DNA-Methylation-Caused Downregulation of miR-30 Contributes to the High Expression of XPO1 and the Aggressive Growth of Tumors in Pancreatic Ductal Adenocarcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
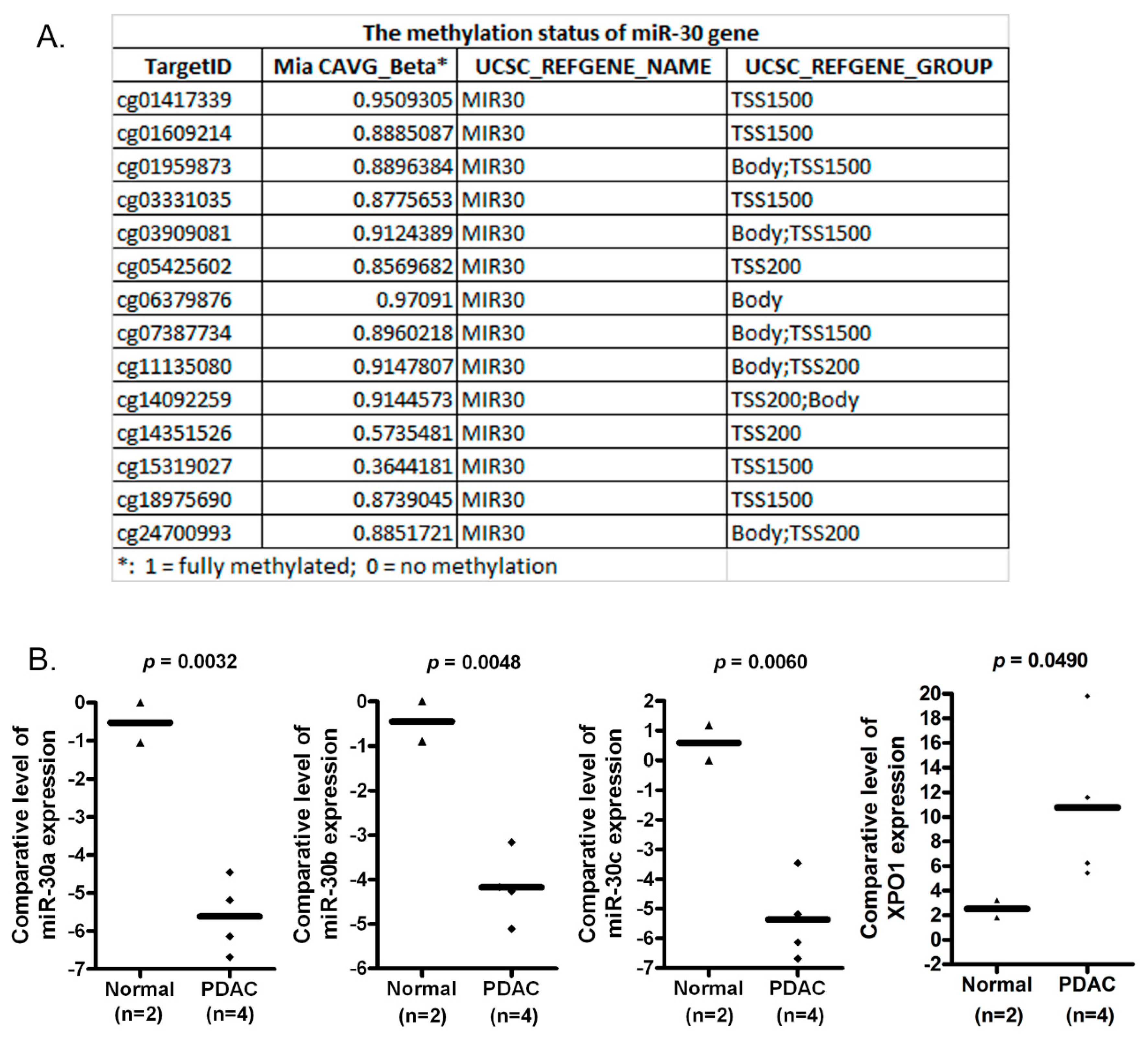
2.1. The miR-30 Gene Is Hypermethylated in MiaPaCa-2 Pancreatic Cancer Cells
2.2. The Expression of miR-30a, miR-30b and miR-30c Is Lower in PDAC Tissues When Compared with Normal Pancreatic Tissues
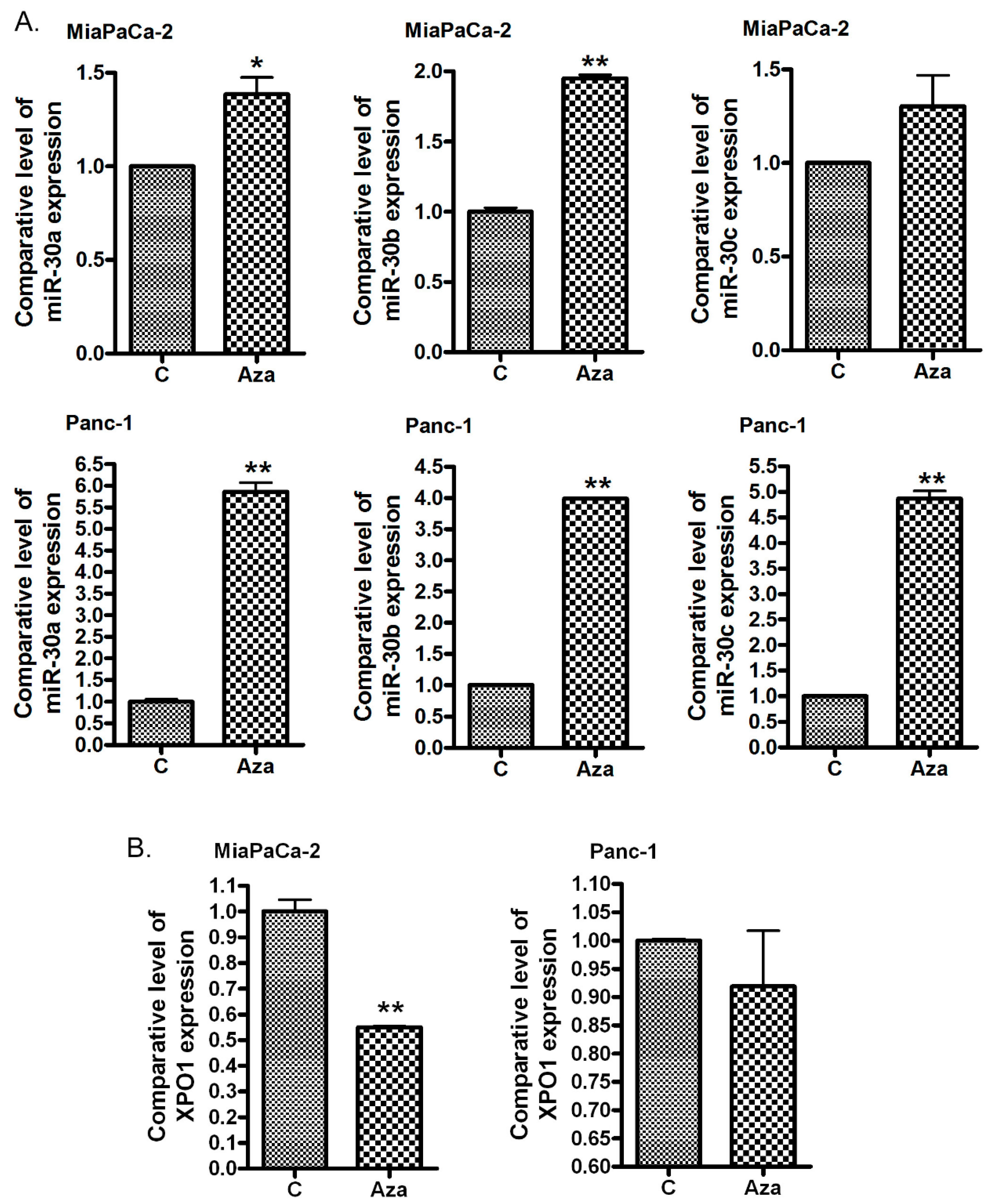
2.3. The Treatment of PDAC Cells with 5-Aza-dC Increases miR-30 Expression and Decreases the Expression of XPO1
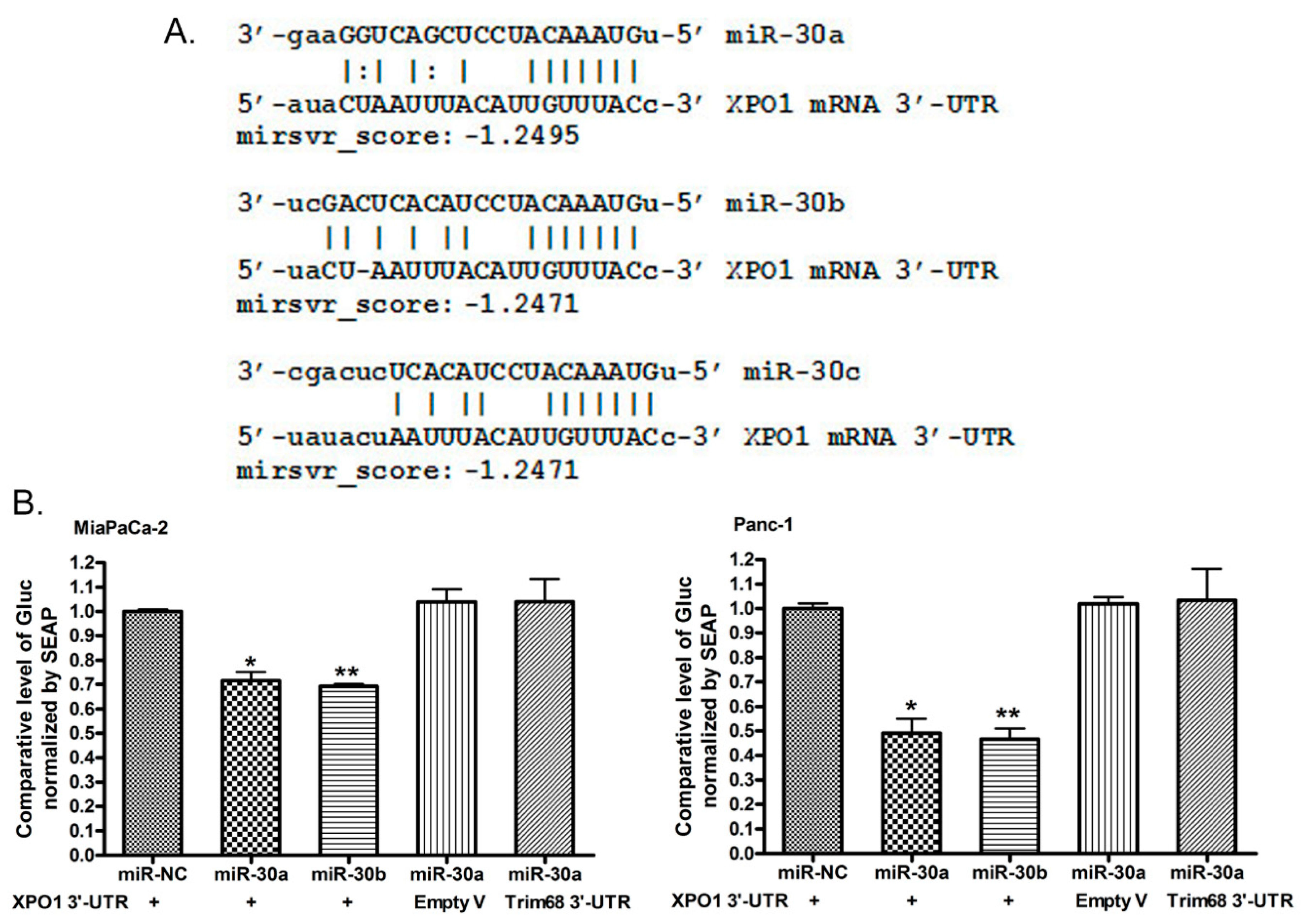
2.4. miR-30a or miR-30b Binds 3′-UTR of XPO1 and Inhibits Its Translational Activity
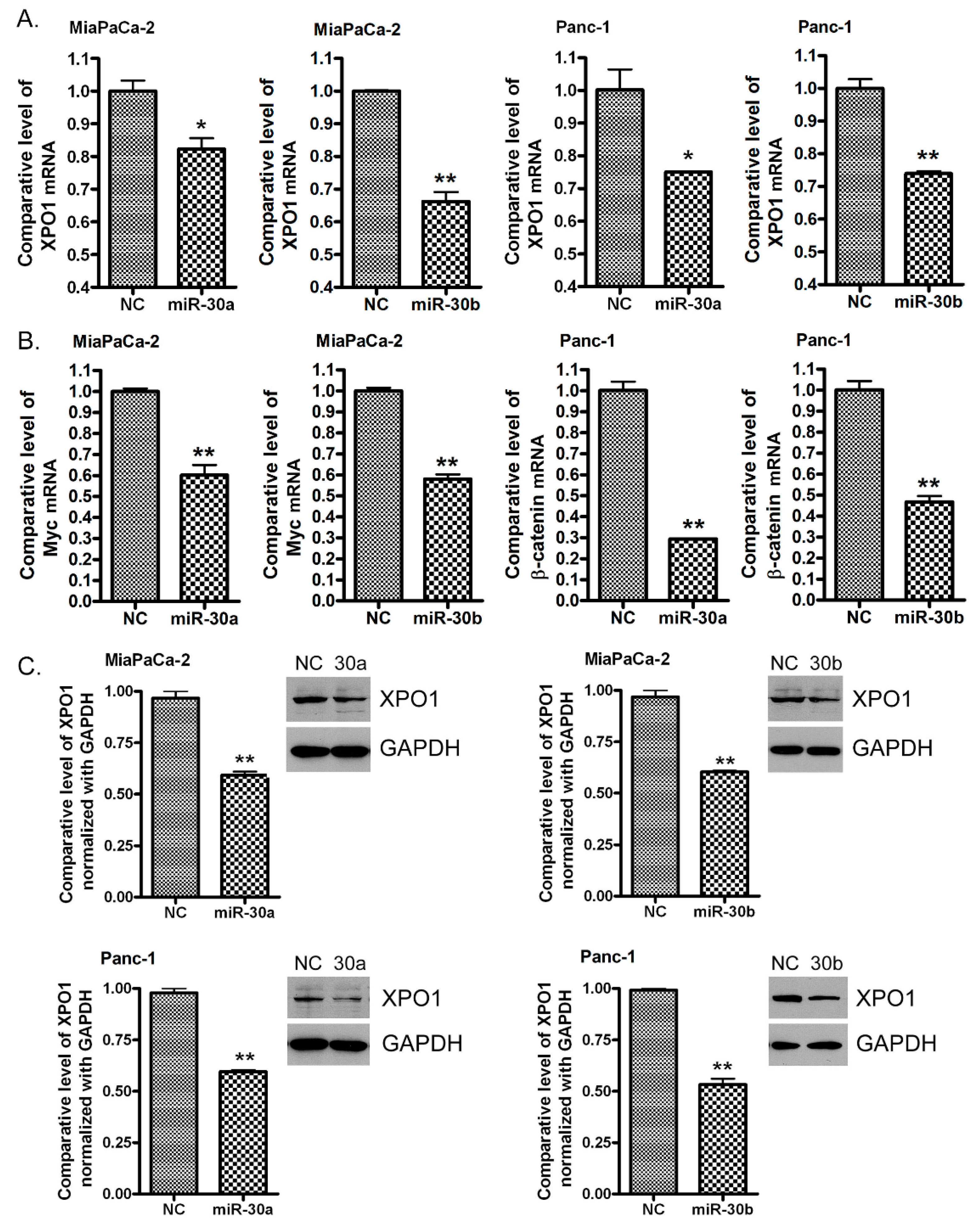
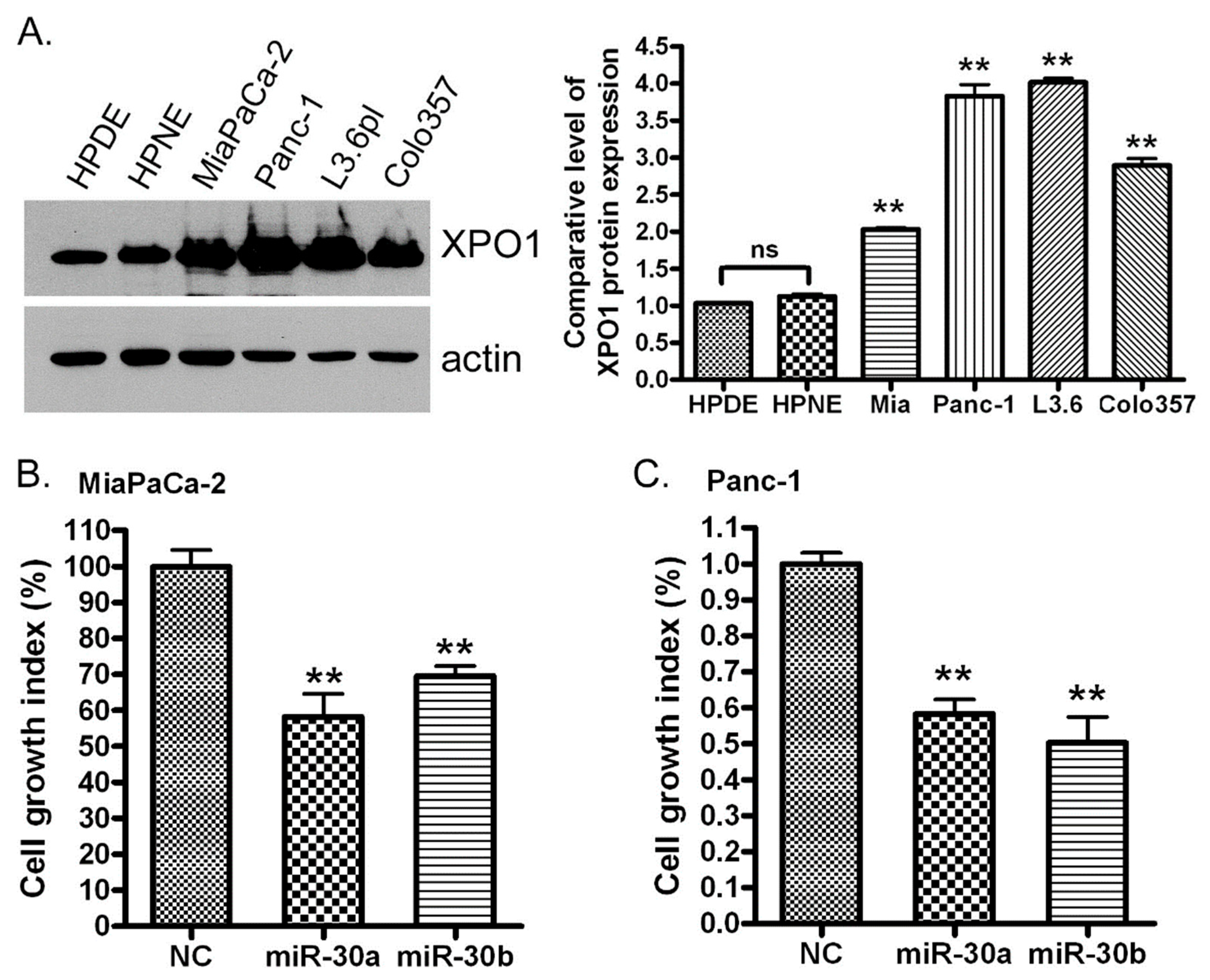
2.5. Re-Expression of miR-30a or miR-30b Inhibits the Expression of XPO1
2.6. miR-30a or miR-30b Transfection Inhibits PDAC Cell Proliferation In Vitro
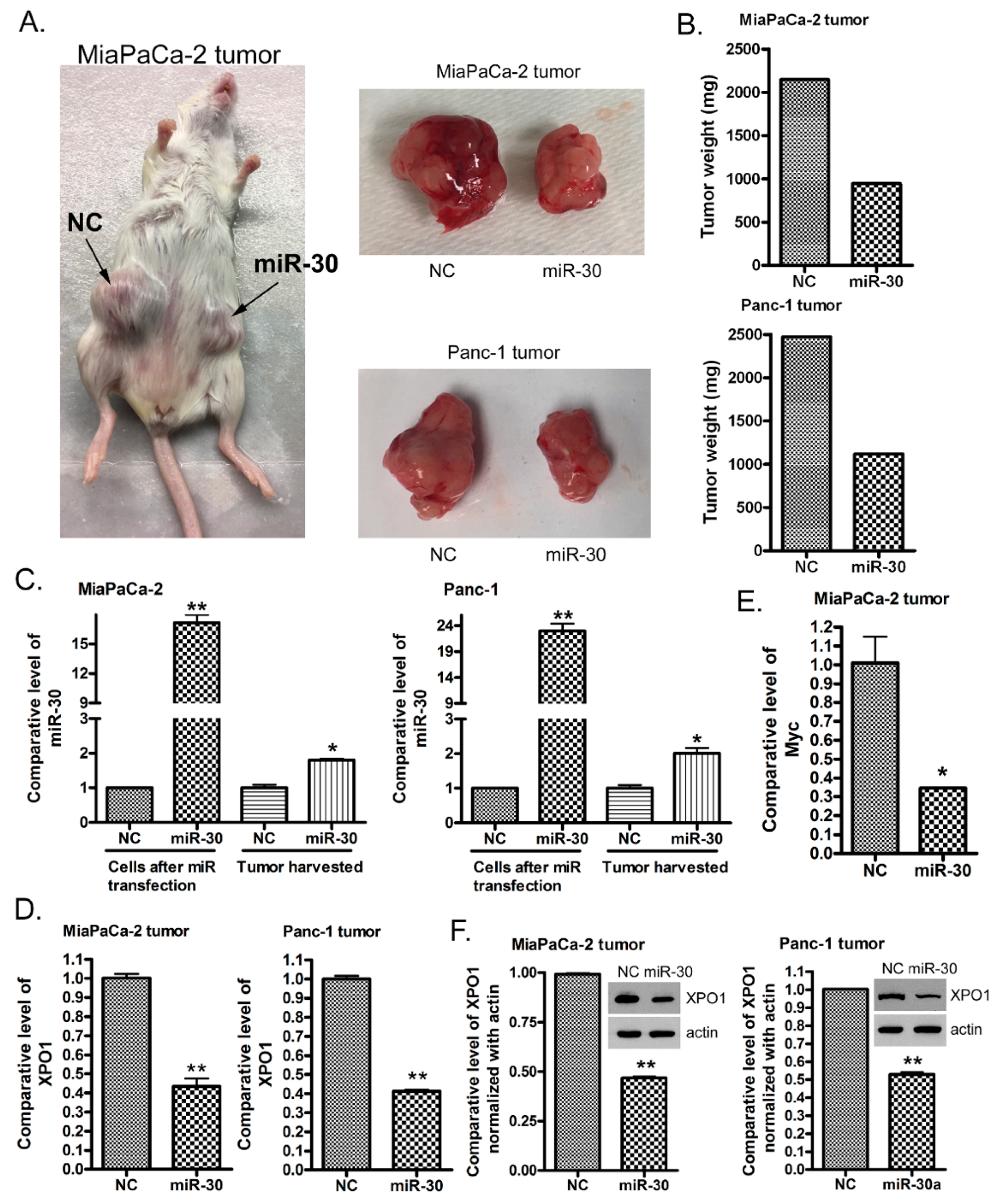
2.7. The miR-30a or miR-30b Transfection Suppresses PDAC Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Tissues, Reagents, and Antibodies
4.2. Genomic DNA Extraction and Methylation Analysis
4.3. RNA Isolation and miRNA Real-Time RT-PCR
4.4. mRNA Real-Time RT-PCR
4.5. Western Blot Analysis
4.6. Re-Expression of miR-30 in PDAC Cells
4.7. Luciferase Activity Assay for Confirming miRNA Binding to Target 3’UTR
4.8. Cell Growth Inhibition Assay (MTT Assay)
4.9. Tumor Growth Inhibition Assay
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Sun, H.; Hattori, N.; Chien, W.; Sun, Q.; Sudo, M.; Ling, G.L.; Ding, L.; Lim, S.L.; Shacham, S.; Kauffman, M.; et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br. J. Cancer 2014, 111, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Holloway, M.P.; Nguyen, K.; McCauley, D.; Landesman, Y.; Kauffman, M.G.; Shacham, S.; Altura, R.A. XPO1 (CRM1) inhibition represses STAT3 activation to drive a survivin-dependent oncogenic switch in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Aboukameel, A.; Bao, B.; Sarkar, F.H.; Philip, P.A.; Kauffman, M.; Shacham, S.; Mohammad, R.M. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 2013, 144, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Muqbil, I.; Aboukameel, A.; Elloul, S.; Carlson, R.; Senapedis, W.; Baloglu, E.; Kauffman, M.; Shacham, S.; Bhutani, D.; Zonder, J.; et al. Anti-tumor activity of selective inhibitor of nuclear export (SINE) compounds, is enhanced in non-Hodgkin lymphoma through combination with mTOR inhibitor and dexamethasone. Cancer Lett. 2016, 383, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, J.; Savona, M.; Baz, R.; Mau-Sorensen, P.M.; Gabrail, N.; Garzon, R.; Stone, R.; Wang, M.; Savoie, L.; Martin, P.; et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood 2017, 129, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chong, Y.; Tu, Y.; Liu, N.; Yue, C.; Qi, Z.; Liu, H.; Yao, Y.; Liu, H.; Gao, S.; et al. CRM1/XPO1 is associated with clinical outcome in glioma and represents a therapeutic target by perturbing multiple core pathways. J. Hematol. Oncol. 2016, 9, 108. [Google Scholar] [CrossRef]
- Noske, A.; Weichert, W.; Niesporek, S.; Roske, A.; Buckendahl, A.C.; Koch, I.; Sehouli, J.; Dietel, M.; Denkert, C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008, 112, 1733–1743. [Google Scholar] [CrossRef]
- Huang, W.Y.; Yue, L.; Qiu, W.S.; Wang, L.W.; Zhou, X.H.; Sun, Y.J. Prognostic value of CRM1 in pancreas cancer. Clin. Investig. Med. 2009, 32, E315. [Google Scholar] [CrossRef]
- Saulino, D.M.; Younes, P.S.; Bailey, J.M.; Younes, M. CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget 2018, 9, 21289–21295. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, D.J.; Finetti, P.; Birnbaum, D.; Mamessier, E.; Bertucci, F. XPO1 Expression Is a Poor-Prognosis Marker in Pancreatic Adenocarcinoma. J. Clin. Med. 2019, 8, 596. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, W.; Claret, F.X. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 2017, 12, 187–197. [Google Scholar] [CrossRef]
- Daniunaite, K.; Dubikaityte, M.; Gibas, P.; Bakavicius, A.; Rimantas, L.J.; Ulys, A.; Jankevicius, F.; Jarmalaite, S. Clinical significance of miRNA host gene promoter methylation in prostate cancer. Hum. Mol. Genet. 2017, 26, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Bastami, M.; Solali, S.; Alivand, M.R. Aberrant miRNA promoter methylation and EMT-involving miRNAs in breast cancer metastasis: Diagnosis and therapeutic implications. J. Cell. Physiol. 2018, 233, 3729–3744. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Jonsson, Z.O.; Dutta, A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J. Biol. Chem. 2003, 278, 44312–44319. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes. Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef]
- Jackson, R.J.; Standart, N. How do microRNAs regulate gene expression? Sci. Stke 2007, 2007, re1. [Google Scholar] [CrossRef]
- Bagga, S.; Bracht, J.; Hunter, S.; Massirer, K.; Holtz, J.; Eachus, R.; Pasquinelli, A.E. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 2005, 122, 553–563. [Google Scholar] [CrossRef]
- Nicoloso, M.S.; Spizzo, R.; Shimizu, M.; Rossi, S.; Calin, G.A. MicroRNAs—The micro steering wheel of tumour metastases. Nat. Rev. Cancer 2009, 9, 293–302. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z. MicroRNAs as oncogenes and tumor suppressors. N. Engl. J. Med. 2005, 353, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Miao, D.; Wang, M.; Lv, J.; Wang, Y.; Tong, J. MiR-30-5p suppresses cell chemoresistance and stemness in colorectal cancer through USP22/Wnt/beta-catenin signaling axis. J. Cell. Mol. Med. 2019, 23, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Lin, J.; Zhu, D.; Wang, X.; Brooks, D.; Chen, M.; Chu, Z.B.; Takada, K.; Ciccarelli, B.; Admin, S.; et al. miR-30-5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/beta-catenin/BCL9 pathway. Cancer Res. 2014, 74, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Yang, S.Y.; Wang, D.D.; Chen, X.; Shen, H.Y.; Zhang, X.H.; Zhong, S.L.; Tang, J.H.; Zhao, J.H. The miR-30 family: Versatile players in breast cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Malik, K.; Brown, K.W. Epigenetic gene deregulation in cancer. Br. J. Cancer 2000, 83, 1583–1588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Kao, C.J.; Martiniez, A.; Shi, X.B.; Yang, J.; Evans, C.P.; Dobi, A.; deVere White, R.W.; Kung, H.J. miR-30 as a tumor suppressor connects EGF/Src signal to ERG and EMT. Oncogene 2014, 33, 2495–2503. [Google Scholar] [CrossRef]
- Saleh, A.D.; Cheng, H.; Martin, S.E.; Si, H.; Ormanoglu, P.; Carlson, S.; Clavijo, P.E.; Yang, X.; Das, R.; Cornelius, S.; et al. Integrated Genomic and Functional microRNA Analysis Identifies miR-30-5p as a Tumor Suppressor and Potential Therapeutic Nanomedicine in Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 2860–2873. [Google Scholar] [CrossRef]
- Wang, C.; Cai, L.; Liu, J.; Wang, G.; Li, H.; Wang, X.; Xu, W.; Ren, M.; Feng, L.; Liu, P.; et al. MicroRNA-30a-5p Inhibits the Growth of Renal Cell Carcinoma by Modulating GRP78 Expression. Cell. Physiol. Biochem. 2017, 43, 2405–2419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, Q.; Qin, D.; Yu, L.; Huang, R.; Lv, G.; Zou, Z.; Jiang, X.C.; Zou, C.; Liu, W.; et al. Role of microRNA 30a targeting insulin receptor substrate 2 in colorectal tumorigenesis. Mol. Cell. Biol. 2015, 35, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhong, K.; Chen, K.; Han, L.; Li, B. MicroRNA-30b/c inhibits non-small cell lung cancer cell proliferation by targeting Rab18. BMC Cancer 2014, 14, 703. [Google Scholar] [CrossRef] [PubMed]
- Croset, M.; Pantano, F.; Kan, C.W.S.; Bonnelye, E.; Descotes, F.; Alix-Panabieres, C.; Lecellier, C.H.; Bachelier, R.; Allioli, N.; Hong, S.S.; et al. miRNA-30 Family Members Inhibit Breast Cancer Invasion, Osteomimicry, and Bone Destruction by Directly Targeting Multiple Bone Metastasis-Associated Genes. Cancer Res. 2018, 78, 5259–5273. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Deng, H.; Yao, H.; Liu, Q.; Su, F.; Song, E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene 2010, 29, 4194–4204. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, Y.; Wang, L.; Huang, Y.; Xu, Y.; Xu, L.; Guo, Y.; Lu, J.; Li, X.; Zhu, M.; et al. MicroRNA-30b targets Snail to impede epithelial-mesenchymal transition in pancreatic cancer stem cells. J. Cancer 2018, 9, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Jeromin, S.; Weissmann, S.; Haferlach, C.; Dicker, F.; Bayer, K.; Grossmann, V.; Alpermann, T.; Roller, A.; Kohlmann, A.; Haferlach, T.; et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014, 28, 108–117. [Google Scholar] [CrossRef]
- Ishizawa, J.; Kojima, K.; Hail, N., Jr.; Tabe, Y.; Andreeff, M. Expression, function, and targeting of the nuclear exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol. Ther. 2015, 153, 25–35. [Google Scholar] [CrossRef]
- Azmi, A.S.; Muqbil, I.; Wu, J.; Aboukameel, A.; Senapedis, W.; Baloglu, E.; Bollig-Fischer, A.; Dyson, G.; Kauffman, M.; Landesman, Y.; et al. Targeting the Nuclear Export Protein XPO1/CRM1 Reverses Epithelial to Mesenchymal Transition. Sci. Rep. 2015, 5, 16077. [Google Scholar] [CrossRef]
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Zonder, J.; Baz, R.; Nooka, A.; Richter, J.; et al. Selective Inhibition of Nuclear Export with Oral Selinexor for Treatment of Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 859–866. [Google Scholar] [CrossRef]
- Tai, Y.T.; Landesman, Y.; Acharya, C.; Calle, Y.; Zhong, M.Y.; Cea, M.; Tannenbaum, D.; Cagnetta, A.; Reagan, M.; Munshi, A.A.; et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 2014, 28, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Sanda, T.; Mansour, M.R.; Kentsis, A.; Montero, J.; Le, B.T.; Christie, A.L.; McCauley, D.; Rodig, S.J.; Kauffman, M.; et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br. J. Haematol. 2013, 161, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Arango, N.P.; Yuca, E.; Zhao, M.; Evans, K.W.; Scott, S.; Kim, C.; Gonzalez-Angulo, A.M.; Janku, F.; Ueno, N.T.; Tripathy, D.; et al. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Camacho, S.C.; Silvers, T.R.; Razak, A.R.; Gabrail, N.Y.; Gerecitano, J.F.; Kalir, E.; Pereira, E.; Evans, B.R.; Ramus, S.J.; et al. Inhibition of the Nuclear Export Receptor XPO1 as a Therapeutic Target for Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. 2017, 23, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [PubMed]
- Kazim, S.; Malafa, M.P.; Coppola, D.; Husain, K.; Zibadi, S.; Kashyap, T.; Crochiere, M.; Landesman, Y.; Rashal, T.; Sullivan, D.M.; et al. Selective Nuclear Export Inhibitor KPT-330 Enhances the Antitumor Activity of Gemcitabine in Human Pancreatic Cancer. Mol. Cancer Ther. 2015, 14, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Savona, M.; Baz, R.; Andreeff, M.; Gabrail, N.; Gutierrez, M.; Savoie, L.; Mau-Sorensen, P.M.; Wagner-Johnston, N.; Yee, K.; et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood 2017, 129, 3165–3174. [Google Scholar] [CrossRef]
- Gounder, M.M.; Zer, A.; Tap, W.D.; Salah, S.; Dickson, M.A.; Gupta, A.A.; Keohan, M.L.; Loong, H.H.; D’Angelo, S.P.; Baker, S.; et al. Phase IB Study of Selinexor, a First-in-Class Inhibitor of Nuclear Export, in Patients with Advanced Refractory Bone or Soft Tissue Sarcoma. J. Clin. Oncol. 2016, 34, 3166–3174. [Google Scholar] [CrossRef]
- Alexander, T.B.; Lacayo, N.J.; Choi, J.K.; Ribeiro, R.C.; Pui, C.H.; Rubnitz, J.E. Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Combination with Fludarabine and Cytarabine, in Pediatric Relapsed or Refractory Acute Leukemia. J. Clin. Oncol. 2016, 34, 4094–4101. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azmi, A.S.; Li, Y.; Aboukameel, A.; Muqbil, I.; Philip, P.A.; Mohammad, R.M. DNA-Methylation-Caused Downregulation of miR-30 Contributes to the High Expression of XPO1 and the Aggressive Growth of Tumors in Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 1101. https://doi.org/10.3390/cancers11081101
Azmi AS, Li Y, Aboukameel A, Muqbil I, Philip PA, Mohammad RM. DNA-Methylation-Caused Downregulation of miR-30 Contributes to the High Expression of XPO1 and the Aggressive Growth of Tumors in Pancreatic Ductal Adenocarcinoma. Cancers. 2019; 11(8):1101. https://doi.org/10.3390/cancers11081101
Chicago/Turabian StyleAzmi, Asfar S., Yiwei Li, Amro Aboukameel, Irfana Muqbil, Philip A. Philip, and Ramzi M. Mohammad. 2019. "DNA-Methylation-Caused Downregulation of miR-30 Contributes to the High Expression of XPO1 and the Aggressive Growth of Tumors in Pancreatic Ductal Adenocarcinoma" Cancers 11, no. 8: 1101. https://doi.org/10.3390/cancers11081101
APA StyleAzmi, A. S., Li, Y., Aboukameel, A., Muqbil, I., Philip, P. A., & Mohammad, R. M. (2019). DNA-Methylation-Caused Downregulation of miR-30 Contributes to the High Expression of XPO1 and the Aggressive Growth of Tumors in Pancreatic Ductal Adenocarcinoma. Cancers, 11(8), 1101. https://doi.org/10.3390/cancers11081101