Altered Expression and Localization of Tumor Suppressive E3 Ubiquitin Ligase SMURF2 in Human Prostate and Breast Cancer

 ,
, {kind=link}
{kind=link}
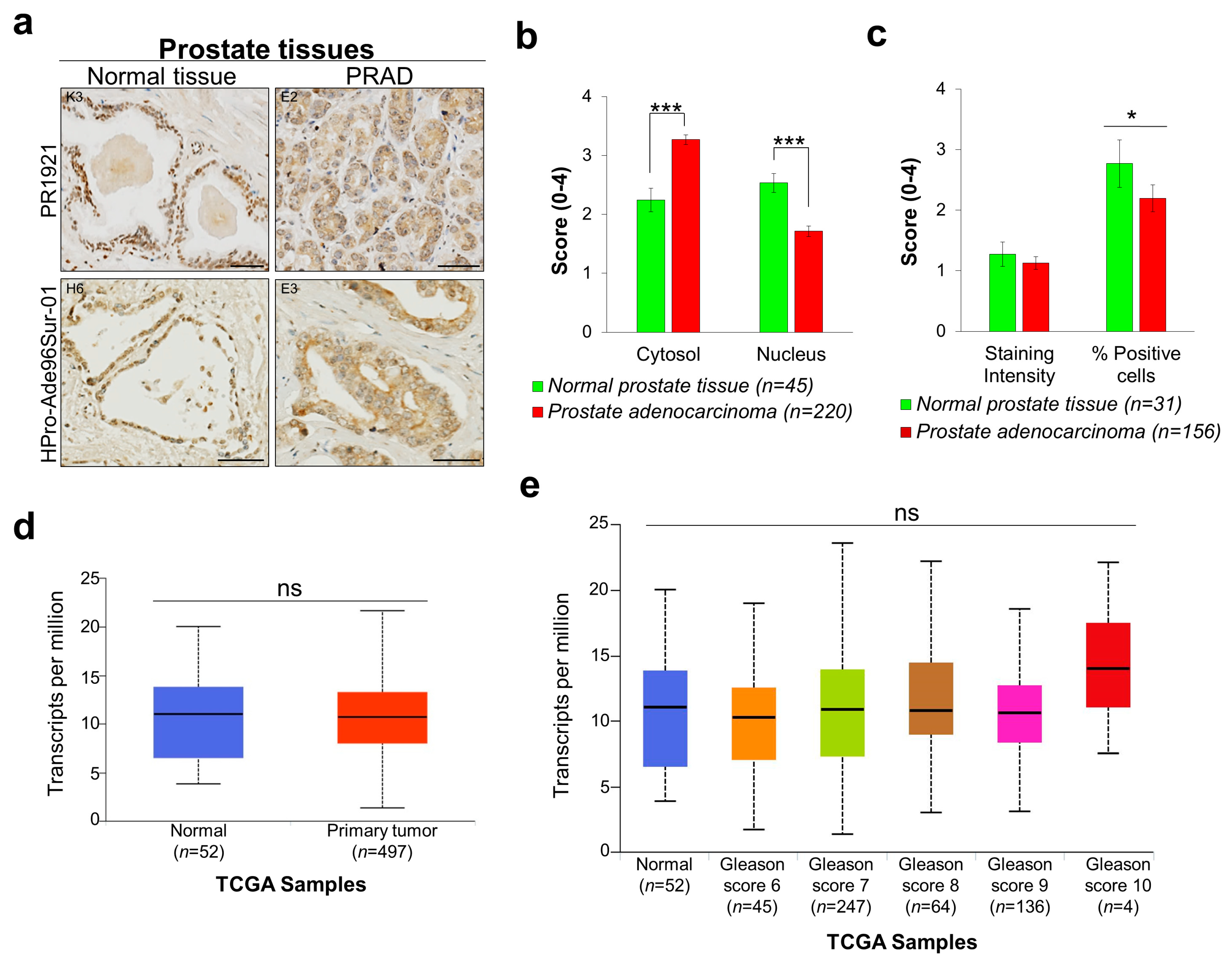{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Validation of the Specificity of Anti-SMURF2 Antibody in IHC Studies
2.2. SMURF2 is Widely Expressed in Different Types of Human Normal Tissues
2.3. Smurf2 Exhibits Altered Localization in Prostate Cancer
2.4. Aberrant Expression of SMURF2 in Human Breast Carcinoma (BRCA) Tissues and Cell Strains
2.5. SMURF2 Cytoplasmic Accumulation Does Not Involve the Nuclear Export Machinery
2.6. The Protein Stability of the Cytoplasmic and Nucelar Pools of SMURF2 is Vastly Different
2.7. Characterization of the SMURF2 Interactome and Identification of 14-3-3 Isotypes as SMURF2 Novel Binding Partners Which Could Potentially Regulate SMURF2 Stability and Localization
3. Discussion
4. Materials and Methods
4.1. Human Tissue Microarrays, Mouse Tissues, Immunohistochemistry (IHC) and Ethics Statement
4.2. SMURF2 Gene Expression Analysis
4.3. Cell Cultures and Reagents
4.4. Protein Extraction, Fractionation and Western Blot Analysis
4.5. Elucidation and Characterization of the SMURF2 Interactome
4.6. Co-Immunoprecipitation (co-IP)
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Jaynes, P.W.; Saei, A.; Iyengar, P.V.; Richard, J.L.C.; Eichhorn, P.J.A. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 456–483. [Google Scholar] [CrossRef]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef]
- Uchida, C.; Kitagawa, M. RING-, HECT-, and RBR-type E3 Ubiquitin Ligases: Involvement in Human Cancer. Curr. Cancer Drug Targets 2016, 16, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.; Tang, Y.; Yamashita, M.; Burkett, S.S.; Cheng, S.Y.; Zhang, Y.E. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat. Med. 2012, 18, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramkumar, C.; Kong, Y.; Cui, H.; Hao, S.; Jones, S.N.; Gerstein, R.M.; Zhang, H. Smurf2 regulates the senescence response and suppresses tumorigenesis in mice. Cancer Res. 2012, 72, 2714–2719. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Levy-Cohen, G.; Blank, M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Koganti, P.; Levy-Cohen, G.; Blank, M. Smurfs in Protein Homeostasis, Signaling, and Cancer. Front. Oncol. 2018, 8, 295. [Google Scholar] [CrossRef] [PubMed]
- Fierz, B.; Chatterjee, C.; McGinty, R.K.; Bar-Dagan, M.; Raleigh, D.P.; Muir, T.W. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 2011, 7, 113–119. [Google Scholar] [CrossRef]
- Minsky, N.; Shema, E.; Field, Y.; Schuster, M.; Segal, E.; Oren, M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat. Cell Biol. 2008, 10, 483–488. [Google Scholar] [CrossRef]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef]
- Tarcic, O.; Granit, R.Z.; Pateras, I.S.; Masury, H.; Maly, B.; Zwang, Y.; Yarden, Y.; Gorgoulis, V.G.; Pikarsky, E.; Ben-Porath, I.; et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. 2017, 24, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Emanuelli, A.; Borroni, A.P.; Apel-Sarid, L.; Shah, P.A.; Manikoth Ayyathan, D.; Koganti, P.; Levy-Cohen, G.; Blank, M. Smurf2-Mediated Stabilization of DNA Topoisomerase IIα Controls Genomic Integrity. Cancer Res. 2017, 77, 4217–4227. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Yamashita, M.; Coussens, N.P.; Tang, Y.; Wang, X.; Li, C.; Deng, C.X.; Cheng, S.Y.; Zhang, Y.E. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011, 30, 4777–4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, D.; Ahsan, A.; Helman, A.; Chen, G.; Hegde, A.; Gurjar, S.R.; Zhao, L.; Kiyokawa, H.; Beer, D.G.; Lawrence, T.S.; et al. Regulation of EGFR protein stability by the HECT-type ubiquitin ligase SMURF2. Neoplasia 2011, 13, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Huang, J.; Sampson, E.; Kim, K.O.; Zuscik, M.J.; O’Keefe, R.J.; Chen, D.; Rosier, R.N. Smurf2 induces degradation of GSK-3β and upregulates β-catenin in chondrocytes: A potential mechanism for Smurf2-induced degeneration of articular cartilage. Exp. Cell Res. 2009, 315, 2386–2398. [Google Scholar] [CrossRef]
- Kim, S.; Jho, E. The Protein Stability of Axin, a Negative Regulator of Wnt Signaling, Is Regulated by Smad Ubiquitination Regulatory Factor 2 (Smurf2). J. Biol. Chem. 2010, 285, 36420–36426. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Xie, P.; Liu, L.; Xing, G.; Chang, Z.; Yin, Y.; Tian, C.; He, F.; Zhang, L. Smad ubiquitylation regulatory factor 1/2 (Smurf1/2) promotes p53 degradation by stabilizing the E3 ligase MDM2. J. Biol. Chem. 2010, 285, 22818–22830. [Google Scholar] [CrossRef]
- Shukla, S.; Allam, U.S.; Ahsan, A.; Chen, G.; Krishnamurthy, P.M.; Marsh, K.; Rumschlag, M.; Shankar, S.; Whitehead, C.; Schipper, M.; et al. KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated β-TrCP1 degradation. Neoplasia 2014, 16, 115–128. [Google Scholar] [CrossRef]
- Borroni, A.P.; Emanuelli, A.; Shah, P.A.; Ilić, N.; Apel-Sarid, L.; Paolini, B.; Manikoth Ayyathan, D.; Koganti, P.; Levy-Cohen, G.; Blank, M. Smurf2 regulates stability and the autophagic-lysosomal turnover of lamin A and its disease-associated form progerin. Aging Cell 2018, 17, e12732. [Google Scholar] [CrossRef]
- Yu, Y.L.; Chou, R.H.; Shyu, W.C.; Hsieh, S.C.; Wu, C.S.; Chiang, S.Y.; Chang, W.J.; Chen, J.N.; Tseng, Y.J.; Lin, Y.H.; et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol. Med. 2013, 5, 531–547. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Nebreda, A.R.; Hanenberg, H.; Kinnebrew, G.H.; Ivan, M.; Yoder, M.C.; Filippi, M.D.; Broxmeyer, H.E.; Kapur, R. P38α/JNK signaling restrains erythropoiesis by suppressing Ezh2-mediated epigenetic silencing of Bim. Nat Commun. 2018, 9, 3518. [Google Scholar] [CrossRef]
- Du, J.X.; Hagos, E.G.; Nandan, M.O.; Bialkowska, A.B.; Yu, B.; Yang, V.W. The E3 ubiquitin ligase SMAD ubiquitination regulatory factor 2 negatively regulates Krüppel-like factor 5 protein. J. Biol. Chem. 2011, 286, 40354–40364. [Google Scholar] [CrossRef]
- Ramkumar, C.; Cui, H.; Kong, Y.; Jones, S.N.; Gerstein, R.M.; Zhang, H. Smurf2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nat. Commun. 2013, 4, 2598. [Google Scholar] [CrossRef]
- Jeong, H.M.; Lee, S.H.; Yum, J.; Yeo, C.Y.; Lee, K.Y. Smurf2 regulates the degradation of YY1. Biochim. Biophys. Acta 2014, 1843, 2005–2011. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Cui, H.; Zhang, H. Smurf2-mediated ubiquitination and degradation of Id1 regulates p16 expression during senescence. Aging Cell 2011, 10, 1038–1046. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Dong, L.; Wang, Y.; Liu, L.; Long, H.; Li, H.; Li, J.; Yang, X.; Liu, Z.; Duan, G.; et al. Reversible Regulation of SATB1 Ubiquitination by USP47 and SMURF2 Mediates Colon Cancer Cell Proliferation and Tumor Progression. Cancer Lett. 2019, 448, 40–51. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Wang, X.; Li, S. Protein mislocalization: Mechanisms, functions and clinical applications in cancer. Biochim. Biophys. Acta 2014, 1846, 13–25. [Google Scholar] [CrossRef] [Green Version]
- El-Tanani, M.; Dakir, E.-H.; Raynor, B.; Morgan, R. Mechanisms of Nuclear Export in Cancer and Resistance to Chemotherapy. Cancers (Basel) 2016, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, Y.; Papadopoulos, V. The role of the 14-3-3 protein family in health, disease, and drug development. Drug Discov. Today 2016, 21, 278–287. [Google Scholar] [CrossRef]
- Pennington, K.L.; Chan, T.Y.; Torres, M.P.; Andersen, J.L. The dynamic and stress-adaptive signaling hub of 14-3-3: Emerging mechanisms of regulation and context-dependent protein-protein interactions. Oncogene 2018, 37, 5587–5604. [Google Scholar] [CrossRef]
- Choi, Y.H.; Kim, Y.J.; Jeong, H.M.; Jin, Y.H.; Yeo, C.Y.; Lee, K.Y. Akt enhances Runx2 protein stability by regulating Smurf2 function during osteoblast differentiation. FEBS J. 2014, 281, 3656–3666. [Google Scholar] [CrossRef] [Green Version]
- Iezaki, T.; Fukasawa, K.; Horie, T.; Park, G.; Robinson, S.; Nakaya, M.; Fujita, H.; Onishi, Y.; Ozaki, K.; Kanayama, T.; et al. The MAPK Erk5 is necessary for proper skeletogenesis involving a Smurf-Smad-Sox9 molecular axis. Development 2018, 145, dev164004. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47(D1), D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Rodriguez, I.P.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Keen, J.C.; Moore, H.M. The Genotype-Tissue Expression (GTEx) Project: Linking Clinical Data with Molecular Analysis to Advance Personalized Medicine. J. Pers. Med. 2015, 5, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Hallstrom, B.M.; Fagerberg, L.; Ponten, F.; Kawaji, H.; Carninci, P.; Forrest, A.R.; Fantom, C.T.; Hayashizaki, Y.; Uhlen, M.; et al. Complementing tissue characterization by integrating transcriptome profiling from the Human Protein Atlas and from the FANTOM5 consortium. Nucleic Acids Res. 2015, 43, 6787–6798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, M.; Lerenthal, Y.; Mittelman, L.; Shiloh, Y. Condensin I recruitment and uneven chromatin condensation precede mitotic cell death in response to DNA damage. J. Cell Biol. 2006, 174, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Mi, H.; Poudel, S.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016, 44, D336–D342. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emanuelli, A.; Manikoth Ayyathan, D.; Koganti, P.; Shah, P.A.; Apel-Sarid, L.; Paolini, B.; Detroja, R.; Frenkel-Morgenstern, M.; Blank, M. Altered Expression and Localization of Tumor Suppressive E3 Ubiquitin Ligase SMURF2 in Human Prostate and Breast Cancer. Cancers 2019, 11, 556. https://doi.org/10.3390/cancers11040556
Emanuelli A, Manikoth Ayyathan D, Koganti P, Shah PA, Apel-Sarid L, Paolini B, Detroja R, Frenkel-Morgenstern M, Blank M. Altered Expression and Localization of Tumor Suppressive E3 Ubiquitin Ligase SMURF2 in Human Prostate and Breast Cancer. Cancers. 2019; 11(4):556. https://doi.org/10.3390/cancers11040556
Chicago/Turabian StyleEmanuelli, Andrea, Dhanoop Manikoth Ayyathan, Praveen Koganti, Pooja Anil Shah, Liat Apel-Sarid, Biagio Paolini, Rajesh Detroja, Milana Frenkel-Morgenstern, and Michael Blank. 2019. "Altered Expression and Localization of Tumor Suppressive E3 Ubiquitin Ligase SMURF2 in Human Prostate and Breast Cancer" Cancers 11, no. 4: 556. https://doi.org/10.3390/cancers11040556