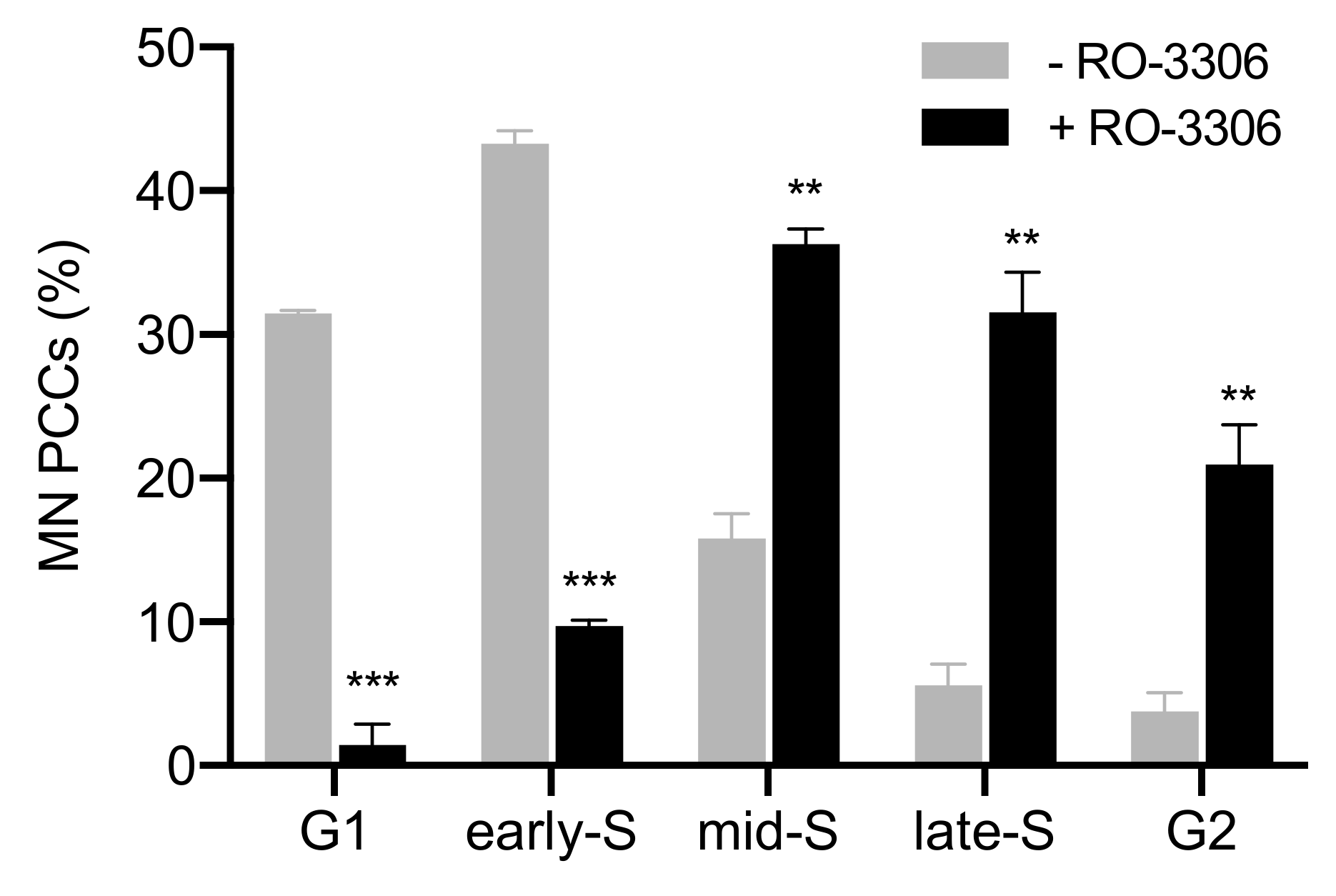
2.4. The Presence of Asynchronous Mitosis in Multinucleated Cells Generated by Fusion of Exponentially Growing Cells Is an Important Determinant of the Shattering of Genetic Material in a Single Catastrophic Event
3. Discussion
4. Materials and Methods
4.1. Lymphocyte Whole Blood Cultures, Production of Micronucleated Cells, and Irradiation Conditions
4.2. Micronucleated Cells Generated by Irradiation of Mitotic CHO Cells
4.3. Heterophasic Multinucleated Cells Produced by Cell Fusion of Exponentially Growing CHO Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boveri, T. Zur Frage der Entstehung Maligner Tumoren. Gustav. Fish. 1914, 40, 1–64. [Google Scholar]
- Duijf, P.H.G.; Nanayakkara, D.; Nones, K.; Srihari, S.; Kalimutho, M.; Khanna, K.K. Mechanisms of Genomic Instability in Breast Cancer. Trends Mol. Med. 2019, 25, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Natarajan, A.T.; Hande, M.P. Chromosomal instability—Mechanisms and consequences. Mutat. Res. Genet. Toxicol. Env. Mutagen. 2015, 793, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.T. Chromosome aberrations: Past, present and future. Mutat. Res. 2002, 504, 3–16. [Google Scholar] [CrossRef]
- Natarajan, A.; Simpson, D.A.; Sanders, G.M. An unusual complication of a Bivona Hyperflex tracheostomy tube. Anaesthesia 2005, 60, 208, discussion 208. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.T.; Palitti, F. DNA repair and chromosomal alterations. Mutat. Res. 2008, 657, 3–7. [Google Scholar] [CrossRef]
- Palumbo, E.; Russo, A. Chromosome Imbalances in Cancer: Molecular Cytogenetics Meets Genomics. Cytogenet. Genome Res. 2016, 150, 176–184. [Google Scholar] [CrossRef]
- Campbell, P.J.; Stephens, P.J.; Pleasance, E.D.; O’Meara, S.; Li, H.; Santarius, T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Hardy, C.; et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008, 40, 722–729. [Google Scholar] [CrossRef]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef] [Green Version]
- Ratnaparkhe, M.; Wong, J.K.L.; Wei, P.C.; Hlevnjak, M.; Kolb, T.; Simovic, M.; Haag, D.; Paul, Y.; Devens, F.; Northcott, P.; et al. Defective DNA damage repair leads to frequent catastrophic genomic events in murine and human tumors. Nat. Commun. 2018, 9, 4760. [Google Scholar] [CrossRef]
- Hair, J.M.; Terzoudi, G.I.; Hatzi, V.I.; Lehockey, K.A.; Srivastava, D.; Wang, W.; Pantelias, G.E.; Georgakilas, A.G. BRCA1 role in the mitigation of radiotoxicity and chromosomal instability through repair of clustered DNA lesions. Chem. Biol. Interact. 2010, 188, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibowitz, M.L.; Zhang, C.Z.; Pellman, D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu. Rev. Genet. 2015, 49, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Marcozzi, A.; Pellestor, F.; Kloosterman, W.P. The Genomic Characteristics and Origin of Chromothripsis. Methods Mol. Biol. 2018, 1769, 3–19. [Google Scholar] [PubMed]
- Cortés-Ciriano, I.; Lee, J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. bioRxiv 2018, 333617. [Google Scholar] [CrossRef] [Green Version]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.M.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline†. Hum. Mol. Genet. 2011, 20, 1916–1924. [Google Scholar] [CrossRef]
- Deng, L.; Wu, R.A.; Sonneville, R.; Kochenova, O.V.; Labib, K.; Pellman, D.; Walter, J.C. Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol. Cell 2019, 73, 915–929. [Google Scholar] [CrossRef]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 2019, 51, 705–715. [Google Scholar] [CrossRef]
- Luijten, M.N.H.; Lee, J.X.T.; Crasta, K.C. Mutational game changer: Chromothripsis and its emerging relevance to cancer. Mutat. Res. Mutat. Res. 2018, 777, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Degrassi, F. Molecular cytogenetics of the micronucleus: Still surprising. Mutat. Res. Toxicol. Environ. Mutagen. 2018, 836, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Terradas, M.; Martín, M.; Genescà, A. Impaired nuclear functions in micronuclei results in genome instability and chromothripsis. Arch. Toxicol. 2016, 90, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Terzoudi, G.I.; Karakosta, M.; Pantelias, A.; Hatzi, V.I.; Karachristou, I.; Pantelias, G. Stress induced by premature chromatin condensation triggers chromosome shattering and chromothripsis at DNA sites still replicating in micronuclei or multinucleate cells when primary nuclei enter mitosis. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, A.; Guo, Y.; Arthanari, H.; Wagner, G.; Halperin, J.A.; Chorev, M. Synthetic studies toward aryl-(4-aryl-4H-[1,2,4]triazole-3-yl)-amine from 1,3-diarylthiourea as urea mimetics. J. Org. Chem. 2005, 70, 6362–6368. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Ni, J.; Liang, Z.; Xue, J.; Fenech, M.F.; Wang, X. The molecular origins and pathophysiological consequences of micronuclei: New insights into an age-old problem. Mutat. Res. Mutat. Res. 2019, 779, 1–35. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Mardin, B.R.; Drainas, A.P.; Waszak, S.M.; Weischenfeldt, J.; Isokane, M.; Stutz, A.M.; Raeder, B.; Efthymiopoulos, T.; Buccitelli, C.; Segura-Wang, M.; et al. A cell-based model system links chromothripsis with hyperploidy. Mol. Syst. Biol. 2015, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Shim, G.; Ricoul, M.; Hempel, W.M.; Azzam, E.I.; Sabatier, L. Crosstalk between telomere maintenance and radiation effects: A key player in the process of radiation-induced carcinogenesis. Mutat. Res. Mutat. Res. 2014, 760, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Teitz, L.S.; Kim, D.H.; Shoshani, O.; Skaletsky, H.; Fachinetti, D.; Page, D.C.; Cleveland, D.W. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 2017, 19, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell 2017, 66, 735–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terradas, M.; Martín, M.; Tusell, L.; Genescà, A. Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat. Res. Mutat. Res. 2010, 705, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Terradas, M.; Iliakis, G.; Tusell, L.; Genesca, A. Breaks invisible to the DNA damage response machinery accumulate in ATM-deficient cells. Genes Chromosom. Cancer 2009, 48, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Pantelias, G.E. Radiation-induced cytogenetic damage in relation to changes in interphase chromosome conformation. Radiat. Res. 1986, 105, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Pantelias, G.E.; Terzoudi, G.I. Functional cell-cycle chromatin conformation changes in the presence of DNA damage result into chromatid breaks: A new insight in the formation of radiation-induced chromosomal aberrations based on the direct observation of interphase chromatin. Mutat. Res. 2010, 701, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Terzoudi, G.I.; Hatzi, V.I.; Donta-Bakoyianni, C.; Pantelias, G.E. Chromatin dynamics during cell cycle mediate conversion of DNA damage into chromatid breaks and affect formation of chromosomal aberrations: Biological and clinical significance. Mutat. Res. 2011, 711, 174–186. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladenov, E.; Saha, J.; Iliakis, G. Processing-Challenges Generated by Clusters of DNA Double-Strand Breaks Underpin Increased Effectiveness of High-LET Radiation and Chromothripsis. Adv. Exp. Med. Biol. 2018, 1044, 149–168. [Google Scholar] [PubMed]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Terradas, M.; Martín, M.; Tusell, L.; Genescà, A. DNA lesions sequestered in micronuclei induce a local defective-damage response. DNA Repair (Amst) 2009, 8, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sandberg, A.A. Chromosome Pulverization in Human Cells With Micronuclei. JNCI J. Natl. Cancer Inst. 1968, 40, 165–179. [Google Scholar]
- Terradas, M.; Martín, M.; Hernández, L.; Tusell, L.; Genescà, A. Nuclear envelope defects impede a proper response to micronuclear DNA lesions. Mutat. Res. Mol. Mech. Mutagen. 2012, 729, 35–40. [Google Scholar] [CrossRef]
- Johnson, R.T.; Rao, P.N. Mammalian cell fusion: Induction of premature chromosome condensation in interphase nuclei. Nature 1970, 226, 717–722. [Google Scholar] [CrossRef]
- Obe, G.; Beek, B. The human leulocyte test system. VII. Further investigations concerning micronucleus-derived premature chromosome condensation. Humangenetik 1975, 30, 143–154. [Google Scholar]
- Falk, M.; Falková, I.; Kopečná, O.; Bačíková, A.; Pagáčová, E.; Šimek, D.; Golan, M.; Kozubek, S.; Pekarová, M.; Follett, S.E.; et al. Chromatin architecture changes and DNA replication fork collapse are critical features in cryopreserved cells that are differentially controlled by cryoprotectants. Sci. Rep. 2018, 8, 14694. [Google Scholar] [CrossRef] [Green Version]
- Terzoudi, G.I.; Donta-Bakoyianni, C.; Iliakis, G.; Pantelias, G.E. Investigation of bystander effects in hybrid cells by means of cell fusion and premature chromosome condensation induction. Radiat. Res. 2010, 173, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Scott, C.D.; Beck, T.M. Premature chromosome condensation in human leukemia. Blood 1976, 47, 687–693. [Google Scholar] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantelias, A.; Karachristou, I.; Georgakilas, A.G.; Terzoudi, G.I. Interphase Cytogenetic Analysis of Micronucleated and Multinucleated Cells Supports the Premature Chromosome Condensation Hypothesis as the Mechanistic Origin of Chromothripsis. Cancers 2019, 11, 1123. https://doi.org/10.3390/cancers11081123
Pantelias A, Karachristou I, Georgakilas AG, Terzoudi GI. Interphase Cytogenetic Analysis of Micronucleated and Multinucleated Cells Supports the Premature Chromosome Condensation Hypothesis as the Mechanistic Origin of Chromothripsis. Cancers. 2019; 11(8):1123. https://doi.org/10.3390/cancers11081123
Chicago/Turabian StylePantelias, Antonio, Ioanna Karachristou, Alexandros G. Georgakilas, and Georgia I. Terzoudi. 2019. "Interphase Cytogenetic Analysis of Micronucleated and Multinucleated Cells Supports the Premature Chromosome Condensation Hypothesis as the Mechanistic Origin of Chromothripsis" Cancers 11, no. 8: 1123. https://doi.org/10.3390/cancers11081123