GSK3-β Stimulates Claspin Degradation via β-TrCP Ubiquitin Ligase and Alters Cancer Cell Survival



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
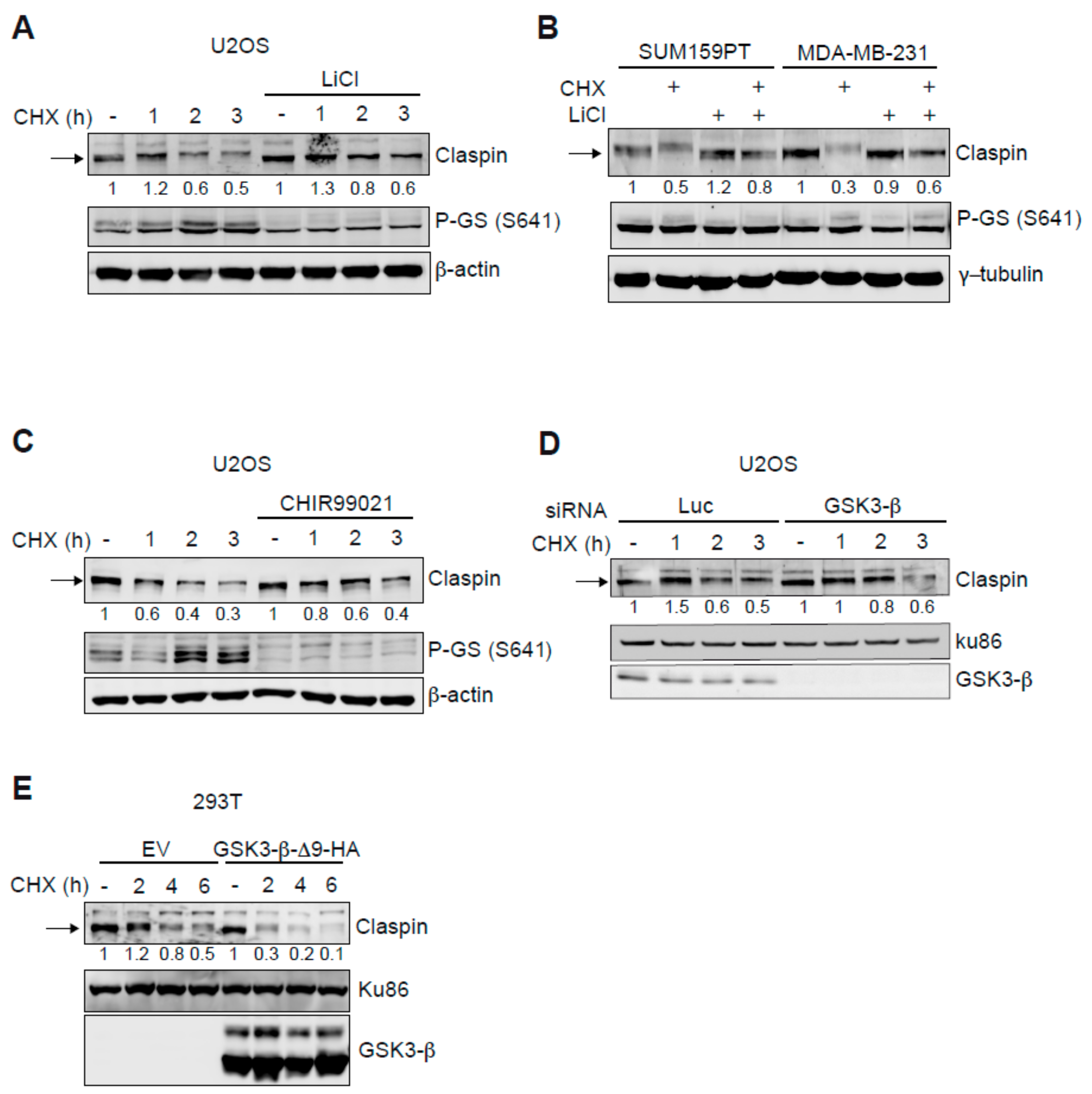
2.1. GSK3-β Modulates Claspin Protein Stability
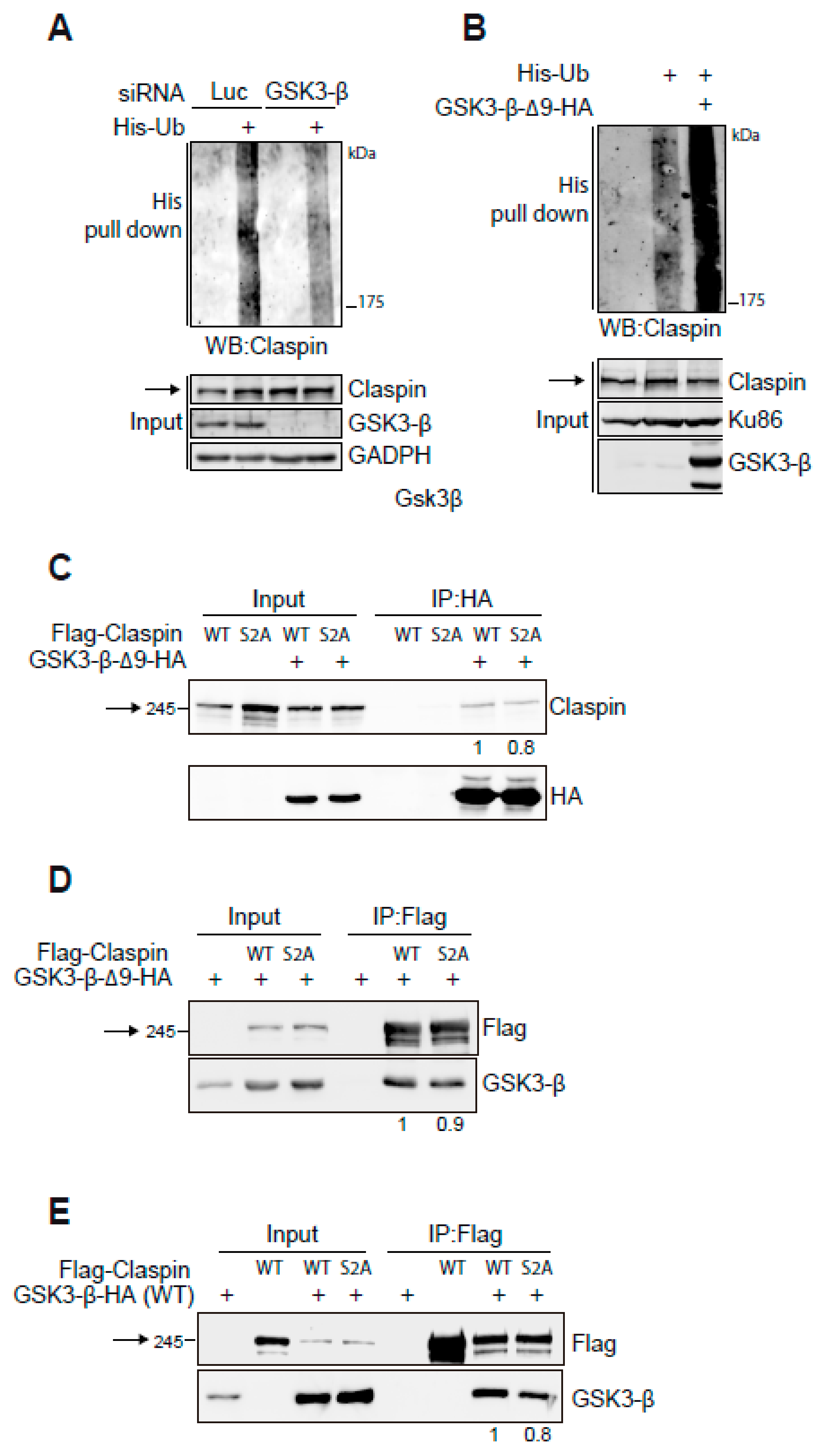
2.2. GSK3-β Interacts with Claspin and Regulates Claspin Ubiquitination
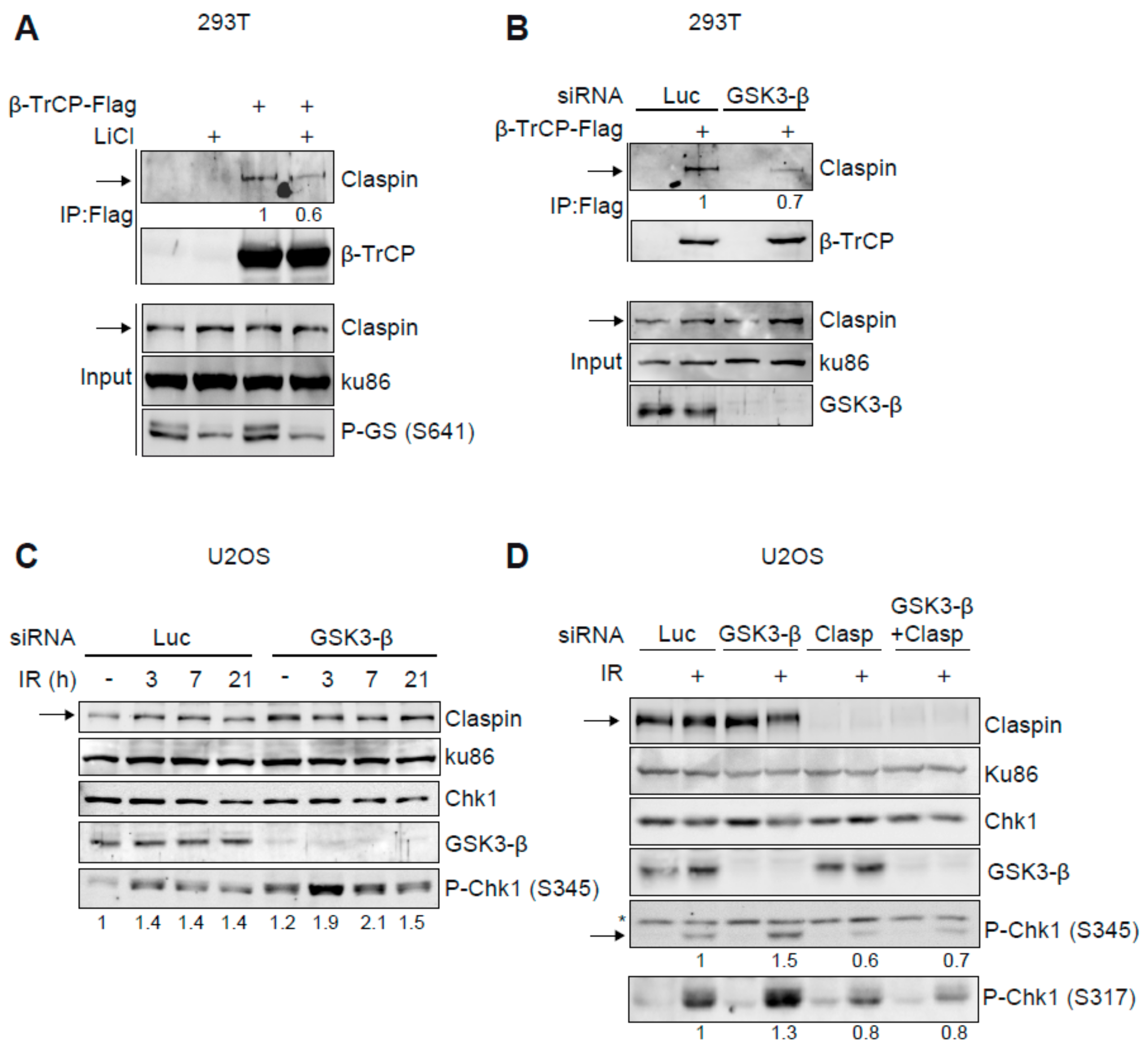
2.3. Claspin-β-TrCP Interaction Is GSK3-β Dependent
2.4. GSK3-β Regulates Chk1 Activation
2.5. Combined Inhibition of GSK3-β and Chk1 Sensitizes Triple Negative Breast Cancer Cell Lines
2.6. Combination Treatment with GSK3-β and Chk1 Sensitizes Breast Cancer Cell Lines in 3D Cultures
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Antibodies, Reagents and Plasmids
4.2. Transfections
| Luciferase | 5′-UCGAAGUAUUCCGCGUACGdTdT-3′ |
| GSK3-β | 5′-GCUAGAUCACUGUAACAUAdTdT-3′ |
4.3. Immunoprecipitations and In Vivo Ubiquitin Assays
4.4. Irradiation
4.5. Cell Proliferation Assays
4.6. Colony Formation Assays
4.7. Apoptotic Assays
4.8. Tumour Spheroid Assays
4.9. Western Blot Quantifications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smits, V.A.J.; Warmerdam, D.O.; Martin, Y.; Freire, R. Mechanisms of ATR-mediated checkpoint signalling. Front. Biosci. 2010, 15, 840–853. [Google Scholar] [CrossRef]
- Smits, V.A.J.; Gillespie, D.A. DNA damage control: Regulation and functions of checkpoint kinase 1. FEBS J. 2015, 282, 3681–3692. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Dunphy, W.G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 2000, 6, 839–849. [Google Scholar] [CrossRef]
- Mamely, I.; van Vugt, M.A.; Smits, V.A.J.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Medema, R.H.; Freire, R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Bartek, J.; Lukas, J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol. Cell 2006, 23, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Dorrello, N.V.; Guardavaccaro, D.; Venere, M.; Halazonetis, T.; Sherman, N.E.; Pagano, M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol. Cell 2006, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T.; Caldecott, K.W. Claspin promotes normal replication fork rates in human cells. Mol. Biol. Cell 2008, 19, 2373–2378. [Google Scholar] [CrossRef]
- Smits, V.A.J.; Cabrera, E.; Freire, R.; Gillespie, D.A. Claspin-checkpoint adaptor and DNA replication factor. FEBS J. 2018, 6, 839. [Google Scholar] [CrossRef] [PubMed]
- Bassermann, F.; Frescas, D.; Guardavaccaro, D.; Busino, L.; Peschiaroli, A.; Pagano, M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008, 134, 256–267. [Google Scholar] [CrossRef]
- Faustrup, H.; Bekker-Jensen, S.; Bartek, J.; Lukas, J.; Mailand, N. USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. J. Cell Biol. 2009, 184, 13–19. [Google Scholar] [CrossRef]
- Martín, Y.; Cabrera, E.; Amoedo, H.; Hernández-Pérez, S.; Domínguez-Kelly, R.; Freire, R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene 2015, 34, 1058. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhao, H.; Liao, J.; Xu, X. HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability. Nucleic Acids Res. 2014, 42, 13074–13081. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Deng, M.; Li, Y.; Yin, P.; Gao, B.; Fang, Y.; Wu, P.; Liu, T.; Lou, Z. HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Res. 2014, 42, 13110–13121. [Google Scholar] [CrossRef] [PubMed]
- McGarry, E.; Gaboriau, D.; Rainey, M.; Restuccia, U.; Bachi, A.; Santocanale, C. The deubiquitinase USP9X maintains DNA replication fork stability and DNA damage checkpoint responses by regulating CLASPIN during S-phase. Cancer Res. 2016, 76, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 2014, 40, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Fiol, C.; Munemitsu, S.; Polakis, P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996, 272, 1023–1026. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Walz, A.; Ugolkov, A.; Chandra, S.; Kozikowski, A.; Carneiro, B.A.; O’Halloran, T.V.; Giles, F.J.; Billadeau, D.D.; Mazar, A.P. Molecular Pathways: Revisiting Glycogen Synthase Kinase-3β as a Target for the Treatment of Cancer. Clin. Cancer Res. 2017, 23, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ugolkov, A.; Gaisina, I.; Zhang, J.-S.; Billadeau, D.D.; White, K.; Kozikowski, A.; Jain, S.; Cristofanilli, M.; Giles, F.; O’Halloran, T.; et al. GSK-3 inhibition overcomes chemoresistance in human breast cancer. Cancer Lett. 2016, 380, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Gallmeier, E.; Hermann, P.C.; Mueller, M.-T.; Machado, J.G.; Ziesch, A.; De Toni, E.N.; Palagyi, A.; Eisen, C.; Ellwart, J.W.; Rivera, J.; et al. Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 2011, 29, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, V.A.; Parsels, L.A.; Parsels, J.D.; Zhao, L.; Zabludoff, S.D.; Simeone, D.M.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia 2012, 14, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1668. [Google Scholar] [CrossRef]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef]
- Murai, H.; Okazaki, M.; Kikuchi, A. Tyrosine dephosphorylation of glycogen synthase kinase-3 is involved in its extracellular signal-dependent inactivation. FEBS Lett. 1996, 392, 153–160. [Google Scholar] [CrossRef]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 2011. [Google Scholar] [CrossRef]
- Freire, R.; van Vugt, M.A.T.M.; Mamely, I.; Medema, R.H. Claspin: Timing the cell cycle arrest when the genome is damaged. Cell Cycle 2006, 5, 2831–2834. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Kim, C.-S.; Lee, J.-H.; Jang, S.J.; Hwang, J.J.; Ro, S.; Choi, J. CG0009, a novel glycogen synthase kinase 3 inhibitor, induces cell death through cyclin D1 depletion in breast cancer cells. PLoS ONE 2013, 8, e60383. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wolgamott, L.; Tcherkezian, J.; Vallabhapurapu, S.; Yu, Y.; Roux, P.P.; Yoon, S.-O. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 2014, 33, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Hinoi, T.; Yamamoto, H.; Kishida, M.; Takada, S.; Kishida, S.; Kikuchi, A. Complex formation of adenomatous polyposis coli gene product and axin facilitates glycogen synthase kinase-3 beta-dependent phosphorylation of beta-catenin and down-regulates beta-catenin. J. Biol. Chem. 2000, 275, 34399–34406. [Google Scholar] [CrossRef]
- Zhou, Z.-R.; Yang, Z.-Z.; Wang, S.-J.; Zhang, L.; Luo, J.-R.; Feng, Y.; Yu, X.-L.; Chen, X.-X.; Guo, X.-M. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacol. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef]
- Grellety, T.; Callens, C.; Richard, E.; Briaux, A.; Vélasco, V.; Pulido, M.; Gonçalves, A.; Gestraud, P.; MacGrogan, G.; Bonnefoi, H.; et al. Enhancing Abiraterone Acetate Efficacy in Androgen Receptor-positive Triple-negative Breast Cancer: Chk1 as a Potential Target. Clin. Cancer Res. 2019, 25, 856–867. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar]
- Semple, J.I.; Smits, V.A.J.; Fernaud, J.-R.; Mamely, I.; Freire, R. Cleavage and degradation of Claspin during apoptosis by caspases and the proteasome. Cell Death Differ. 2007, 14, 1433–1442. [Google Scholar] [CrossRef][Green Version]
- Pérez-Castro, A.J.; Freire, R. Rad9B responds to nucleolar stress through ATR and JNK signalling, and delays the G1-S transition. J. Cell Sci. 2012, 125, 1152–1164. [Google Scholar] [CrossRef]
- Kalimutho, M.; Sinha, D.; Jeffery, J.; Nones, K.; Srihari, S.; Fernando, W.C.; Duijf, P.H.; Vennin, C.; Raninga, P.; Nanayakkara, D.; et al. CEP55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol. Med. 2018, 10, e8566. [Google Scholar] [CrossRef]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance in multiple myeloma through NF-кβ inhibition. Cell Cycle 2016, 15, 559–572. [Google Scholar] [CrossRef]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera, E.; Raninga, P.; Khanna, K.K.; Freire, R. GSK3-β Stimulates Claspin Degradation via β-TrCP Ubiquitin Ligase and Alters Cancer Cell Survival. Cancers 2019, 11, 1073. https://doi.org/10.3390/cancers11081073
Cabrera E, Raninga P, Khanna KK, Freire R. GSK3-β Stimulates Claspin Degradation via β-TrCP Ubiquitin Ligase and Alters Cancer Cell Survival. Cancers. 2019; 11(8):1073. https://doi.org/10.3390/cancers11081073
Chicago/Turabian StyleCabrera, Elisa, Prahlad Raninga, Kum Kum Khanna, and Raimundo Freire. 2019. "GSK3-β Stimulates Claspin Degradation via β-TrCP Ubiquitin Ligase and Alters Cancer Cell Survival" Cancers 11, no. 8: 1073. https://doi.org/10.3390/cancers11081073
APA StyleCabrera, E., Raninga, P., Khanna, K. K., & Freire, R. (2019). GSK3-β Stimulates Claspin Degradation via β-TrCP Ubiquitin Ligase and Alters Cancer Cell Survival. Cancers, 11(8), 1073. https://doi.org/10.3390/cancers11081073