TP53 DNA Binding Domain Mutations Predict Progression-Free Survival of Bevacizumab Therapy in Metastatic Colorectal Cancer

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Patients
2.2. Genetic Alterations in Oncogenic Pathways and Progression-Free Survival in Bevacizumab-Treated Patients
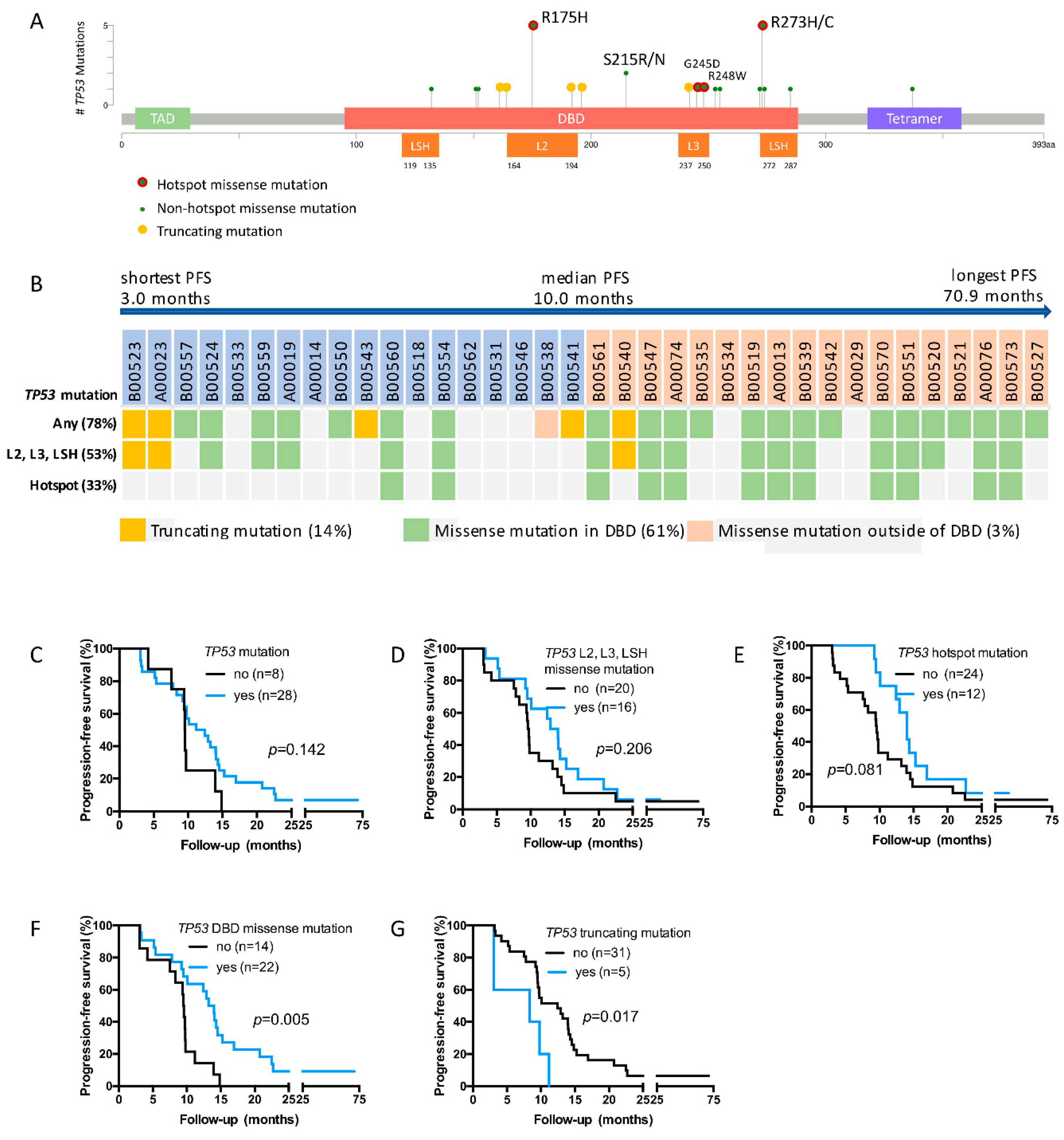
2.3. TP53 Mutations Detected in the Study Cohort
2.4. TP53 DBD Missense Mutations Are Associated with Prolonged PFS, Whereas Truncating Mutations Are Associated with Short PFS
2.5. TP53 DBD Missense Mutations Are an Independent Predictor of PFS
3. Discussion
4. Methods
4.1. Patients, Treatment and Next-Generation Sequencing
4.2. Variant Classification
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer WHO. GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. World Fact Sheet. 2012 (Last Update 2012). Available online: http://globocan.iarc.fr/Pages/fact_sheets_ population.aspx (accessed on 8 September 2016).
- National Cancer Institute. SEER Cancer Statistics Factsheets: Colon and Rectum Cancer. 2016 (Last Update 2016). Available online: http://seer.cancer.gov/ statfacts/ html/ colorect.html (accessed on 23 August 2016).
- Bockelman, C.; Engelmann, B.E.; Kaprio, T.; Hansen, T.F.; Glimelius, B. Risk of recurrence in patients with colon cancer stage II and III: A systematic review and meta-analysis of recent literature. Acta Oncol. 2015, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Molecular and biological properties of vascular endothelial growth factor. J. Mol. Med. 1999, 77, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.C.; Wilson, K.; Gebski, V.J.; Cummins, M.M.; Zannino, D.; Van Hazel, G.A.; Robinson, B.; Broad, A.; Ganju, V.; Ackland, S.P.; et al. Capecitabine, bevacizumab, and mitomycin in first-line treatment of metastatic colorectal cancer: Results of the Australasian Gastrointestinal Trials Group Randomized Phase III MAX Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3191–3198. [Google Scholar] [CrossRef]
- Kabbinavar, F.; Irl, C.; Zurlo, A.; Hurwitz, H. Bevacizumab improves the overall and progression-free survival of patients with metastatic colorectal cancer treated with 5-fluorouracil-based regimens irrespective of baseline risk. Oncology 2008, 75, 215–223. [Google Scholar] [CrossRef]
- Cunningham, D.; Lang, I.; Marcuello, E.; Lorusso, V.; Ocvirk, J.; Shin, D.B.; Jonker, D.; Osborne, S.; Andre, N.; Waterkamp, D.; et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): An open-label, randomised phase 3 trial. Lancet Oncol. 2013, 14, 1077–1085. [Google Scholar] [CrossRef]
- Smeets, D.; Miller, I.S.; O’Connor, D.P.; Das, S.; Moran, B.; Boeckx, B.; Gaiser, T.; Betge, J.; Barat, A.; Klinger, R.; et al. Copy number load predicts outcome of metastatic colorectal cancer patients receiving bevacizumab combination therapy. Nat. Commun. 2018, 9, 4112. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Yi, J.; Ince, W.; Novotny, W.F.; Rosen, O. The clinical benefit of bevacizumab in metastatic colorectal cancer is independent of K-ras mutation status: Analysis of a phase III study of bevacizumab with chemotherapy in previously untreated metastatic colorectal cancer. Oncologist 2009, 14, 22–28. [Google Scholar] [CrossRef]
- Ince, W.L.; Jubb, A.M.; Holden, S.N.; Holmgren, E.B.; Tobin, P.; Sridhar, M.; Hurwitz, H.I.; Kabbinavar, F.; Novotny, W.F.; Hillan, K.J.; et al. Association of k-ras, b-raf, and p53 status with the treatment effect of bevacizumab. J. Natl. Cancer Inst. 2005, 97, 981–989. [Google Scholar] [CrossRef]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum and taxanebased standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef]
- Hsu, H.C.; Lapke, N.; Chen, S.J.; Lu, Y.-J.; Jhou, R.-S.; Yeh, C.-Y.; Tsai, W.-S.; Hung, H.-Y.; Hsieh, J.C.-H.; Yang, T.-S.; et al. PTPRT and PTPRD Deleterious Mutations and Deletion Predict Bevacizumab Resistance in Metastatic Colorectal Cancer Patients. Cancers 2018, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Hedrick, E.E.; Mass, R.D.; Sarkar, S.; Suzuki, S.; Ramanathan, R.K.; Hurwitz, H.I.; Goldberg, R.M.; Sargent, D.J. Response-independent survival benefit in metastatic colorectal cancer: A comparative analysis of N9741 and AVF2107. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Marshall, J.; Mitchell, E.; Wierzbicki, R.; Ganju, V.; Jeffery, M.; Schulz, J.; Richards, D.; Soufi-Mahjoubi, R.; Wang, B.; et al. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: Results from the BICC-C Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 4779–4786. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.C.; Shi, Y.; Wang, Y.R.; Lv, Y.; Yan, H.; Mao, H.; Wang, Z.-K.; Wu, Z.-Y.; Shi, W.-W.; Dai, G.-H. KRAS mutation and primary tumor location do not affect efficacy of bevacizumab-containing chemotherapy in stagae IV colorectal cancer patients. Sci. Rep. 2017, 7, 14368. [Google Scholar] [CrossRef] [PubMed]
- Said, R.; Hong, D.S.; Warneke, C.L.; Lee, J.J.; Wheler, J.J.; Janku, F.; Naing, A.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; et al. P53 mutations in advanced cancers: Clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget 2013, 4, 705–714. [Google Scholar] [CrossRef]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. TP53 Alterations Correlate with Response to VEGF/VEGFR Inhibitors: Implications for Targeted Therapeutics. Mol. Cancer Ther. 2016, 15, 2475–2485. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Lopez, I.; Oliveira, L.P.; Tucci, P.; Alvarez-Valin, F.; Coudry, R.A.; Marin, M. Different mutation profiles associated to P53 accumulation in colorectal cancer. Gene 2012, 499, 81–87. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136e3. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Curtin, K.; Ma, K.N.; Edwards, S.; Schaffer, D.; Leppert, M.F.; Slattery, M.L. Prognostic significance of p53 mutations in colon cancer at the population level. Int. J. Cancer 2002, 99, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Vegran, F.; Rebucci, M.; Chevrier, S.; Cadouot, M.; Boidot, R.; Lizard-Nacol, S. Only missense mutations affecting the DNA binding domain of p53 influence outcomes in patients with breast carcinoma. PLoS ONE 2013, 8, e55103. [Google Scholar] [CrossRef] [PubMed]
- Lapke, N.; Lu, Y.J.; Liao, C.T.; Lee, L.-Y.; Lin, C.-Y.; Wang, H.-M.; Ng, S.-H.; Chen, S.-J.; Yen, T.-C. Missense mutations in the TP53 DNA-binding domain predict outcomes in patients with advanced oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 44194–44210. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Kishore, R.; Dolan, C.M.; Silver, M.; Wecker, A.; Luedemann, C.N.; Thorne, T.; Hanley, A.; Curry, C.; Heyd, L.; et al. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc. Natl. Acad. Sci. USA 2006, 103, 11015–11020. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Datta, K.; Mukhopadhyay, D. Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer Res. 2001, 61, 6952–6957. [Google Scholar] [PubMed]
- Schwaederle, M.; Lazar, V.; Validire, P.; Hansson, J.; Lacroix, L.; Soria, J.-C.; Pawitan, Y.; Kurzrock, R. VEGF-A Expression Correlates with TP53 Mutations in Non-Small Cell Lung Cancer: Implications for Antiangiogenesis Therapy. Cancer Res. 2015, 75, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xiao, Z.; Hong, Z.; Jiao, H.; Zhu, S.; Zhao, Y.; Bi, J.; Qiu, J.; Zhang, D.; Yan, J.; et al. FOXF1 promotes angiogenesis and accelerates bevacizumab resistance in colorectal cancer by transcriptionally activating VEGFA. Cancer Lett. 2018, 439, 78–90. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Aschauer, L.; Muller, P.A. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem. Soc. Trans. 2016, 44, 460–466. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Feng, Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009, 16, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Vacarezza, N.; Alonso, I.; Arroyo, G.; Martínez, J.; De Andrés, F.; Llerena, A.; Estévez-Carrizo, F. Predictive biomarkers candidates for patients with metastatic colorectal cancer treated with bevacizumab-containing regimen. Drug Metab. Personal. Ther. 2016, 31, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.S.; Atreya, C.E.; Niedzwiecki, D.; Weinberg, V.K.; Donner, D.B.; Mayer, R.J.; Goldberg, R.M.; Compton, C.C.; Zuraek, M.B.; Ye, C.; et al. Association of TP53 mutational status and gender with survival after adjuvant treatment for stage III colon cancer: Results of CALGB 89803. Clin. Cancer Res. 2013, 19, 5777–5787. [Google Scholar] [CrossRef]
- Xu, J.; Wang, J.; Hu, Y.; Qian, J.; Xu, B.; Chen, H.; Zou, W.; Fang, J.-Y. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014, 5, e1108. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genetic Alterations | n (%) | Median PFS (Months) | HR (95% CI) | p Value | |
|---|---|---|---|---|---|
| All patients | 36 (100) | 10.0 | |||
| Gene | |||||
| TP53 | no | 7 (19) | 9.5 | 1.00 | 0.188 |
| yes | 29 (81) | 11.2 | 0.58 (0.19–1.37) | ||
| KRAS | no | 11 (31) | 11.2 | 1.00 | 0.863 |
| yes | 25 (69) | 9.8 | 1.06 (0.52–2.17) | ||
| APC | no | 14 (39) | 9.7 | 1.00 | 0.645 |
| yes | 22 (61) | 10.7 | 0.85 (0.42–1.70) | ||
| SMAD4 | no | 16 (44) | 9.8 | 1.00 | 0.728 |
| yes | 20 (56) | 11.1 | 0.89 (0.45–1.74) | ||
| SMAD2 | no | 20 (56) | 9.9 | 1.00 | 0.375 |
| yes | 16 (44) | 11.1 | 0.74 (0.38–1.44) | ||
| NF2 | no | 28 (78) | 10.0 | 1.00 | 0.502 |
| yes | 8 (22) | 11.9 | 1.30 (0.57–3.17) | ||
| KMT2C | no | 29 (81) | 10.1 | 1.00 | 0.125 |
| yes | 7 (19) | 9.8 | 0.51 (0.24–1.13) | ||
| ERBB2 | no | 30 (83) | 9.8 | 1.00 | 0.915 |
| yes | 6 (17) | 11.8 | 1.05 (0.43–2.57) | ||
| FGFR1 | no | 31 (86) | 9.8 | 1.00 | 0.986 |
| yes | 5 (14) | 10.1 | 0.99 (0.38–2.56) | ||
| TRRAP | no | 31 (86) | 9.8 | 1.00 | 0.571 |
| yes | 5 (14) | 11.2 | 0.74 (0.30–1.94) | ||
| CDKN2A | no | 31 (86) | 12.4 | 1.00 | 0.017 |
| yes | 5 (14) | 9.2 | 2.93 (1.44–27.10) | ||
| Pathway | |||||
| p53 | no | 7 (19) | 9.5 | 1.00 | 0.188 |
| yes | 29 (81) | 11.2 | 0.58 (0.19–1.34) | ||
| MAPK/ERK | no | 10 (28) | 12.1 | 1.00 | 0.642 |
| yes | 26 (72) | 9.8 | 1.19 (0.58–2.43) | ||
| TGFβ | no | 13 (36) | 10.1 | 1.00 | 0.947 |
| yes | 23 (64) | 9.8 | 0.98 (0.49–1.95) | ||
| Wnt | no | 13 (36) | 9.8 | 1.00 | 0.815 |
| yes | 23 (64) | 10.1 | 0.92 (0.46–1.85) | ||
| Receptor tyrosine kinases | no | 15 (42) | 9.8 | 1.00 | 0.594 |
| yes | 21 (58) | 10.1 | 0.83 (0.42–1.65) | ||
| PI3K/AKT/mTOR | no | 17 (47) | 10.1 | 1.00 | 0.981 |
| yes | 19 (53) | 9.8 | 1.01 (0.51–1.98) | ||
| Chromatin remodeling | no | 23 (64) | 12.4 | 1.00 | 0.771 |
| yes | 13 (36) | 9.5 | 0.90 (0.44–1.82) | ||
| Cell cycle | no | 28 (78) | 13.1 | 1.00 | 0.017 |
| yes | 8 (22) | 9.6 | 2.44 (1.35–11.40) | ||
| n (%) | Median PFS (Months) | HR (95% CI) | p Value | Responders n (%) | p Value | Median Tumor Change from Baseline | p Value | |
|---|---|---|---|---|---|---|---|---|
| All patients | 36 (100) | 10.0 | 18 (50) | −25% | ||||
| TP53 mutation | ||||||||
| no | 8 (22) | 9.5 | 1.00 | 0.142 | 4 (50) | 1.000 | −25% | 0.759 |
| yes | 28 (78) | 11.8 | 0.57 (0.19–1.24) | 14 (50) | −25% | |||
| TP53 L2, L3, LSH missense mutation | ||||||||
| no | 20 (56) | 9.6 | 1 | 0.206 | 9 (45) | 0.738 | −17% | 0.260 |
| yes | 16 (44) | 13.5 | 0.65 (0.33–1.27) | 9 (56) | −32% | |||
| TP53 hotspot missense mutation | ||||||||
| no | 24 (67) | 9.5 | 1 | 0.081 | 10 (42) | 0.289 | −15% | 0.029 |
| yes | 12 (33) | 14.0 | 0.54 (0.28–1.07) | 8 (67) | −35% | |||
| TP53 DBD missense mutation | ||||||||
| no | 14 (39) | 9.5 | 1.00 | 0.005 | 6 (43) | 0.733 | −17% | 0.482 |
| yes | 22 (61) | 13.6 | 0.41 (0.13–0.65) | 12 (55) | −30% | |||
| TP53 truncating mutation | ||||||||
| no | 31 (86) | 12.4 | 1.00 | 0.017 | 16 (52) | 1.000 | −30% | 0.761 |
| yes | 5 (14) | 8.3 | 2.95 (1.45–27.50) | 2 (40) | −16% | |||
| Factors | n | HR (95% CI) | p Value |
|---|---|---|---|
| Clinical | |||
| Primary tumor site (rectum/colon) | 15/21 | 2.36 (1.05–5.31) | 0.037 |
| Genetic | |||
| TP53 truncating mutations (yes/no) | 5/31 | 1.26 (0.35–4.55) | 0.725 |
| TP53 DBD missense mutations (yes/no) | 22/14 | 0.31 (0.13–0.77) | 0.011 |
| PTPRT/PTPRD deleterious alteration (yes/no) | 10/26 | 3.87 (1.66–9.00) | 0.002 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, H.-C.; You, J.-F.; Chen, S.-J.; Chen, H.-C.; Yeh, C.-Y.; Tsai, W.-S.; Hung, H.-Y.; Yang, T.-S.; Lapke, N.; Tan, K.T. TP53 DNA Binding Domain Mutations Predict Progression-Free Survival of Bevacizumab Therapy in Metastatic Colorectal Cancer. Cancers 2019, 11, 1079. https://doi.org/10.3390/cancers11081079
Hsu H-C, You J-F, Chen S-J, Chen H-C, Yeh C-Y, Tsai W-S, Hung H-Y, Yang T-S, Lapke N, Tan KT. TP53 DNA Binding Domain Mutations Predict Progression-Free Survival of Bevacizumab Therapy in Metastatic Colorectal Cancer. Cancers. 2019; 11(8):1079. https://doi.org/10.3390/cancers11081079
Chicago/Turabian StyleHsu, Hung-Chih, Jeng-Fu You, Shu-Jen Chen, Hua-Chien Chen, Chien-Yuh Yeh, Wen-Sy Tsai, Hsin-Yuan Hung, Tsai-Sheng Yang, Nina Lapke, and Kien Thiam Tan. 2019. "TP53 DNA Binding Domain Mutations Predict Progression-Free Survival of Bevacizumab Therapy in Metastatic Colorectal Cancer" Cancers 11, no. 8: 1079. https://doi.org/10.3390/cancers11081079
APA StyleHsu, H.-C., You, J.-F., Chen, S.-J., Chen, H.-C., Yeh, C.-Y., Tsai, W.-S., Hung, H.-Y., Yang, T.-S., Lapke, N., & Tan, K. T. (2019). TP53 DNA Binding Domain Mutations Predict Progression-Free Survival of Bevacizumab Therapy in Metastatic Colorectal Cancer. Cancers, 11(8), 1079. https://doi.org/10.3390/cancers11081079