Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology


Abstract
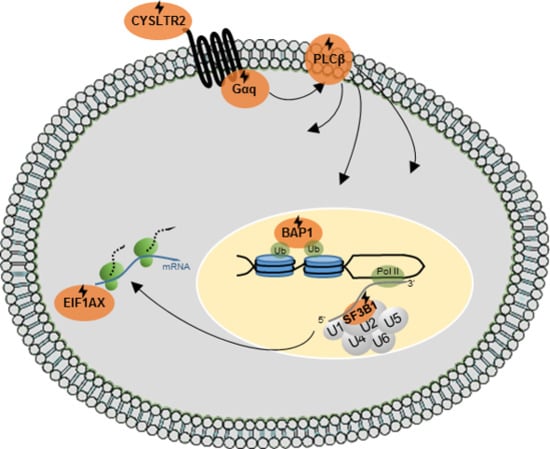
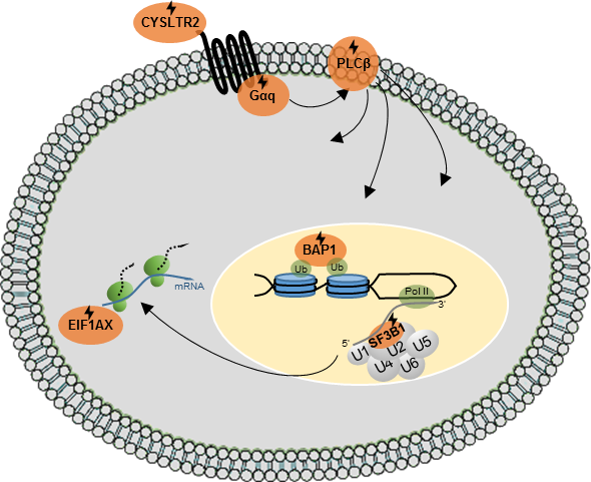{kind=link}
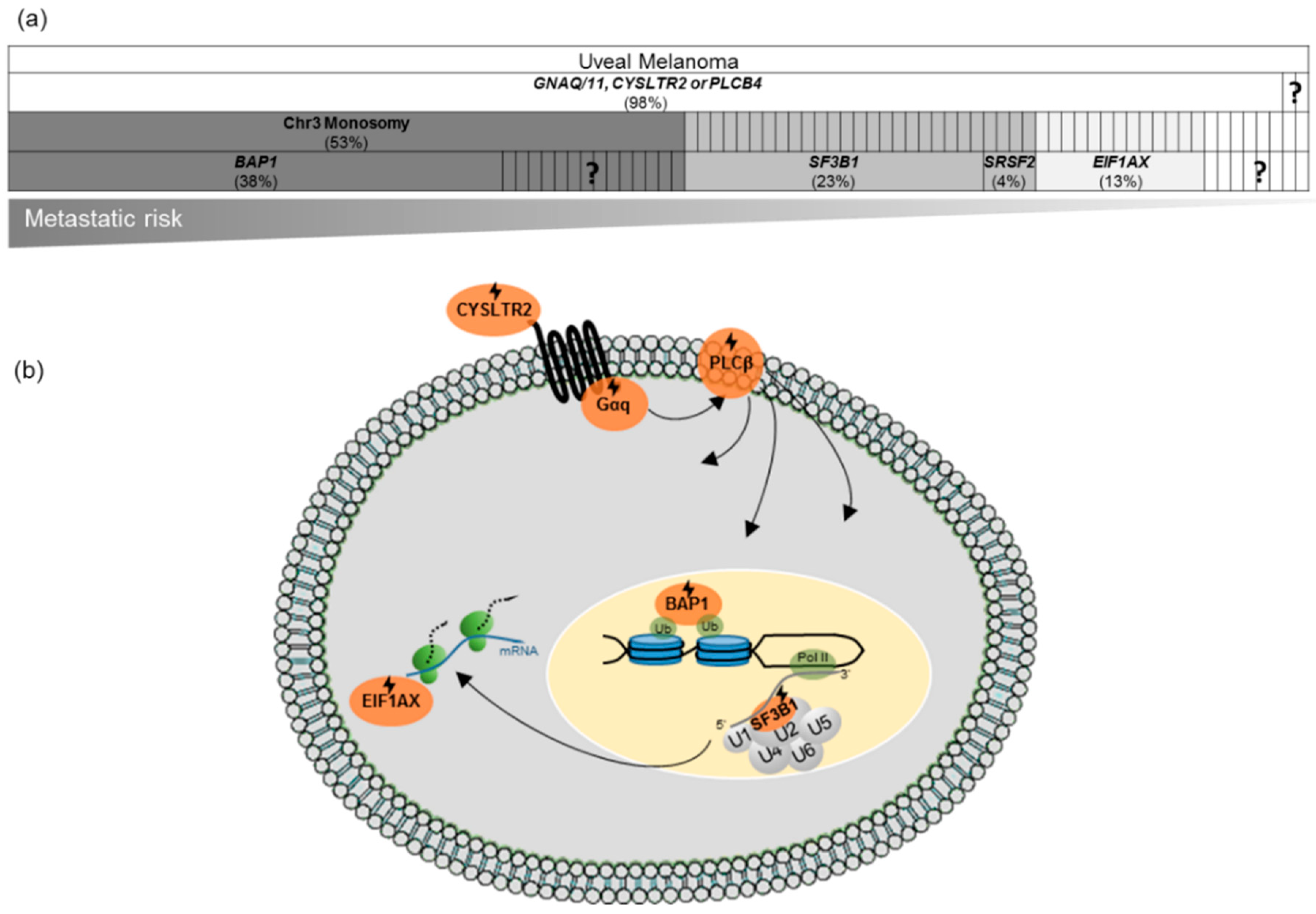{kind=link}
{kind=link}
1. Introduction
2. Uveal Melanoma Risk and Prognostic Factors
3. Biology-Based Therapeutic Strategies in Uveal Melanoma
3.1. Dysregulated Signaling Pathways
3.1.1. Apoptosis and Cell Cycle
3.1.2. Hypoxia-Induced Response
3.1.3. cMET-PI3K Pathway
3.1.4. NF-κB Proinflammatory Signaling
3.2. Genomic Aberrations and Mutational Burden
3.3. Mutational Landscape and Related Therapeutic Perspectives
3.3.1. Gαq-Pathway Activating Mutations
Gαq-Corresponding Therapeutic Strategies
3.3.2. BAP1, SF3B1, SRSF2 or EIF1AX Mutations
BAP1
EIF1AX
SF3B1 and SRSF2
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mahendraraj, K.; Lau, C.S.M.; Lee, I.; Chamberlain, R.S. Trends in Incidence, Survival, and Management of Uveal Melanoma: A Population-Based Study of 7516 Patients from the Surveillance, Epidemiology, and End Results Database (1973–2012). Clin. Ophthalmol. 2016, 10, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal Melanoma: Trends in Incidence, Treatment, and Survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Pandiani, C.; Béranger, G.E.; Leclerc, J.; Ballotti, R.; Bertolotto, C. Focus on Cutaneous and Uveal Melanoma Specificities. Genes Dev. 2017, 31, 724–743. [Google Scholar] [CrossRef] [PubMed]
- Mathis, T.; Cassoux, N.; Tardy, M.; Piperno, S.; Gastaud, L.; Dendale, R.; Maschi, C.; Nguyen, A.; Meyer, L.; Bonnin, N.; et al. Management of Uveal Melanomas, Guidelines for Oncologists. Bull. Cancer 2018, 105, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of Uveal Melanoma: Where Are We Now? Ther. Adv. Med. Oncol. 2018, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.A.; Salama, A.K. New NCCN Guidelines for Uveal Melanoma and Treatment of Recurrent or Progressive Distant Metastatic Melanoma. JNCCN J. Natl. Compr. Cancer Netw. 2018, 16, 646–650. [Google Scholar] [CrossRef]
- Kujala, E.; Mäkitie, T.; Kivelä, T. Very Long-Term Prognosis of Patients with Malignant Uveal Melanoma. Investig. Ophtalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Schoumacher, M.; Stern, M.-H.; Cassoux, N.; Desjardins, L.; Piperno-Neumann, S.; Lantz, O.; Roman-Roman, S. Upcoming Translational Challenges for Uveal Melanoma. Br. J. Cancer 2015, 113, 1249–1253. [Google Scholar] [CrossRef]
- Patel, M.; Smyth, E.; Chapman, P.B.; Wolchok, J.D.; Schwartz, G.K.; Abramson, D.H.; Carvajal, R.D. Therapeutic Implications of the Emerging Molecular Biology of Uveal Melanoma. Clin. Cancer Res. 2011, 17, 2087–2100. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2018, 32, 204–220. [Google Scholar] [CrossRef]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 Mutations Are Associated with Alternative Splicing in Uveal Melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Royer-bertrand, B.; Torsello, M.; Rimoldi, D.; Zaoui, I.E.; Cisarova, K.; Pescini-gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Klaus, G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I. Germline Mutations in BAP1 Predispose to Melanocytic Tumors. Nat. Genet. 2012, 43, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Ewens, K.G.; Lalonde, E.; Shields, C.L.; Ganguly, A. Comparison of Germline versus Somatic BAP1 Mutations for Risk of Metastasis in Uveal Melanoma. BMC Cancer 2018, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Njauw, C.N.J.; Kim, I.; Piris, A.; Gabree, M.; Taylor, M.; Lane, A.M.; DeAngelis, M.M.; Gragoudas, E.; Duncan, L.M.; Tsao, H. Germline BAP1 Inactivation Is Preferentially Associated with Metastatic Ocular Melanoma and Cutaneous-Ocular Melanoma Families. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.H.; Pilarski, R.; Cebulla, C.M.; Massengill, J.B.; Christopher, B.N.; Boru, G.; Hovland, P.; Davidorf, F.H. Germline BAP1 Mutation Predisposes to Uveal Melanoma, Lung Adenocarcinoma, Meningioma, and Other Cancers. J. Med. Genet. 2011, 48. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Fiévet, A.; Gardrat, S.; Barnhill, R.L.; Popova, T.; Servois, V.; Rampanou, A.; Mouton, A.; et al. Outlier Response to Anti-PD1 in Uveal Melanoma Reveals Germline MBD4 Mutations in Hypermutated Tumors. Nat. Commun. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Johansson, P.A.; Stark, A.; Palmer, J.M.; Bigby, K.; Brooks, K.; Rolfe, O.; Pritchard, A.L.; Whitehead, K.; Warrier, S.; Glasson, W.; et al. Prolonged Stable Disease in a Uveal Melanoma Patient with Germline MBD4 Nonsense Mutation Treated with Pembrolizumab and Ipilimumab. Immunogenetics 2019, 4, 1–4. [Google Scholar] [CrossRef]
- Bellacosa, A. Role of MED1 (MBD4) Gene in DNA Repair and Human Cancer. J. Cell. Physiol. 2001, 187, 137–144. [Google Scholar] [CrossRef]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jöckel, K.; Becher, R. Prognostic Implications of Monosomy 3 in Uveal Melanoma. Lancet 1996, 347, 1222–1225. [Google Scholar] [CrossRef]
- White, V.A.; Chambers, J.D.; Courtright, P.D.; Chang, W.Y.; Horsman, D.E. Correlation of Cytogenetic Abnormalities with the Outcome of Patients with Uveal Melanoma. Cancer 1998, 83, 354–359. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Char, D.H.; Augsburger, J.J.; Correa, Z.M.; Nudleman, E.; Aaberg, T.M., Jr.; Altaweel, M.M.; Bardenstein, D.S.; Finger, P.T.; et al. Collaborative Ocular Oncology Group Report No. 1: Prospective Validation of a Multi-Gene Prognostic Assay in Uveal Melanoma. Ophtalmology 2012, 119, 1596–1603. [Google Scholar] [CrossRef]
- Worley, L.A.; Onken, M.D.; Person, E.; Robirds, D.; Branson, J.; Char, D.H.; Perry, A.; Harbour, J.W. Transcriptomic versus Chromosomal Prognostic Markers and Clinical Outcome in Uveal Melanoma. Clin. Cancer Res. 2007, 13, 1466–1471. [Google Scholar] [CrossRef]
- Bove, R.; Char, D.H. Nondiagnosed Uveal Melanomas. Ophthalmology 2004, 111, 554–557. [Google Scholar] [CrossRef]
- Shields, C.L.; Kaliki, S.; Rojanaporn, D.; Ferenczy, S.R.; Shields, J.A. Enhanced Depth Imaging Optical Coherence Tomography of Small Choroidal Melanoma. Arch. Ophthalmol. 2012, 130, 850–856. [Google Scholar] [CrossRef]
- Coupland, S.E.; Bechrakis, N.; Schüler, A.; Anagnostopoulos, I.; Hummel, M.; Bornfeld, N.; Stein, H. Expression Patterns of Cyclin D1 and Related Proteins Regulating G1-S Phase Transition in Uveal Melanoma and Retinoblastoma. Br. J. Ophthalmol. 1998, 82, 961–970. [Google Scholar] [CrossRef]
- Brantley, M.A.; Harbour, J.W. Deregulation of the Rb and P53 Pathways in Uveal Melanoma. Nat. Cell Biol. 2000, 157, 1795–1801. [Google Scholar] [CrossRef]
- Helgadottir, H.; Höiom, V. The Genetics of Uveal Melanoma: Current Insights. Appl. Clin. Genet. 2016, 9, 147–155. [Google Scholar] [CrossRef]
- Némati, F.; de Montrion, C.; Lang, G.; Kraus-Berthier, L.; Carita, G.; Sastre-Garau, X.; Berniard, A.; Vallerand, D.; Geneste, O.; de Plater, L.; et al. Targeting Bcl Bcl-X L Induces Antitumor Activity in Uveal Melanoma Patient-Derived Xenografts. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Carita, G.; Frisch-Dit-Leitz, E.; Dahmani, A.; Raymondie, C.; Cassoux, N.; Piperno-Neumann, S.; Némati, F.; Laurent, C.; De Koning, L.; Halilovic, E.; et al. Dual Inhibition of Protein Kinase C and P53-MDM2 or PKC and MTORC1 Are Novel Efficient Therapeutic Approaches for Uveal Melanoma. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Heijkants, R.; Willekens, K.; Schoonderwoerd, M.; Teunisse, A.; Nieveen, M.; Radaelli, E.; Hawinkels, L.; Marine, J.-C.; Jochemsen, A. Combined Inhibition of CDK and HDAC as a Promising Therapeutic Strategy for Both Cutaneous and Uveal Metastatic Melanoma. Oncotarget 2018, 9, 6174–6187. [Google Scholar] [CrossRef]
- Marine, J.C.; Jochemsen, A.G. MDMX (MDM4), a Promising Target for P53 Reactivation Therapy and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Brantley, M.A.; Harbour, J.W. Inactivation of Retinoblastoma Protein in Uveal Melanoma by Phosphorylation of Sites in the COOH-Terminal Region. Cancer Res. 2000, 60, 4320–4323. [Google Scholar]
- An, J.; Wan, H.; Zhou, X.; Hu, D.N.; Wang, L.; Hao, L.; Yan, D.; Shi, F.; Zhou, Z.; Wang, J.; et al. A Comparative Transcriptomic Analysis of Uveal Melanoma and Normal Uveal Melanocyte. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Van der Veiden, P.A.; Metzelaar-Blok, J.A.W.; Bergman, W.; Hurks, H.M.H.; Frants, R.R.; Gruis, N.A.; Jager, M.J. Promoter Hypermethylation: A Common Cause of Reduced P16(INK4a) Expression in Uveal Melanoma. Cancer Res. 2001, 61, 5303–5306. [Google Scholar]
- Semenza, G.L. HIF-1: Upstream and Downstream of Cancer Metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Dong, L.; You, S.; Zhang, Q.; Osuka, S.; Devi, N.S.; Kaluz, S.; Ferguson, J.H.; Yang, H.; Chen, G.; Wang, B.; et al. Arylsulfonamide 64B Inhibits Hypoxia/HIF-Induced Expression of c-Met and CXCR4 and Reduces Primary Tumor Growth and Metastasis of Uveal Melanoma. Clin. Cancer Res. 2018, 25, 2206–2219. [Google Scholar] [CrossRef]
- Cheng, H.; Chua, V.; Liao, C.; Purwin, T.J.; Terai, M.; Kageyama, K.; Davies, M.A.; Sato, T.; Aplin, A.E. Co-Targeting HGF-CMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol. Cancer Ther. 2017, 118, 6072–6078. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Eng, C.; Singh, A.D. Targeted Therapy for Uveal Melanoma. Cancer Treat. Rev. 2008, 34, 247–258. [Google Scholar] [CrossRef]
- Simpson, L.; Parsons, R. PTEN: Life as a Tumor Suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.H.; Yang, Y.; Zhou, X.P.; Craig, E.L.; Davidorf, F.H.; Eng, C. High Frequency of Submicroscopic Hemizygous Deletion Is a Major Mechanism of Loss of Expression of PTEN in Uveal Melanoma. J. Clin. Oncol. 2006, 24, 288–295. [Google Scholar] [CrossRef]
- Ling, J.; Lu, P.; Zhang, Y.; Jiang, S.; Zhang, Z. MiR-367 Promotes Uveal Melanoma Cell Proliferation and Migration by Regulating PTEN. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Q.; Gao, X.; Shi, D.; Mi, S.; Han, Q. MicroRNA-454 Functions as an Oncogene by Regulating PTEN in Uveal Melanoma. FEBS Lett. 2015, 589, 2791–2796. [Google Scholar] [CrossRef]
- Ambrosini, G.; Do, C.; Tycko, B.; Realubit, R.B.; Karan, C.; Musi, E.; Carvajal, R.D.; Chua, V.; Aplin, A.E.; Schwartz, G.K. Inhibition of NF-ΚB-Dependent Signaling Enhances Sensitivity and Overcomes Resistance to BET Inhibition in Uveal Melanoma. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Wadelin, F.R.; Fulton, J.; Collins, H.M.; Tertipis, N.; Bottley, A.; Spriggs, K.A.; Falcone, F.H.; Heery, D.M. PRAME Is a Golgi-Targeted Protein That Associates with the Elongin BC Complex and Is Upregulated by Interferon-Gamma and Bacterial PAMPs. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; Van Der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an Independent Biomarker for Metastasis in Uveal Melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef]
- Griffioen, M.; Heemskerk, M.H.M.; van Loenen, M.M.; de Boer, R.; van Kooten, C.; Lugthart, G.-J.; van Veelen, P.A.; van der Steen, D.M.; Falkenburg, J.H.F.; Amir, A.L.; et al. PRAME-Specific Allo-HLA-Restricted T Cells with Potent Antitumor Reactivity Useful for Therapeutic T-Cell Receptor Gene Transfer. Clin. Cancer Res. 2011, 17, 5615–5625. [Google Scholar] [CrossRef]
- Gezgin, G.; Luk, S.J.; Cao, J.; Dogrusöz, M.; Van Der Steen, D.M.; Hagedoorn, R.S.; Krijgsman, D.; Van Der Velden, P.A.; Field, M.G.; Luyten, G.P.M.; et al. PRAME as a Potential Target for Immunotherapy in Metastatic Uveal Melanoma. JAMA Ophthalmol. 2017, 135, 541–549. [Google Scholar] [CrossRef]
- van den Bosch, T.; van Beek, J.G.M.; Vaarwater, J.; Verdijk, R.M.; Naus, N.C.; Paridaens, D.; de Klein, A.; Kiliç, E. Higher Percentage of FISH-Determined Monosomy 3 and 8q Amplification in Uveal Melanoma Cells Relate to Poor Patient Prognosis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2668–2674. [Google Scholar] [CrossRef]
- Cassoux, N.; Rodrigues, M.J.; Plancher, C.; Asselain, B.; Levy-Gabriel, C.; Lumbroso-Le Rouic, L.; Piperno-Neumann, S.; Dendale, R.; Sastre, X.; Desjardins, L.; et al. Genome-Wide Profiling Is a Clinically Relevant and Affordable Prognostic Test in Posterior Uveal Melanoma. Br. J. Ophthalmol. 2014, 98, 769–774. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent Somatic Mutations of GNAQ in Uveal Melanoma and Blue Naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep Sequencing of Uveal Melanoma Identifies a Recurrent Mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Taylor, B.S.; Huber, T.; Chi, P.; et al. Recurrent Activating Mutations of G-Protein-Coupled Receptor CYSLTR2 in Uveal Melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; Van Bodegom, A.; Paridaens, D.; Kiliç, E.; De Klein, A. Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Gao, J.; Arman Aksoy, B.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2014, 6, 1–34. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Chua, V.; Lapadula, D.; Randolph, C.; Benovic, J.L.; Wedegaertner, P.B.; Aplin, A.E. Dysregulated GPCR Signaling and Therapeutic Options in Uveal Melanoma. Mol. Cancer Res. 2017, 15, 501–506. [Google Scholar] [CrossRef]
- Koopmans, A.E.; Vaarwater, J.; Paridaens, D.; Naus, N.C.; Kilic, E.; De Klein, A. Patient Survival in Uveal Melanoma Is Not Affected by Oncogenic Mutations in GNAQ and GNA11. Br. J. Cancer 2013, 109, 493–496. [Google Scholar] [CrossRef]
- Bakalian, S.; Marshall, J.C.; Logan, P.; Faingold, D.; Maloney, S.; Di Cesare, S.; Martins, C.; Fernandes, B.F.; Burnier, M.N. Molecular Pathways Mediating Liver Metastasis in Patients with Uveal Melanoma. Clin. Cancer Res. 2008, 14, 951–956. [Google Scholar] [CrossRef]
- Khalili, J.S.; Yu, X.; Wang, J.; Hayes, B.C.; Davies, M.A.; Lizee, G.; Esmaeli, B.; Woodman, S.E. Combination Small Molecule MEK and PI3K Inhibition Enhances Uveal Melanoma Cell Death in a Mutant GNAQ- and GNA11-Dependent Manner. Clin. Cancer Res. 2012, 18, 4345–4355. [Google Scholar] [CrossRef]
- Huang, J.L.; Urtatiz, O.; Van Raamsdonk, C.D. Oncogenic G Protein GNAQ Induces Uveal Melanoma and Intravasation in Mice. Cancer Res. 2015, 75, 3384–3398. [Google Scholar] [CrossRef]
- Moore, A.R.; Ran, L.; Guan, Y.; Sher, J.J.; Hitchman, T.D.; Zhang, J.Q.; Hwang, C.; Walzak, E.G.; Shoushtari, A.N.; Monette, S.; et al. GNA11 Q209L Mouse Model Reveals RasGRP3 as an Essential Signaling Node in Uveal Melanoma. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef]
- Taniguchi, M.; Suzumura, K.; Nagai, K.; Kawasaki, T.; Takasaki, J.; Sekiguchi, M.; Moritani, Y.; Saito, T.; Hayashi, K.; Fujita, S.; et al. YM-254890 Analogues, Novel Cyclic Depsipeptides with Gαq/11 Inhibitory Activity from Chromobacterium Sp. QS3666. Bioorg. Med. Chem. 2004, 12, 3125–3133. [Google Scholar] [CrossRef]
- Nishimura, A.; Kitano, K.; Takasaki, J.; Taniguchi, M.; Mizuno, N.; Tago, K.; Hakoshima, T.; Itoh, H. Structural Basis for the Specific Inhibition of Heterotrimeric Gq Protein by a Small Molecule. Proc. Natl. Acad. Sci. USA 2010, 107, 13666–13671. [Google Scholar] [CrossRef]
- Takasaki, J.; Saito, T.; Taniguchi, M.; Kawasaki, T.; Moritani, Y.; Hayashi, K.; Kobori, M. A Novel Gαq/11-Selective Inhibitor. J. Biol. Chem. 2004, 279, 47438–47445. [Google Scholar] [CrossRef]
- Schrage, R.; Schmitz, A.-L.; Gaffal, E.; Annala, S.; Kehraus, S.; Wenzel, D.; Büllesbach, K.M.; Bald, T.; Inoue, A.; Shinjo, Y.; et al. The Experimental Power of FR900359 to Study Gq-Regulated Biological Processes. Nature 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Lapadula, D.; Farias, E.; Randolph, C.; Purwin, T.; McGrath, D.; Charpentier, T. Effects of Oncogenic Gαq and Gα11 Inhibition by FR900359 in Uveal Melanoma. Mol. Cancer Res. 2018, 17, 963–973. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of Selumetinib vs Chemotherapy on Progression-Free Survival in Uveal Melanoma: A Randomized Clinical Trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Kapitejin, E.; Larkin, J.M.G.; Carvajal, R.D.; Luke, J.J.; SeifertInge Roozen, H.; Zoubir, M.; Yang, L.; Choudhury, S.; Yerramilli-Rao, P.; et al. Phase I Dose-Escalation Study of the Protein Kinase C (PKC) Inhibitor AEB071 in Patients with Metastatic Uveal Melanoma. J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, M.; Fletcher, J.A.; Giobbie-Hurder, A.; Hodi, F.S. The Protein Kinase C Inhibitor Enzastaurin Exhibits Antitumor Activity against Uveal Melanoma. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Nelson, E.E.; Guyer, A.E. The Phosphoinositide 3-Kinaseα Selective Inhibitor, BYL719, Enhances the Effect of the Protein Kinase C Inhibitor, AEB071, in GNAQ/GNA11 Mutant Uveal Melanoma Cells. Mol. Cancer Ther. 2015, 13, 1044–1053. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Frisch-Dit-Leitz, E.; Carita, G.; Dahmani, A.; Raymondie, C.; Liot, G.; Gentien, D.; Némati, F.; Decaudin, D.; Roman-Roman, S.; et al. The MTOR Inhibitor Everolimus Synergizes with the PI3K Inhibitor GDC0941 to Enhance Anti-Tumor Efficacy in Uveal Melanoma. Oncotarget 2016, 7, 23633–23646. [Google Scholar] [CrossRef][Green Version]
- Heijkants, R.C.; Nieveen, M.; Hart, K.C.T.; Teunisse, A.F.A.S.; Jochemsen, A.G. Targeting MDMX and PKCδ to Improve Current Uveal Melanoma Therapeutic Strategies. Oncogenesis 2018, 7. [Google Scholar] [CrossRef]
- Yoo, J.H.; Shi, D.S.; Grossmann, A.H.; Sorensen, L.K.; Tong, Z.; Mleynek, T.M.; Rogers, A.; Zhu, W.; Richards, J.R.; Winter, J.M.; et al. ARF6 Is an Actionable Node That Orchestrates Oncogenic GNAQ Signaling in Uveal Melanoma. Cancer Cell 2017, 29, 889–904. [Google Scholar] [CrossRef]
- Otsuka, Y.; Oikawa, T.; Yoshino, H.; Hashimoto, S.; Handa, H.; Yamamoto, H.; Hashimoto, A.; Sabe, H. Frequent Overexpression of AMAP1, an Arf6 Effector in Cell Invasion, Is Characteristic of the MMTV-PyMT Rather than the MMTV-Neu Human Breast Cancer Model. Cell Commun. Signal. 2018, 16, 1–9. [Google Scholar] [CrossRef]
- Morishige, M.; Hashimoto, S.; Ogawa, E.; Toda, Y.; Kotani, H.; Hirose, M.; Wei, S.; Hashimoto, A.; Yamada, A.; Yano, H.; et al. GEP100 Links Epidermal Growth Factor Receptor Signalling to Arf6 Activation to Induce Breast Cancer Invasion. Nat. Cell Biol. 2008, 10, 85–92. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Zuidervaart, W.; Pavey, S.; Van Nieuwpoort, F.A.; Packer, L.; Out, C.; Maat, W.; Jager, M.J.; Gruis, N.A.; Hayward, N.K. Expression of Wnt5a and Its Downstream Effector β-Catenin in Uveal Melanoma. Melanoma Res. 2007, 17, 380–386. [Google Scholar] [CrossRef]
- Zheng, L.; Liu, Y.; Pan, J. Inhibitory Effect of Pyrvinium Pamoate on Uveal Melanoma Cells Involves Blocking of Wnt/β-Catenin Pathway. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 890–898. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Feng, X.; Degese, M.S.; Iglesias-bartolome, R.; Vaque, J.P.; Molinolo, A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Chen, Q.; et al. Hippo-Independent Activation of YAP by the GNAQ Uveal Melanoma Oncogene through a Trio-Regulated Rho GTPase Signaling Circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef]
- Feng, X.; Arang, N.; Cosimo Rigiracciolo, D.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; König, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals That the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 2019, 35, 457–472. [Google Scholar] [CrossRef]
- Yu, F.-X.; Luo, J.; Mo, J.-S.; Liu, G.; Chul Kim, Y.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 Promote Uveal Melanoma Tumorigenesis by Activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Liu-chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD–YAP Complex Suppresses the Oncogenic Activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Faião-Flores, F.; Emmons, M.F.; Durante, M.A.; Kinose, F.; Saha, B.; Fang, B.; Koomen, J.M.; Chellappan, S.P.; Maria-Engler, S.S.; Rix, U.; et al. HDAC Inhibition Enhances the in Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Bressler, N.M.; Bressler, S.B. Photodynamic Therapy with Verteporfin (Visudyne): Impact on Ophthalmology and Visual Sciences. IOVS 2000, 41, 624–628. [Google Scholar]
- Martin, M.; Mabhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; et al. Exome Sequencing Identifies Recurrent Somatic Mutations in EIF1AX and SF3B1 in Uveal Melanoma with Disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef]
- Aoude, L.G.; Wadt, K.; Bojesen, A.; Crüger, D.; Borg, A.; Trent, J.M.; Brown, K.M.; Gerdes, A.M.; Jönsson, G.; Hayward, N.K. A BAP1 Mutation in a Danish Family Predisposes to Uveal Melanoma and Other Cancers. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Ismail, I.H.; Davidson, R.; Gagné, J.P.; Xu, Z.Z.; Poirier, G.G.; Hendzel, M.J. Germline Mutations in BAP1 Impair Its Function in DNA Double-Strand Break Repair. Cancer Res. 2014, 74, 4282–4294. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2011, 330, 1410–1413. [Google Scholar] [CrossRef]
- Van De Nes, J.A.P.; Nelles, J.; Kreis, S.; Metz, C.H.D.; Hager, T.; Lohmann, D.R.; Zeschnigk, M. Comparing the Prognostic Value of BAP1 Mutation Immunohistochemistry in Uveal Melanoma. Am. J. Pathol. 2016, 40, 796–805. [Google Scholar] [CrossRef]
- Sahtoe, D.D.; Van Dijk, W.J.; Ekkebus, R.; Ovaa, H.; Sixma, T.K. BAP1/ASXL1 Recruitment and Activation for H2A Deubiquitination. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Scheuerman, J.; Gaytán de Ayala Alonso, A.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S. Histone H2A Deubiquitinase Activity of the Polycomb Repressive Complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Jin, Y.; Jiang, S.; Pan, J. In Vitro and in Vivo Anti-Uveal Melanoma Activity of JSL-1, a Novel HDAC Inhibitor. Cancer Lett. 2017, 400, 47–60. [Google Scholar] [CrossRef]
- Landreville, S.; Agapova, O.A.; Matatall, K.A.; Kneass, Z.T.; Onken, M.D.; Lee, R.S.; Bowcock, A.M.; Harbour, J.W. Histone Deacetylase Inhibitors Induce Growth Arrest and Differentiation in Uveal Melanoma. Clin. Cancer Res. 2012, 18, 408–416. [Google Scholar] [CrossRef]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel Histone Deacetylase Inhibitors in Clinical Trials as Anti-Cancer Agents. J. Hematol. Oncol. 2010, 3, 1–13. [Google Scholar] [CrossRef]
- Lafave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Micol, J.; et al. Loss of BAP1 Function Leads to EZH2-Dependent Transformation. Nat. Med. 2016, 21, 1344–1349. [Google Scholar] [CrossRef]
- Schoumacher, M.; Le Corre, S.; Houy, A.; Mulugeta, E.; Stern, M.H.; Roman-Roman, S.; Margueron, R. Uveal Melanoma Cells Are Resistant to EZH2 Inhibition Regardless of BAP1 Status. Nat. Med. 2016, 22, 577–578. [Google Scholar] [CrossRef]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hebert, J.; Drobetsky, E.; et al. Tumor Suppressor and Deubiquitinase BAP1 Promotes DNA Double-Strand Break Repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef]
- Parrotta, R.; Okonska, A.; Ronner, M.; Weder, W.; Stahel, R.; Penengo, L.; Felley-Bosco, E. A Novel BRCA1-Associated Protein-1 Isoform Affects Response of Mesothelioma Cells to Drugs Impairing BRCA1-Mediated DNA Repair. J. Thorac. Oncol. 2017, 12, 1309–1319. [Google Scholar] [CrossRef]
- de Koning, L.; Decaudin, D.; Botty, R.E.; Nicolas, A.; Carita, G.; Schuller, M.; Naguez, A.; Fleury, J.; Cooke, V.; Wylie, A.; et al. PARP Inhibition Increases the Response to Chemotherapy in Uveal Melanoma. Cancers 2019, 11, 751. [Google Scholar] [CrossRef]
- Pestova, T.V.; Borukhov, S.I.; Hellen, C.U.T. Eukaryotic Ribosomes Require Initiation Factors 1 and 1A to Locate Initiation Codons. Nature 1998, 394, 854–859. [Google Scholar] [CrossRef]
- Ali, M.U.; Ur Rahman, M.S.; Jia, Z.; Jiang, C. Eukaryotic Translation Initiation Factors and Cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Yu, C.; Luo, C.; Qu, B.; Khudhair, N.; Gu, X.; Zang, Y.; Wang, C.; Zhang, N.; Li, Q.; Gao, X. Molecular Network Including EIF1AX, RPS7, and 14-3-3γ Regulates Protein Translation and Cell Proliferation in Bovine Mammary Epithelial Cells. Arch. Biochem. Biophys. 2014, 564, 142–155. [Google Scholar] [CrossRef]
- Grawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2015, 159, 184–199. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; Azar, W.J.; Lei, Y.; Moujaber, T.; Garsed, D.W.; Kennedy, C.J.; Fereday, S.; Mitchell, C.; Chiew, Y.E.; Hendley, J.; et al. EIF1AX and NRAS Mutations Co-Occur and Cooperate in Low-Grade Serous Ovarian Carcinomas. Cancer Res. 2017, 77, 4268–4278. [Google Scholar] [CrossRef]
- Krishnamoorthy, G.P.; Davidson, N.R.; Leach, S.D.; Zhao, Z.; Lowe, S.W.; Lee, G.; Landa, I.; Nagarajah, J.; Saqcena, M.; Singh, K.; et al. EIF1AX and RAS Mutations Cooperate to Drive Thyroid Tumorigenesis through ATF4 and C-MYC. Cancer Discov. 2019, 9, 264–281. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and Other Novel Cancer Genes in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Cara, L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; David, E.; et al. Recurrent Mutations in the U2AF1 Splicing Factor in Myelodysplastic Syndromes. Nat. Genet. 2012, 44, 53–57. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Wu, J.; Chang, D.K.; et al. Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.Y.; Sapino, A.; Salomon, A.V.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 Mutations Constitute a Novel Therapeutic Target in Breast Cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Loo, P.V.; Wedge, D.C.; Nik-zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; Yates, L.R.; et al. The Landscape of Cancer Genes and Mutational Processes in Breast Cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Harbour, J.W.; Roberson, E.D.O.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent Mutations at Codon 625 of the Splicing Factor SF3B1 in Uveal Melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-neumann, S.; Roman-Roman, S.; et al. Cancer-Associated SF3B1 Mutations Affect Alternative Splicing by Promoting Alternative Branchpoint Usage. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M., Jr.; Fu, X.-D. Genome-Wide Analysis Reveals SR Protein Cooperation and Competition in Regulated Splicing. Mol. Cell 2013, 50, 223–225. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Roller, A.; Haferlach, T.; Eder, C.; Dicker, F.; Grossman, V.; Kohlmann, A.; Alpermann, T.; Yoshida, K.; Ogawa, S.; et al. SRSF2 Mutations in 275 Cases with Chronic Myelomonocytic Leukemia (CMML). Blood 2012, 120, 3080–3088. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-wahab, O.; Bradley, R.K. RNA Splicing Factors as Oncoproteins and Tumor Suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Gentien, D.; Kosmider, O.; Nguyen-Khac, F.; Albaud, B.; Rapinat, A.; Dumont, A.G.; Damm, F.; Popova, T.; Marais, R.; Fontenay, M.; et al. A Common Alternative Splicing Signature Is Associated with SF3B1 Mutations in Malignancies from Different Cell Lineages. Leukemia 2014, 28, 1355–1357. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Stanley, C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.; Murphy, M.; et al. SRSF2 Mutations Contribute to Myelodysplasia Through Mutant-Specific Effects on Exon Recognition. Cancer Cell 2016, 27, 617–630. [Google Scholar] [CrossRef]
- Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-Associated Mutation in SRSF2 Misregulates Splicing by Altering RNA-Binding Affinities. Proc. Natl. Acad. Sci. USA 2015, 112, E4726–E4734. [Google Scholar] [CrossRef]
- Muto, T.; Sashida, G.; Oshima, M.; Wendt, G.R.; Mochizuki-Kashio, M.; Nagata, Y.; Sanada, M.; Miyagi, S.; Saraya, A.; Kamio, A.; et al. Concurrent Loss of Ezh2 and Tet2 Cooperates in the Pathogenesis of Myelodysplastic Disorders. J. Exp. Med. 2013, 210, 2627–2639. [Google Scholar] [CrossRef]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-trillos, A.; et al. Exome Sequencing Identifies Recurrent Mutations of the Splicing Factor SF3B1 Gene in Chronic Lymphocytic Leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.M.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome Sequencing Reveals Potential Mechanism of Cryptic 3’ Splice Site Selection in SF3B1-Mutated Cancers. PLoS Comput. Biol. 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An Alternative Splicing Network Links Cell Cycle Control to Apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef]
- Larrayoz, M.; Blakemore, S.J.; Dobson, R.C.; Blunt, M.D.; Rose-Zerilli, M.J.J.; Walewska, R.; Duncombe, A.; Oscier, D.; Koide, K.; Forconi, F.; et al. The SF3B1 Inhibitor Spliceostatin A (SSA) Elicits Apoptosis in Chronic Lymphocytic Leukaemia Cells through Downregulation of Mcl-1. Leukemia 2016, 30, 351–360. [Google Scholar] [CrossRef]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A Targets SF3b and Inhibits Both Splicing and Nuclear Retention of Pre-MRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef]
- Gao, Y.; Koide, K. Chemical Perturbation of Mcl-1 Pre-MRNA Splicing to Induce Apoptosis in Cancer Cells. ACS Chem. Biol. 2014, 8, 895–900. [Google Scholar] [CrossRef]
- Gao, Y.; Trivedi, S.; Ferris, R.L.; Koide, K. Regulation of HPV16 E6 and MCL1 by SF3B1 Inhibitor in Head and Neck Cancer Cells. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef]
- Eskens, F.A.L.M.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; De Jonge, M.J.A.; Mizui, Y.; Wiemer, E.A.C.; Carreras, M.J.; Baselga, J.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the First-in-Class Spliceosome Inhibitor E7107 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-MRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef]
- Xargay-Torrent, S.; López-Guerra, M.; Rosich, L.; Montraveta, A.; Roldán, J.; Rodríguez, V.; Villamor, N.; Aymerich, M.; Lagisetti, C.; Webb, T.R.; et al. The Splicing Modulator Sudemycin Induces a Specific Antitumor Response and Cooperates with Ibrutinib in Chronic Lymphocytic Leukemia. Oncotarget 2015, 6, 22734–22749. [Google Scholar] [CrossRef]
- Pawellek, A.; McElroy, S.; Samatov, T.; Mitchell, L.; Woodland, A.; Ryder, U.; Gray, D.; Lührmann, R.; Lamond, A.I. Identification of Small Molecule Inhibitors of Pre-MRNA Splicing. J. Biol. Chem. 2014, 289, 34683–34698. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an Orally Available Small-Molecule Splicing Modulator, Induces Lethality in Spliceosome-Mutant Cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K. Van; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Caesar-Johnson, S.J.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef]
 .
.
.
. .
.
.
.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vivet-Noguer, R.; Tarin, M.; Roman-Roman, S.; Alsafadi, S. Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology. Cancers 2019, 11, 1019. https://doi.org/10.3390/cancers11071019
Vivet-Noguer R, Tarin M, Roman-Roman S, Alsafadi S. Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology. Cancers. 2019; 11(7):1019. https://doi.org/10.3390/cancers11071019
Chicago/Turabian StyleVivet-Noguer, Raquel, Malcy Tarin, Sergio Roman-Roman, and Samar Alsafadi. 2019. "Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology" Cancers 11, no. 7: 1019. https://doi.org/10.3390/cancers11071019
APA StyleVivet-Noguer, R., Tarin, M., Roman-Roman, S., & Alsafadi, S. (2019). Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology. Cancers, 11(7), 1019. https://doi.org/10.3390/cancers11071019