NECTIN4 (PVRL4) as Putative Therapeutic Target for a Specific Subtype of High Grade Serous Ovarian Cancer—An Integrative Multi-Omics Approach


 , and
, and
Abstract
:
1. Introduction
2. Results
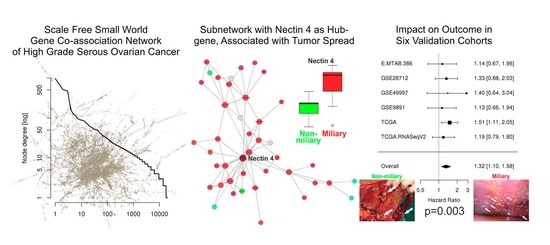
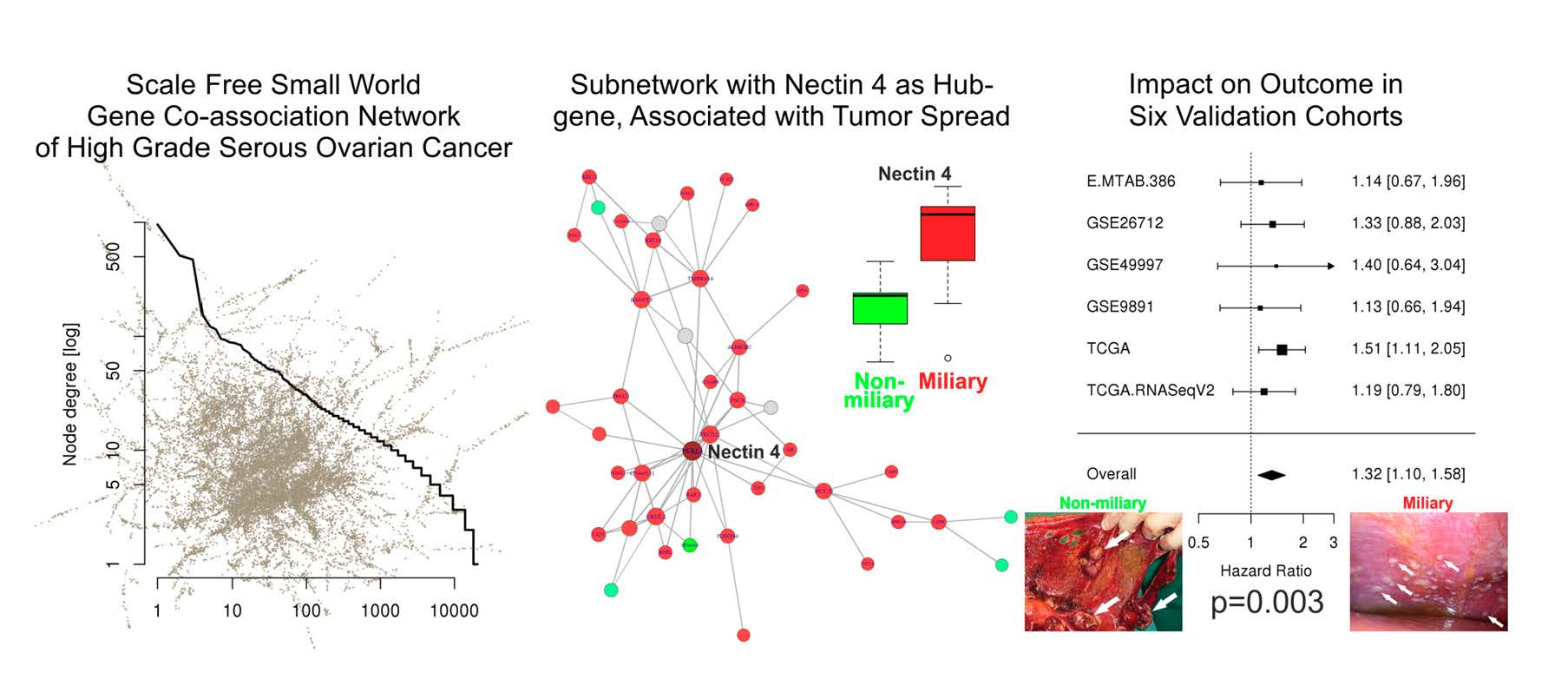
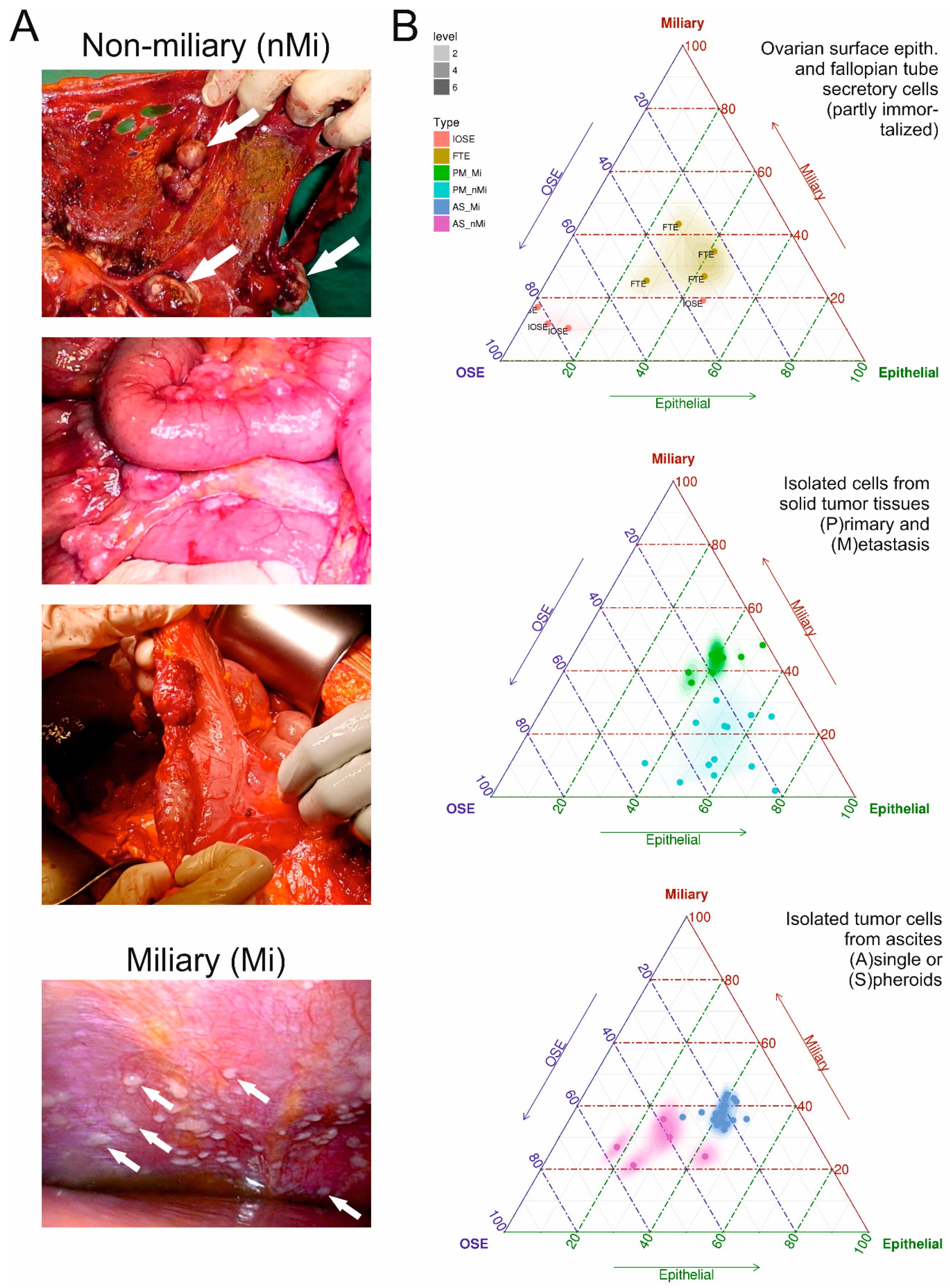
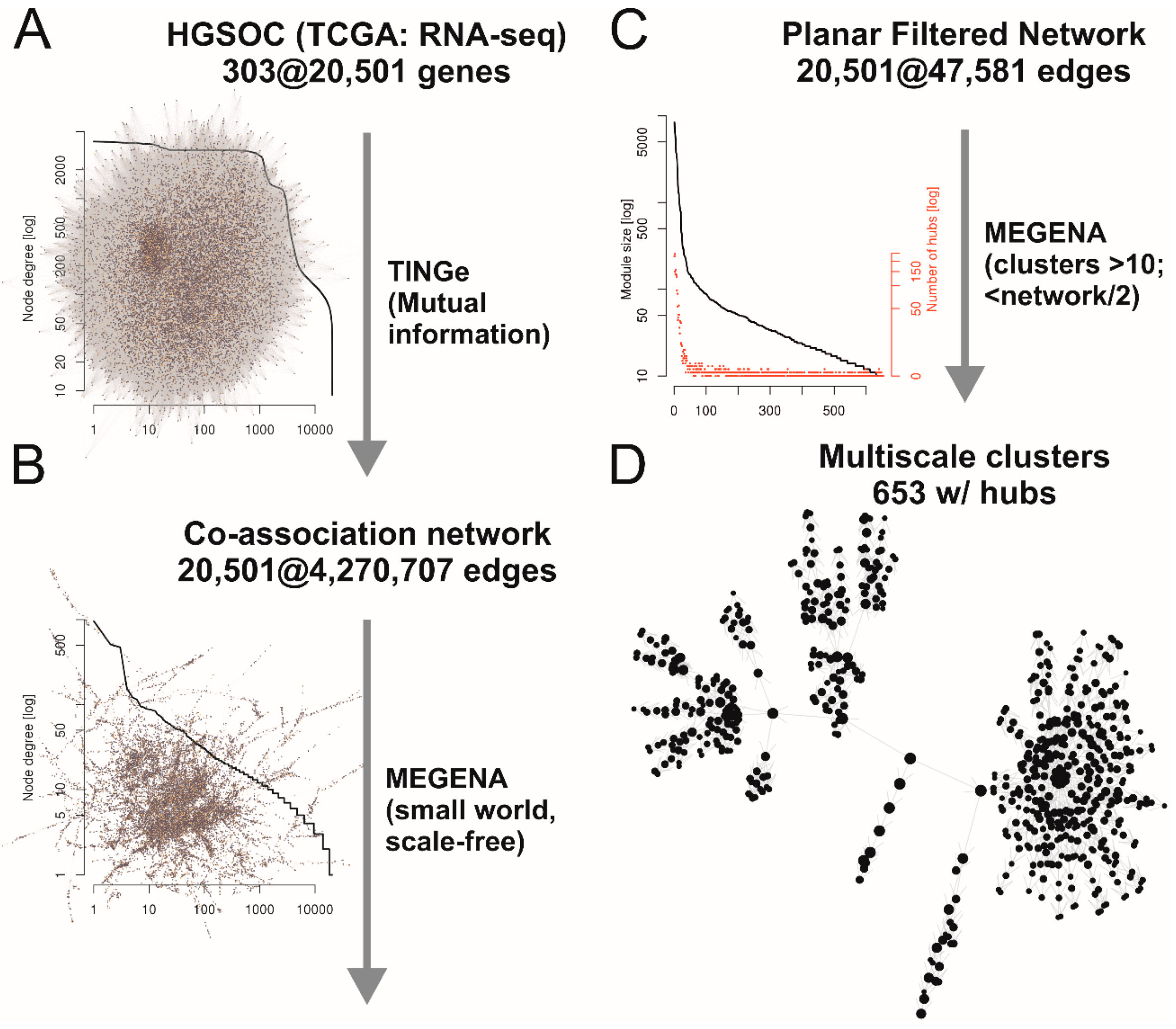
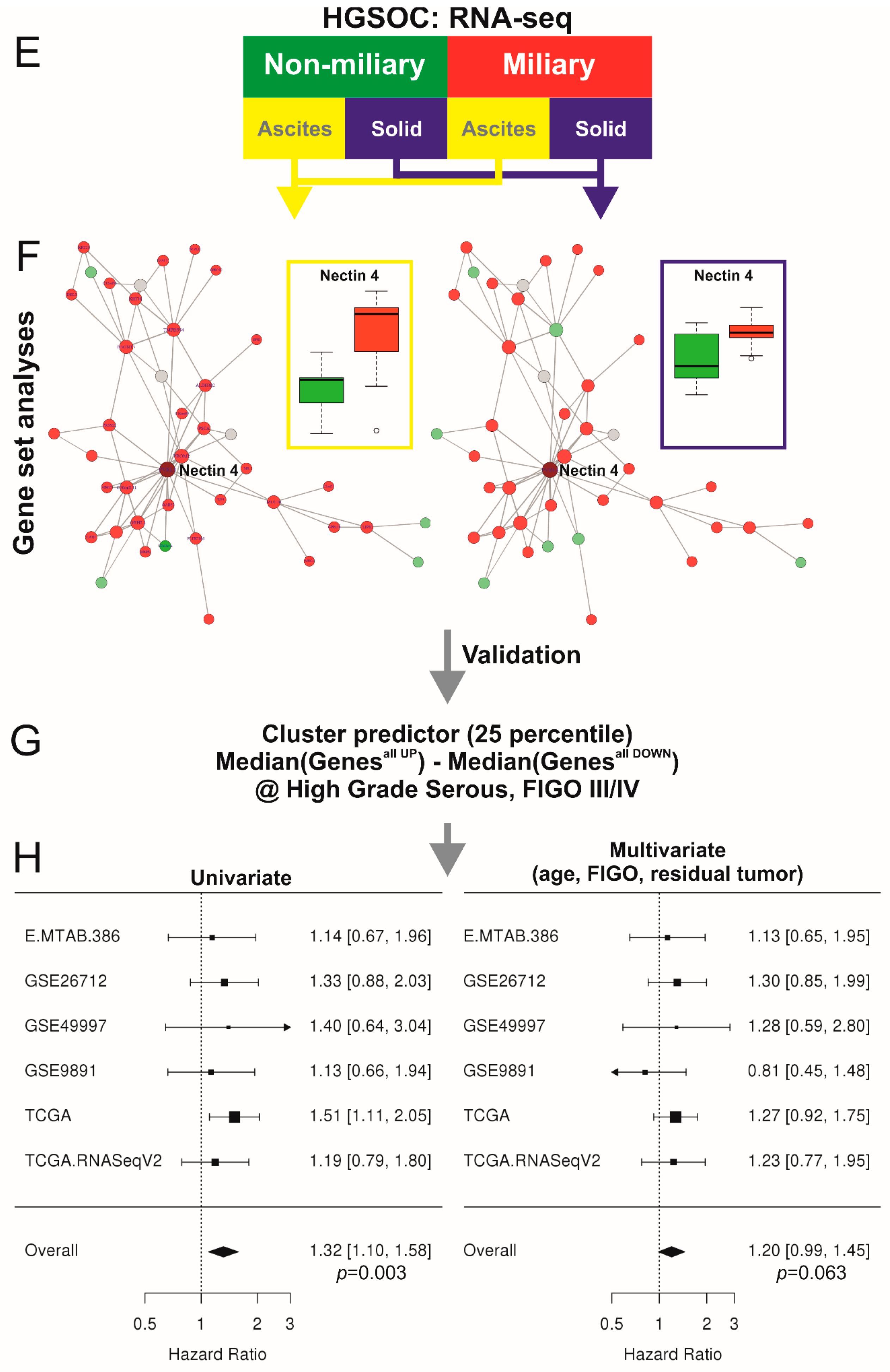
2.1. Co-Association Gene Expression Clusters Associated with Tumor Spread Types
2.2. HGSOC Cell Line Models
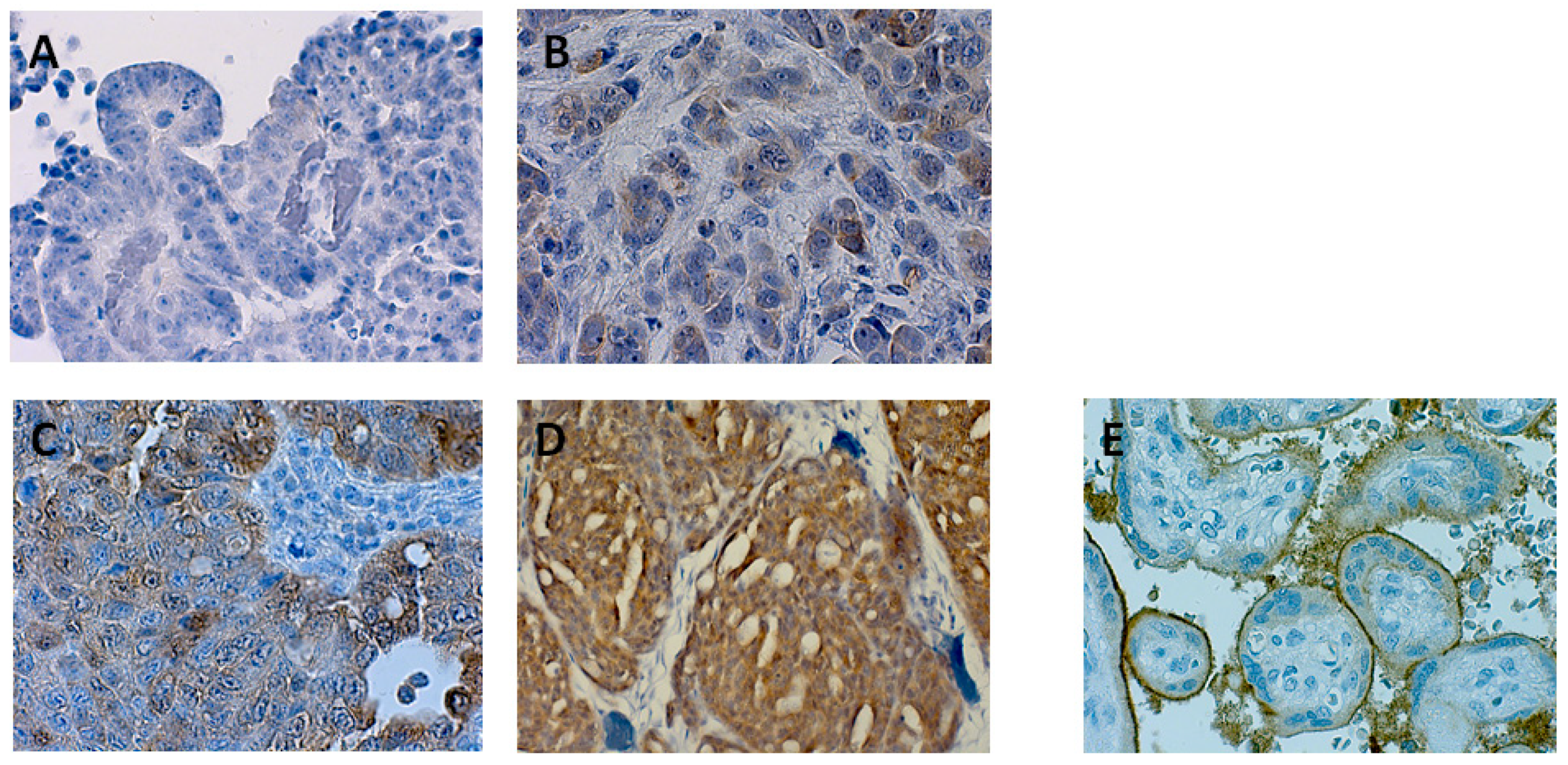
2.3. Protein Expression of Nectin 4 (Assessed by Immunohistochemistry) and Impact on Overall Survival
2.4. Network of Associations of the Nectin 4 Driven Expression Clusters with Omics Data and Biological Interpretation
2.5. NECTIN4 Dependent Expression Differences in HGSOC Cell Lines
3. Discussion
4. Material and Methods
4.1. Planar Filtered Gene Co-Association Network and Differential Cluster Expression Analyses
4.2. Cell Line Analyses
4.3. Overall Survival Meta-Analyses
4.4. Immunohistochemistry of Nectin 4 and Impact on Overall Survival
4.5. Untargeted Proteomics of Cell Free Ascites
4.6. Correlations to Omics and Other Data and Network Construction for Biological Interpretation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yoshihara, K.; Tajima, A.; Komata, D.; Yamamoto, T.; Kodama, S.; Fujiwara, H.; Suzuki, M.; Onishi, Y.; Hatae, M.; Sueyoshi, K.; et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009, 100, 1421–1428. [Google Scholar] [CrossRef]
- Crijns, A.P.; Fehrmann, R.S.; de Jong, S.; Gerbens, F.; Meersma, G.J.; Klip, H.G.; Hollema, H.; Hofstra, R.M.; te Meerman, G.J.; de Vries, E.G.; et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS Med. 2009, 6, e24. [Google Scholar] [CrossRef]
- Pils, D.; Hager, G.; Tong, D.; Aust, S.; Heinze, G.; Kohl, M.; Schuster, E.; Wolf, A.; Sehouli, J.; Braicu, I.; et al. Validating the impact of a molecular subtype in ovarian cancer on outcomes: A study of the OVCAD Consortium. Cancer Sci. 2012. [Google Scholar] [CrossRef]
- TCGA. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Auer, K.; Bachmayr-Heyda, A.; Aust, S.; Sukhbaatar, N.; Reiner, A.T.; Grimm, C.; Horvat, R.; Zeillinger, R.; Pils, D. Peritoneal tumor spread in serous ovarian cancer-epithelial mesenchymal status and outcome. Oncotarget 2015, 6, 17261–17275. [Google Scholar] [CrossRef]
- Auer, K.; Bachmayr-Heyda, A.; Sukhbaatar, N.; Aust, S.; Schmetterer, K.G.; Meier, S.M.; Gerner, C.; Grimm, C.; Horvat, R.; Pils, D. Role of the immune system in the peritoneal tumor spread of high grade serous ovarian cancer. Oncotarget 2016, 7, 61336–61354. [Google Scholar] [CrossRef]
- Bachmayr-Heyda, A.; Auer, K.; Sukhbaatar, N.; Aust, S.; Deycmar, S.; Reiner, A.T.; Polterauer, S.; Dekan, S.; Pils, D. Small RNAs and the competing endogenous RNA network in high grade serous ovarian cancer tumor spread. Oncotarget 2016, 7, 39640–39653. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Bachmayr-Heyda, A.; Auer, K.; Aust, S.; Deycmar, S.; Horvat, R.; Pils, D. Two different, mutually exclusively distributed, TP53 mutations in ovarian and peritoneal tumor tissues of a serous ovarian cancer patient: Indicative for tumor origin? Cold Spring Harb. Mol. Case Stud. 2017, 3. [Google Scholar] [CrossRef]
- Bachmayr-Heyda, A.; Aust, S.; Auer, K.; Meier, S.M.; Schmetterer, K.G.; Dekan, S.; Gerner, C.; Pils, D. Integrative Systemic and Local Metabolomics with Impact on Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2017, 23, 2081–2092. [Google Scholar] [CrossRef]
- Aust, S.; Knogler, T.; Pils, D.; Obermayr, E.; Reinthaller, A.; Zahn, L.; Radlgruber, I.; Mayerhoefer, M.E.; Grimm, C.; Polterauer, S. Skeletal Muscle Depletion and Markers for Cancer Cachexia Are Strong Prognostic Factors in Epithelial Ovarian Cancer. PLoS ONE 2015, 10, e0140403. [Google Scholar] [CrossRef] [PubMed]
- Auer, K.; Bachmayr-Heyda, A.; Aust, S.; Grunt, T.W.; Pils, D. Comparative transcriptome analysis links distinct peritoneal tumor spread types, miliary and non-miliary, with putative origin, tubes and ovaries, in high grade serous ovarian cancer. Cancer Lett. 2016. [Google Scholar] [CrossRef]
- Karst, A.M.; Drapkin, R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat. Protoc. 2012, 7, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Eng, K.H.; Morrell, K.; Starbuck, K.; Spring-Robinson, C.; Khan, A.; Cleason, D.; Akman, L.; Zsiros, E.; Odunsi, K.; Szender, J.B. Prognostic value of miliary versus non-miliary sub-staging in advanced ovarian cancer. Gynecol. Oncol. 2017, 146, 52–57. [Google Scholar] [CrossRef]
- Miow, Q.H.; Tan, T.Z.; Ye, J.; Lau, J.A.; Yokomizo, T.; Thiery, J.P.; Mori, S. Epithelial-mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015, 34, 1899–1907. [Google Scholar] [CrossRef]
- Takai, Y.; Irie, K.; Shimizu, K.; Sakisaka, T.; Ikeda, W. Nectins and nectin-like molecules: Roles in cell adhesion, migration, and polarization. Cancer Sci. 2003, 94, 655–667. [Google Scholar] [CrossRef]
- Fuchs, A.; Colonna, M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin. Cancer Biol. 2006, 16, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Klinkebiel, D.; Zhang, W.; Akers, S.N.; Odunsi, K.; Karpf, A.R. DNA Methylome Analyses Implicate Fallopian Tube Epithelia as the Origin for High-grade Serous Ovarian Cancer. Mol. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pils, D.; Bachmayr-Heyda, A.; Auer, K.; Svoboda, M.; Auner, V.; Hager, G.; Obermayr, E.; Reiner, A.; Reinthaller, A.; Speiser, P.; et al. Cyclin E1 (CCNE1) as independent positive prognostic factor in advanced stage serous ovarian cancer patients—A study of the OVCAD consortium. Eur. J. Cancer 2014, 50, 99–110. [Google Scholar] [CrossRef]
- Bentink, S.; Haibe-Kains, B.; Risch, T.; Fan, J.B.; Hirsch, M.S.; Holton, K.; Rubio, R.; April, C.; Chen, J.; Wickham-Garcia, E.; et al. Angiogenic mRNA and microRNA gene expression signature predicts a novel subtype of serous ovarian cancer. PLoS ONE 2012, 7, e30269. [Google Scholar] [CrossRef]
- Bonome, T.; Levine, D.A.; Shih, J.; Randonovich, M.; Pise-Masison, C.A.; Bogomolniy, F.; Ozbun, L.; Brady, J.; Barrett, J.C.; Boyd, J.; et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008, 68, 5478–5486. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef]
- Royston, P.; Sauerbrei, W. Building multivariable regression models with continuous covariates in clinical epidemiology—With an emphasis on fractional polynomials. Methods Inf. Med. 2005, 44, 561–571. [Google Scholar] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Poli, A.; Michel, T.; Theresine, M.; Andres, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef]
- Abdel Azim, S.; Duggan-Peer, M.; Sprung, S.; Reimer, D.; Fiegl, H.; Soleiman, A.; Marth, C.; Zeimet, A.G. Clinical impact of L1CAM expression measured on the transcriptome level in ovarian cancer. Oncotarget 2016, 7, 37205–37214. [Google Scholar] [CrossRef] [PubMed]
- Bondong, S.; Kiefel, H.; Hielscher, T.; Zeimet, A.G.; Zeillinger, R.; Pils, D.; Schuster, E.; Castillo-Tong, D.C.; Cadron, I.; Vergote, I.; et al. Prognostic significance of L1CAM in ovarian cancer and its role in constitutive NF-kappaB activation. Ann. Oncol. 2012, 23, 1795–1802. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Derycke, M.S.; Pambuccian, S.E.; Gilks, C.B.; Kalloger, S.E.; Ghidouche, A.; Lopez, M.; Bliss, R.L.; Geller, M.A.; Argenta, P.A.; Harrington, K.M.; et al. Nectin 4 overexpression in ovarian cancer tissues and serum: Potential role as a serum biomarker. Am. J. Clin. Pathol. 2010, 134, 835–845. [Google Scholar] [CrossRef]
- Nabih, E.S.; Abdel Motaleb, F.I.; Salama, F.A. The diagnostic efficacy of nectin 4 expression in ovarian cancer patients. Biomarkers 2014, 19, 498–504. [Google Scholar] [CrossRef]
- Boylan, K.L.; Buchanan, P.C.; Manion, R.D.; Shukla, D.M.; Braumberger, K.; Bruggemeyer, C.; Skubitz, A.P. The expression of Nectin-4 on the surface of ovarian cancer cells alters their ability to adhere, migrate, aggregate, and proliferate. Oncotarget 2017, 8, 9717–9738. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Pallasch, C.; Elia, A.E.; Braun, C.J.; Westbrook, T.F.; Hemann, M.; Elledge, S.J. A role for PVRL4-driven cell-cell interactions in tumorigenesis. Elife 2013, 2, e00358. [Google Scholar] [CrossRef]
- M-Rabet, M.; Cabaud, O.; Josselin, E.; Finetti, P.; Castellano, R.; Farina, A.; Agavnian-Couquiaud, E.; Saviane, G.; Collette, Y.; Viens, P.; et al. Nectin-4: A new prognostic biomarker for efficient therapeutic targeting of primary and metastatic triple-negative breast cancer. Ann. Oncol. 2017, 28, 769–776. [Google Scholar] [CrossRef]
- Ma, J.; Sheng, Z.; Lv, Y.; Liu, W.; Yao, Q.; Pan, T.; Xu, Z.; Zhang, C.; Xu, G. Expression and clinical significance of Nectin-4 in hepatocellular carcinoma. Onco Targets Ther. 2016, 9, 183–190. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Shen, Q.; Yin, W.; Huang, H.; Liu, Y.; Ni, Q. High expression of Nectin-4 is associated with unfavorable prognosis in gastric cancer. Oncol. Lett. 2018, 15, 8789–8795. [Google Scholar] [CrossRef]
- Aust, S.; Felix, S.; Auer, K.; Bachmayr-Heyda, A.; Kenner, L.; Dekan, S.; Meier, S.M.; Gerner, C.; Grimm, C.; Pils, D. Absence of PD-L1 on tumor cells is associated with reduced MHC I expression and PD-L1 expression increases in recurrent serous ovarian cancer. Sci. Rep. 2017, 7, 42929. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Loessner, D.; Irving-Rodgers, H.; Obermair, A.; Nicklin, J.L.; Clements, J.A. Metastasis of ovarian cancer is mediated by kallikrein related peptidases. Clin. Exp. Metastasis 2014, 31, 135–147. [Google Scholar] [CrossRef]
- Loessner, D.; Goettig, P.; Preis, S.; Felber, J.; Bronger, H.; Clements, J.A.; Dorn, J.; Magdolen, V. Kallikrein-related peptidases represent attractive therapeutic targets for ovarian cancer. Expert Opin. Ther. Targets 2018. [Google Scholar] [CrossRef] [PubMed]
- Planaguma, J.; Liljestrom, M.; Alameda, F.; Butzow, R.; Virtanen, I.; Reventos, J.; Hukkanen, M. Matrix metalloproteinase-2 and matrix metalloproteinase-9 codistribute with transcription factors RUNX1/AML1 and ETV5/ERM at the invasive front of endometrial and ovarian carcinoma. Hum. Pathol. 2011, 42, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, A.; Cardaci, S.; Lamba, S.; Oddo, D.; Marchio, C.; Cassoni, P.; Amoreo, C.A.; Corti, G.; Testori, A.; Bussolino, F.; et al. BCAM and LAMA5 Mediate the Recognition between Tumor Cells and the Endothelium in the Metastatic Spreading of KRAS-Mutant Colorectal Cancer. Clin. Cancer Res. 2016, 22, 4923–4933. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Han, Q.; Deng, Y.; Chen, S.; Chen, R.; Yang, M.; Zhang, Z.; Sun, X.; Wang, W.; He, Y.; Wang, F.; et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci. Rep. 2017, 7, 4759. [Google Scholar] [CrossRef]
- Cho, D.H.; Jo, Y.K.; Kim, S.C.; Park, I.J.; Kim, J.C. Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res. 2012, 32, 4091–4096. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Aluru, M.; Zola, J.; Nettleton, D.; Aluru, S. Reverse engineering and analysis of large genome-scale gene networks. Nucleic Acids Res. 2013, 41, e24. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Zhang, B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Lim, E.; Vaillant, F.; Asselin-Labat, M.L.; Visvader, J.E.; Smyth, G.K. ROAST: Rotation gene set tests for complex microarray experiments. Bioinformatics 2010, 26, 2176–2182. [Google Scholar] [CrossRef]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef]
- Liu, R.; Holik, A.Z.; Su, S.; Jansz, N.; Chen, K.; Leong, H.S.; Blewitt, M.E.; Asselin-Labat, M.L.; Smyth, G.K.; Ritchie, M.E. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 2015, 43, e97. [Google Scholar] [CrossRef]
- Ganzfried, B.F.; Riester, M.; Haibe-Kains, B.; Risch, T.; Tyekucheva, S.; Jazic, I.; Wang, X.V.; Ahmadifar, M.; Birrer, M.J.; Parmigiani, G.; et al. curatedOvarianData: Clinically annotated data for the ovarian cancer transcriptome. Database 2013, 2013, bat013. [Google Scholar] [CrossRef]
- Aust, S.; Bachmayr-Heyda, A.; Pateisky, P.; Tong, D.; Darb-Esfahani, S.; Denkert, C.; Chekerov, R.; Sehouli, J.; Mahner, S.; Van Gorp, T.; et al. Role of TRAP1 and estrogen receptor alpha in patients with ovarian cancer—A study of the OVCAD consortium. Mol. Cancer 2012, 11, 69. [Google Scholar] [CrossRef]
- Muqaku, B.; Eisinger, M.; Meier, S.M.; Tahir, A.; Pukrop, T.; Haferkamp, S.; Slany, A.; Reichle, A.; Gerner, C. Multi-omics Analysis of Serum Samples Demonstrates Reprogramming of Organ Functions Via Systemic Calcium Mobilization and Platelet Activation in Metastatic Melanoma. Mol. Cell. Proteom. 2017, 16, 86–99. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed]
- Yaari, G.; Bolen, C.R.; Thakar, J.; Kleinstein, S.H. Quantitative set analysis for gene expression: A method to quantify gene set differential expression including gene-gene correlations. Nucleic Acids Res. 2013, 41, e170. [Google Scholar] [CrossRef]
- Fang, H.; Gough, J. The ‘dnet’ approach promotes emerging research on cancer patient survival. Genome Med. 2014, 6, 64. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Giraud, C.; Huet, S.; Verzelen, N. Graph selection with GGMselect. Stat. Appl. Genet. Mol. Biol. 2012, 11, 3. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | N Genes | Direction AS | P (AS) | Direction PM | P (PM) |
|---|---|---|---|---|---|
| c1_160 | 37 | Up | 3.32 × 10−1 | Up | 3.92 × 10−14 |
| c1_32 | 165 | Up | 1.00 × 10−5 | Up | 3.62 × 10−9 |
| c1_143 | 38 | Up | 1.00 × 10−5 | Up | 7.98 × 10−9 |
| c1_400 | 22 | Up | 1.00 × 10−5 | Up | 2.26 × 10−8 |
| c1_297 | 86 | Down | 4.00 × 10−2 | Down | 9.31 × 10−12 |
| c1_42 | 115 | Up | 6.51 × 10−3 | Up | 9.22 × 10−11 |
| c1_54 | 72 | Down | 9.47 × 10−1 | Down | 7.50 × 10−13 |
| c1_247 | 16 | Up | 1.00 × 10−5 | Up | 1.06 × 10−7 |
| c1_39 | 96 | Up | 4.49 × 10−2 | Up | 2.66 × 10−11 |
| c1_75 | 69 | Up | 1.00 × 10−5 | Up | 2.28 × 10−7 |
| c1_134 | 30 | Up | 1.26 × 10−1 | Up | 8.37 × 10−11 |
| Clinicopathologic Characteristics | n = 90 | Nectin 4 Abundance | p (Overall) | p (Trend) | ||
|---|---|---|---|---|---|---|
| Negative (n = 42) | ≤50% (n = 36) | >50% (n = 12) | ||||
| Tumor stage 1 | 0.257 | 0.156 | ||||
| FIGO III | 69 | 34 (81.0%) | 28 (77.8%) | 7 (58.3%) | ||
| FIGO IV | 21 | 8 (19.0%) | 8 (22.2%) | 5 (41.7%) | ||
| Histological grade | 0.090 | 0.454 | ||||
| G2 | 18 | 8 (19.0%) | 10 (27.8%) | 0 (0.0%) | ||
| G3 | 72 | 34 (81.0%) | 26 (72.2%) | 12 (100.0%) | ||
| Residual disease | 0.027 | 0.020 | ||||
| R0 | 40 | 23 (54.8%) | 16 (44.4%) | 1 (8.3%) | ||
| R1 | 48 | 18 (42.9%) | 19 (52.8%) | 11 (91.7%) | ||
| Missing | 2 | 1 (2.3%) | 1 (2.8%) | 0 (0.0%) | ||
| Age, diagnosis [y] 2 | 60.0 (11.9) | 58.2 (12.5) | 61.1 (11.3) | 62.6 (11.5) | 0.398 | 0.181 |
| Progression free survival | 0.120 | 0.037 | ||||
| Without progression | 19 | 13 (31.0%) | 5 (13.9%) | 1 (8.3%) | ||
| With progression | 71 | 29 (69.0%) | 31 (86.1%) | 11 (91.7%) | ||
| Status | 0.235 | 0.315 | ||||
| Alive | 50 | 24 (57.1%) | 22 (61.1%) | 4 (33.3%) | ||
| Died | 40 | 18 (42.9%) | 14 (38.9%) | 8 (66.7%) | ||
| Overall Survival | ||||||
|---|---|---|---|---|---|---|
| Cox Regression Analyses | Univariate 1 | Multiple 2 | ||||
| HR | CI95 | p | HR | CI95 | p | |
| Age (decades) | 2.25 | 1.61–3.16 | <0.001 | 2.37 | 1.68–3.33 | <0.001 |
| FIGO stage (IV vs. III) | 1.59 | 0.79–3.20 | 0.198 | 1.55 | 0.75–3.20 | 0.240 |
| Histological grade (G3 vs. G2) | 0.85 | 0.41–1.79 | 0.672 | 0.50 | 0.22–1.14 | 0.099 |
| Residual tumor (R1 vs. R0) | 1.71 | 0.91–3.24 | 0.096 | 1.61 | 0.84–3.09 | 0.155 |
| Nectin 4 (>50% vs.≤50%) 3 | 3.03 | 1.37–6.68 | 0.006 | 3.62 | 1.52–8.63 | 0.004 |
| Analytes Type | Tissue | N (Analytes) | FDR < 5% (<10%) | UP | Correlations FDR < 0% | Positive | Name of Table 1 | Reference |
|---|---|---|---|---|---|---|---|---|
| RNA sequencing | Floating (ascites) and solid tumor cells | 34,034 genes | 908 | 643 | n.a. | sign_RNAseq | [6,12] | |
| --KEGG pathways | 199 | 8 (9) | 0 (0) | n.a. | sign_KEGG | - | ||
| --TCGA clusters | 653 | 20 (58) | 12 (42) | n.a. | sign_TCGA | - | ||
| --Gene-sets | 17,810 | 1828 | 133 | n.a. | sign_GS | - | ||
| Immune cells (FACS) | Ascites | 43 cell populations | 0 | 0 | 9 | 7 | sign_A_FACS | [7] |
| Blood | 43 cell populations | 0 (5) | (2) | 44 | 10 | sign_B_FACS | [7] | |
| Solid tumor tissue | 43 cell populations | 0 | 0 | 37 | 8 | sign_T_FACS | [7] | |
| Immune cells (IF) | Ascites | 8 cell populations | 0 (2) | 0 | 8 | 4 | sign_A_IF | [7] |
| Chemo/cytokines | Ascites | 56 | 0 | 0 | 15 | 4 | sign_A_Lum | [7] |
| Serum | 56 | 0 | 0 | 35 | 6 | sign_S_Lum | [7] | |
| Proteomics | Ascites | 852 | 3 (4) | 1 (2) | 65 | 24 | sign_A_Prot | - |
| RNA seq. | Cell lines | 18,054 genes | 57 (912) 2 | 53 (718) 2 | n.a. | sign_RNAseq _CLs | [25] | |
| KEGG pathways | Cell lines | 199 | 4 (7) 2 | 4 (7) 2 | n.a. | sign_KEGG _CLs | - | |
| Network | Direction/ Hub Genes | Network Function/Gene Names | Description |
|---|---|---|---|
| c1_143 | UP | Epithelial cell development; Adhesion | |
| NECTIN4 | Nectin cell adhesion molecule 4 | involved in cell adhesion through trans-homophilic and -heterophilic interactions | |
| PROM2 | Prominin 2 | localizes to basal epithelial cells and may be involved in the organization of plasma membrane microdomains | |
| GRHL2 | Grainyhead-like protein 2 homolog | primary neurulation and in epithelial development | |
| B3GNT3 | UDP-GlcNAc:BetaGal Beta-1,3-N-Acetylglucosaminyltransferase 3 | L-selectin ligand biosynthesis, lymphocyte homing and lymphocyte trafficking | |
| c1_180 | UP | Neuronal | |
| PYCR2 | Pyrroline-5-carboxylate reductase family | mutations identified as cause of a unique syndrome characterized by postnatal microcephaly, hypomyelination, and reduced cerebral white-matter volume | |
| MRPL55 | Mammalian mitochondrial ribosomal proteins | protein synthesis within the mitochondrion | |
| SNAP47 | Synaptosome Associated Protein 47 | syntaxin binding and SNAP receptor activity | |
| c1_363 | UP | Apoptosis and energy metabolism (Mitochondrion) | |
| TIMM50 | translocase of inner mitochondrial membrane 50 | maintaining membrane permeability barrier, knockdown of this gene results in the release of cytochrome c and apoptosis | |
| MRPS12 | Mitochondrial Ribosomal Protein S12 | key component of the ribosomal small subunit, controls the decoding fidelity and susceptibility to aminoglycoside antibiotics | |
| ECH1 | Enoyl-CoA Hydratase 1 | essential to metabolizing fatty acids to produce both acetyl CoA and energy | |
| c1_747 | UP | G protein signal-transducing/-1 frameshifting at translation | |
| PNMA3 | paraneoplastic Ma antigen | present in sera from patients suffering of paraneoplastic neurological disorders, promotes -1 frameshifting | |
| GPRIN2 | G Protein Regulated Inducer of Neurite Outgrowth | interacted specifically with G-alpha-o and G-alpha-z bound to GTP-gamma-S and GDP-AlF4(−) | |
| c1_782 | UP | Epithelial proliferation; Neuronal? | |
| PNMA3 | paraneoplastic Ma antigen | present in sera from patients suffering of paraneoplastic neurological disorders, promotes-1 frameshifting | |
| KCNIP3 | Potassium Voltage-Gated Channel Interacting Protein 3 | Calsenilin, member of the family of voltage-gated potassium (Kv) channel-interacting proteins, belong to the neuronal calcium sensor family of proteins | |
| RSPO4 | R-spondin 4 | induced epithelial proliferation | |
| c1_335 | UP | Transcription regulation; Cell death | |
| BEX3 | Brain Expressed X-Linked 3 | role in the pathogenesis of neurogenetic diseases | |
| TCEAL8 | Transcription Elongation Factor A Like 8 | modulate transcription in a promoter context-dependent manner. | |
| c1_376 | UP | Transcriptional regulation | |
| ZNF574 | Zinc finger protein 574 | transcriptional regulation | |
| ZNF526 | Zinc finger protein 526 (paralog to ZNF574) | highest expression in ovary | |
| GSK3A | Glycogen synthase kinase-3 alpha | type 2 diabetes, control of glucose homeostasis, Wnt signaling and regulation of transcription factors and microtubules, by phosphorylating and inactivating glycogen synthase | |
| BCAM | Basal cell adhesion molecule | may play a role in epithelial cell cancer; was shown to be overexpressed in ovarian carcinomas; BCAM-AKT2 fusion gene in 7% HGSOC cases | |
| c1_760 | UP | Transcriptional regulation | |
| PHKG2 | Phosphorylase Kinase Catalytic Subunit Gamma 2 | mediates the neural and hormonal regulation of glycogenolysis by phosphorylating and thereby activating glycogen phosphorylase | |
| TBC1D10B | TBC1 Domain family, member 10B | Small G proteins of the RAB family | |
| 7 ZNF genes | ZNF785, ZNF764, ZNF768, ZNF689, ZNF747, ZNF48, ZNF668 | ZNF689: conferred anchorage-independent cell growth | |
| c1_496 | DOWN | Autophagy and splicing | 20/23 genes from chromosomal band 6q21 and 2 further from 6q16 and 6q22 |
| CDC40 | Cell Division Cycle 40 | essential for the catalytic step II in pre-mRNA splicing process | |
| ATG5 | Autophagy related 5 | key regulator of autophagy mutations in the Atg5 gene have also been linked with prostate, gastrointestinal and colorectal cancers as ATG5 plays a role in both cell apoptosis and cell cycle arrest | |
| c1_358 | UP | 11 KLK genes; Aberrant levels of kallikrein-related peptidases have been linked to cancer cell proliferation, invasion and metastasis | |
| KLK10 | Kallikrein-10 | may play a role in suppression of tumorigenesis in breast and prostate cancers; shorter OS in HGSOC | |
| KLK6 | Kallikrein-6 | carcinogenesis, involvement in Alzheimer’s disease; shorter OS in HGSOC | |
| c1_315 | DOWN | Expressed on cells specialized for antigen presentation | |
| CD1A-C; CD1E | Group 1 Thymocyte antigens | CD1 family of transmembrane glycoproteins, which are structurally related to the major histocompatibility complex (MHC) proteins; present lipid and glycolipid antigens to T cells | |
| FCER1A | Fc fragment of IgE, high affinity I, receptor for; alpha polypeptide | ||
| KEGG | UP | Growth factors and cAMP -> MAPK signaling | |
| EGFR | Epidermal Growth Factor Receptor | initiates several signal transduction cascades, i.e., the MAPK, Akt and JNK pathways, leading to DNA synthesis and cell proliferation. | |
| MAPK1 | Mitogen-Activated Protein Kinase 1 | intracellular signaling network that regulates many cellular machines, including the cell cycle machinery and autocrine/paracrine factor synthesizing machinery | |
| PRKACB | Protein Kinase CAMP-Activated Catalytic Subunit Beta | cAMP signalling towards the MAPK complex | |
| CREB5 | CAMP Responsive Element Binding Protein 5 | binds to CRE as a homodimer or a heterodimer with c-Jun or CRE-BP1, and functions as a CRE-dependent trans-activator | |
| TCGAnet | DOWN | Immune cell infiltration? | |
| CD53 | Tetraspanin-25 | expressed from several immune cells, B- and T-cells, monocytes, neutrophils, and NK cells | |
| HAVCR2 | Hepatitis A virus cellular receptor 2 | HAVCR2 is an immune checkpoint mediate the CD8+ T-cell exhaustion | |
| GLIPR1 | Glioma pathogenesis-related protein 1 | tumor suppressor | |
| PTPRC | CD45; receptor-type tyrosine-protein phosphatase C | pan immune cell marker | |
| GPR183 | G protein-coupled receptor 183 | expressed in B cells | |
| STRING | UP | MAPK signaling and insuline pathway | |
| MAPK1 | Mitogen-Activated Protein Kinase 1 | intracellular signaling network that regulates many cellular machines, including the cell cycle machinery and autocrine/paracrine factor synthesizing machinery | |
| CRKL | Crk-like protein | oncogene; participates in the Reelin signaling cascade downstream of DAB1 | |
| AKT2 | RAC-beta serine/threonine-protein kinase | oncogene; amplified and overexpressed in primary ovarian tumors | |
| CREB5 | CAMP Responsive Element Binding Protein 5 | binds to CRE as a homodimer or a heterodimer with c-Jun or CRE-BP1, and functions as a CRE-dependent trans-activator | |
| PPI | UP | Transcription and splicing | |
| TSGA10IP | Testis specific 10 interacting protein | ||
| MDFI | MyoD family inhibitor | transcription factor that negatively regulates other myogenic family proteins | |
| MEOX2 | Homeobox protein MOX-2 | transcription factor | |
| TFIP11 | Tuftelin-interacting protein 11 | associated with RNA and may play a role in splicing | |
| BCAM | basal cell adhesion molecule | overexpressed in ovarian carcinomas in vivo and upregulated following malignant transformation | |
| MAPK1 | Mitogen-Activated Protein Kinase 1 | intracellular signaling network that regulates many cellular machines, including the cell cycle machinery and autocrine/paracrine factor synthesizing machinery | |
| CREB5 | CAMP Responsive Element Binding Protein 5 | binds to CRE as a homodimer or a heterodimer with c-Jun or CRE-BP1, and functions as a CRE-dependent trans-activator |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekos, C.; Muqaku, B.; Dekan, S.; Horvat, R.; Polterauer, S.; Gerner, C.; Aust, S.; Pils, D. NECTIN4 (PVRL4) as Putative Therapeutic Target for a Specific Subtype of High Grade Serous Ovarian Cancer—An Integrative Multi-Omics Approach. Cancers 2019, 11, 698. https://doi.org/10.3390/cancers11050698
Bekos C, Muqaku B, Dekan S, Horvat R, Polterauer S, Gerner C, Aust S, Pils D. NECTIN4 (PVRL4) as Putative Therapeutic Target for a Specific Subtype of High Grade Serous Ovarian Cancer—An Integrative Multi-Omics Approach. Cancers. 2019; 11(5):698. https://doi.org/10.3390/cancers11050698
Chicago/Turabian StyleBekos, Christine, Besnik Muqaku, Sabine Dekan, Reinhard Horvat, Stephan Polterauer, Christopher Gerner, Stefanie Aust, and Dietmar Pils. 2019. "NECTIN4 (PVRL4) as Putative Therapeutic Target for a Specific Subtype of High Grade Serous Ovarian Cancer—An Integrative Multi-Omics Approach" Cancers 11, no. 5: 698. https://doi.org/10.3390/cancers11050698