Genetic Factors Associated with a Poor Outcome in Head and Neck Cancer Patients Receiving Definitive Chemoradiotherapy

 and
and
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics and Sequencing
2.2. “HNSCC Driver Genes”
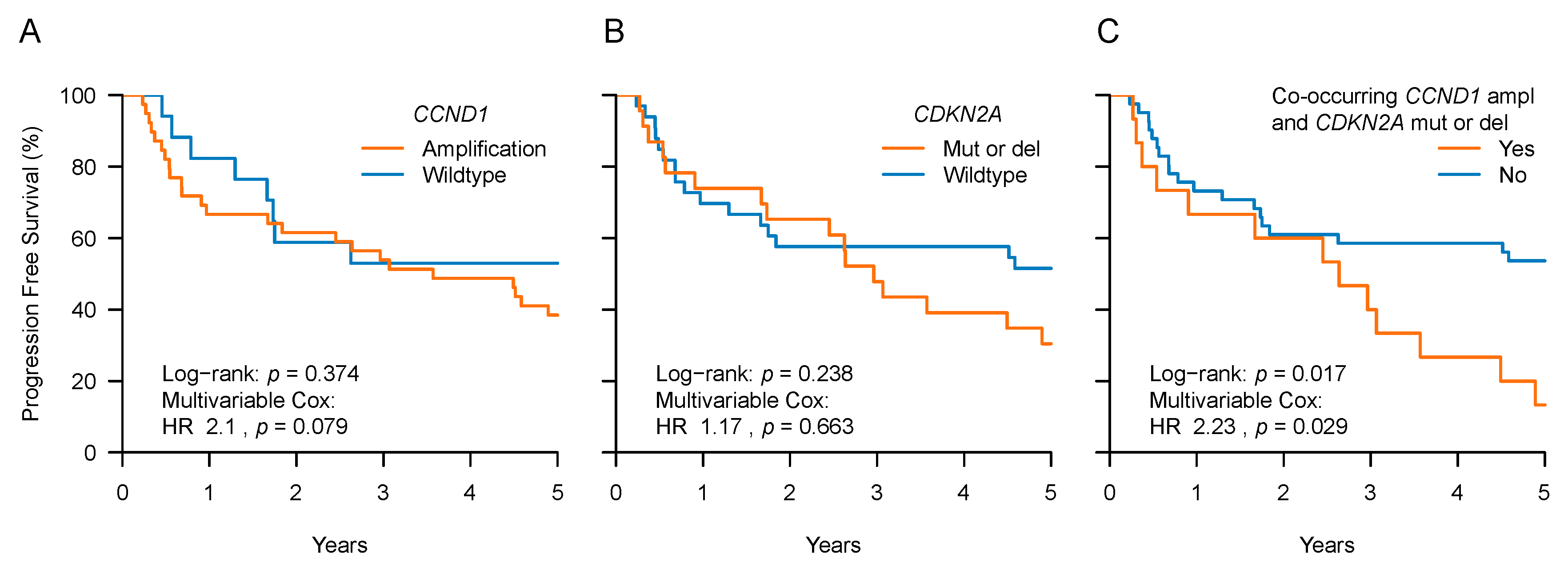
2.3. CCND1 and CDKN2A Expression and Patient Outcome
2.4. Mutational Burden and CIN
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Sequencing and Bioinformatics Protocol
4.3. “HNSCC Driver Genes”
4.4. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pignon, J.P.; Maître, A.L.; Maillard, E.; Bourhis, J. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): An update on 93 randomised trials and 17,346 patients. Radiother. Oncol. 2009, 92, 4–14. [Google Scholar] [CrossRef]
- Lydiatt, W.M.; Patel, S.G.; O’Sullivan, B.; Brandwein, M.S.; Ridge, J.A.; Migliacci, J.C.; Loomis, A.M.; Shah, J.P. Head and Neck cancers-major changes in the American Joint Committee on cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 122–137. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, N.G.; Li, Y.Y.; Jo, V.Y.; Rabinowits, G.; Lorch, J.H.; Tishler, R.B.; Margalit, D.N.; Schoenfeld, J.D.; Annino, D.J.; Goguen, L.A.; et al. Incorporation of Next-Generation Sequencing into Routine Clinical Care to Direct Treatment of Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2016, 22, 2939–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-J.; Liu, H.; Liao, C.-T.; Huang, P.-J.; Huang, Y.; Hsu, A.; Tang, P.; Chang, Y.-S.; Chen, H.-C.; Yen, T.-C. Ultra-deep targeted sequencing of advanced oral squamous cell carcinoma identifies a mutation-based prognostic gene signature. Oncotarget 2015, 6, 18066–18080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubot, C.; Bernard, V.; Sablin, M.P.; Vacher, S.; Chemlali, W.; Schnitzler, A.; Pierron, G.; Ait Rais, K.; Bessoltane, N.; Jeannot, E.; et al. Comprehensive genomic profiling of head and neck squamous cell carcinoma reveals FGFR1 amplifications and tumour genomic alterations burden as prognostic biomarkers of survival. Eur. J. Cancer 2018, 91, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ock, C.-Y.; Son, B.; Keam, B.; Lee, S.-Y.; Moon, J.; Kwak, H.; Kim, S.; Kim, T.M.; Jeon, Y.K.; Kwon, S.K.; et al. Identification of genomic mutations associated with clinical outcomes of induction chemotherapy in patients with head and neck squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 873–883. [Google Scholar] [CrossRef]
- Tinhofer, I.; Budach, V.; Saki, M.; Konschak, R.; Niehr, F.; Jöhrens, K.; Weichert, W.; Linge, A.; Lohaus, F.; Krause, M.; et al. Targeted next-generation sequencing of locally advanced squamous cell carcinomas of the head and neck reveals druggable targets for improving adjuvant chemoradiation. Eur. J. Cancer 2016, 57, 78–86. [Google Scholar] [CrossRef]
- Tinhofer, I.; Stenzinger, A.; Eder, T.; Konschak, R.; Niehr, F.; Endris, V.; Distel, L.; Hautmann, M.G.; Mandic, R.; Stromberger, C.; et al. Targeted next-generation sequencing identifies molecular subgroups in squamous cell carcinoma of the head and neck with distinct outcome after concurrent chemoradiation. Ann. Oncol. 2016, 27, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Vettore, A.L.; Ramnarayanan, K.; Poore, G.; Lim, K.; Ong, C.K.; Huang, K.K.; Leong, H.S.; Chong, F.T.; Lim, T.K.-H.; Lim, W.K.; et al. Mutational landscapes of tongue carcinoma reveal recurrent mutations in genes of therapeutic and prognostic relevance. Genome Med. 2015, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossen, D.M.; Verhagen, C.V.M.; Verheij, M.; Wessels, L.F.A.; Vens, C.; van den Brekel, M.W.M. Comparative genomic analysis of oral versus laryngeal and pharyngeal cancer. Oral Oncol. 2018, 81, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, C.V.M.; Vossen, D.M.; Borgmann, K.; Hageman, F.; Grénman, R.; Verwijs-Janssen, M.; Mout, L.; Kluin, R.J.C.; Nieuwland, M.; Severson, T.M.; et al. Fanconi anemia and homologous recombination gene variants are associated with functional DNA repair defects in vitro and poor outcome in patients with advanced head and neck squamous cell carcinoma. Oncotarget 2018, 9, 18198–18213. [Google Scholar] [CrossRef] [Green Version]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef]
- Zámborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Póti, Á.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2017, 36, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Peduzzi, P.; Concato, J.; Feinstein, A.R.; Holford, T.R. Importance of events per independent variable in proportional hazards regression analysis II. Accuracy and precision of regression estimates. J. Clin. Epidemiol. 1995, 48, 1503–1510. [Google Scholar] [CrossRef]
- Concato, J.; Peduzzi, P.; Holford, T.R.; Feinstein, A.R. Importance of events per independent variable in proportional hazards analysis. I. Background, goals, and general strategy. J. Clin. Epidemiol. 1995, 48, 1495–1501. [Google Scholar] [CrossRef]
- Shi, J.; Hua, X.; Zhu, B.; Ravichandran, S.; Wang, M.; Nguyen, C.; Brodie, S.A.; Palleschi, A.; Alloisio, M.; Pariscenti, G.; et al. Somatic Genomics and Clinical Features of Lung Adenocarcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002162. [Google Scholar] [CrossRef]
- Choma, D.; Daurès, J.P.; Quantin, X.; Pujol, J.L. Aneuploidy and prognosis of non-small-cell lung cancer: A meta-analysis of published data. Br. J. Cancer 2001, 85, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Lennartz, M.; Minner, S.; Brasch, S.; Wittmann, H.; Paterna, L.; Angermeier, K.; Öztürk, E.; Shihada, R.; Ruge, M.; Kluth, M.; et al. The Combination of DNA Ploidy Status and PTEN/6q15 Deletions Provides Strong and Independent Prognostic Information in Prostate Cancer. Clin. Cancer Res. 2016, 22, 2802–2811. [Google Scholar] [CrossRef]
- Walther, A.; Houlston, R.; Tomlinson, I. Association between chromosomal instability and prognosis in colorectal cancer: A meta-analysis. Gut 2008, 57, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Pierssens, D.D.C.G.; Borgemeester, M.C.; van der Heijden, S.J.H.; Peutz-Kootstra, C.J.; Ruland, A.M.; Haesevoets, A.M.; Kessler, P.A.W.H.; Kremer, B.; Speel, E.-J.M. Chromosome instability in tumor resection margins of primary OSCC is a predictor of local recurrence. Oral Oncol. 2017, 66, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Mroz, E.A.; Tward, A.D.; Pickering, C.R.; Myers, J.N.; Ferris, R.L.; Rocco, J.W. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 2013, 119, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Compton, D.A. Chromosomal instability and cancer: A complex relationship with therapeutic potential. J. Clin. Investig. 2012, 122, 1138–1143. [Google Scholar] [CrossRef]
- Thompson, L.L.; Jeusset, L.M.P.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers (Basel) 2017, 9, 151. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Wang, T.; Laajala, T.D.; Winner, K.K.; Bare, J.C.; Neto, E.C.; Khan, S.A.; Peddinti, G.; Airola, A.; Pahikkala, T.; et al. Prediction of overall survival for patients with metastatic castration-resistant prostate cancer: Development of a prognostic model through a crowdsourced challenge with open clinical trial data. Lancet Oncol. 2017, 18, 132–142. [Google Scholar] [CrossRef]
- Rothenberg, S.M.; Ellisen, L.W. The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Investig. 2012, 122, 1951–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okami, K.; Reed, A.L.; Cairns, P.; Koch, W.M.; Westra, W.H.; Wehage, S.; Jen, J.; Sidransky, D. Cyclin D1 amplification is independent of p16 inactivation in head and neck squamous cell carcinoma. Oncogene 1999, 18, 3541–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, J.; Aagaard, L.; Strauss, M.; Bartek, J. Oncogenic aberrations of p16INK4/CDKN2 and cyclin D1 cooperate to deregulate G1 control. Cancer Res. 1995, 55, 4818–4823. [Google Scholar]
- Jadayel, D.M.; Lukas, J.; Nacheva, E.; Bartkova, J.; Stranks, G.; De Schouwer, P.J.; Lens, D.; Bartek, J.; Dyer, M.J.; Kruger, A.R.; et al. Potential role for concurrent abnormalities of the cyclin D1, p16CDKN2 and p15CDKN2B genes in certain B cell non-Hodgkin’s lymphomas. Functional studies in a cell line (Granta 519). Leukemia 1997, 11, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bova, R.J.; Quinn, D.I.; Nankervis, J.S.; Cole, I.E.; Sheridan, B.F.; Jensen, M.J.; Morgan, G.J.; Hughes, C.J.; Sutherland, R.L. Cyclin D1 and p16INK4A expression predict reduced survival in carcinoma of the anterior tongue. Clin. Cancer Res. 1999, 5, 2810–2819. [Google Scholar] [PubMed]
- Jayasurya, R.; Francis, G.; Kannan, S.; Lekshminarayanan, K.; Nalinakumari, K.R.; Abraham, T.; Abraham, E.K.; Nair, M.K. p53, p16 and cyclin D1: Molecular determinants of radiotherapy treatment response in oral carcinoma. Int. J. Cancer 2004, 109, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namazie, A.; Alavi, S.; Olopade, O.I.; Pauletti, G.; Aghamohammadi, N.; Aghamohammadi, M.; Gornbein, J.A.; Calcaterra, T.C.; Slamon, D.J.; Wang, M.B.; et al. Cyclin D1 amplification and p16(MTS1/CDK4I) deletion correlate with poor prognosis in head and neck tumors. Laryngoscope 2002, 112, 472–481. [Google Scholar] [CrossRef]
- Akervall, J.; Bockmühl, U.; Petersen, I.; Yang, K.; Carey, T.E.; Kurnit, D.M. The gene ratios c-MYC:cyclin-dependent kinase (CDK)N2A and CCND1:CDKN2A correlate with poor prognosis in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2003, 9, 1750–1755. [Google Scholar] [PubMed]
- Suzuki, A.; Hayashida, M.; Ito, T.; Kawano, H.; Nakano, T.; Miura, M.; Akahane, K.; Shiraki, K. Survivin initiates cell cycle entry by the competitive interaction with Cdk4/p16(INK4a) and Cdk2/cyclin E complex activation. Oncogene 2000, 19, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Guan, Z.; Wang, C.; Feng, L.; Zheng, Y.; Caicedo, E.; Bearth, E.; Peng, J.-R.; Gaffney, P.; Ondrey, F.G. Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways. Clin. Cancer Res. 2010, 16, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, A.; Mascitti, M.; Rubini, C.; Bambini, F.; Giannatempo, G.; Lo Russo, L.; Sartini, D.; Emanuelli, M.; Procaccini, M.; Lo Muzio, L. Nuclear Survivin as a Prognostic Factor in Squamous-Cell Carcinoma of the Oral Cavity. Appl. Immunohistochem. Mol. Morphol. AIMM 2017, 25, 566–570. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef]
- Michel, L.; Ley, J.; Wildes, T.M.; Schaffer, A.; Robinson, A.; Chun, S.-E.; Lee, W.; Lewis, J.; Trinkaus, K.; Adkins, D. Phase I trial of palbociclib, a selective cyclin dependent kinase 4/6 inhibitor, in combination with cetuximab in patients with recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016, 58, 41–48. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [Green Version]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Riester, M.; Singh, A.P.; Brannon, A.R.; Yu, K.; Campbell, C.D.; Chiang, D.Y.; Morrissey, M.P. PureCN: Copy number calling and SNV classification using targeted short read sequencing. Source Code Biol. Med. 2016, 11, 13. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Shuster, J.J. Median follow-up in clinical trials. J. Clin. Oncol. 1991, 9, 191–192. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variable | All Patients | HPV-Negative | HPV-Positive | p-Value |
|---|---|---|---|---|
| N = 77 | N = 56 | N = 21 | ||
| N (%) | N (%) | N (%) | ||
| Gender | ||||
| Male | 22 (29) | 17 (30) | 5 (24) | 0.778 |
| Female | 55 (71) | 39 (70) | 16 (76) | |
| Tumor site | ||||
| Hypopharynx | 28 (36) | 28 (50) | 0 (0) | <0.001 |
| Oropharynx | 49 (64) | 28 (50) | 21 (100) | |
| Disease stage | ||||
| III | 11 (14) | 9 (16) | 2 (10) | 0.717 |
| IV | 66 (86) | 47 (84) | 19 (90) | |
| cT classification | ||||
| T1-T3 | 49 (64) | 33 (59) | 16 (76) | 0.192 |
| T4 | 28 (36) | 23 (41) | 5 (24) | |
| cN classification | ||||
| N0-N2a | 24 (31) | 20 (36) | 4 (19) | 0.181 |
| N2b-N3 | 53 (69) | 36 (64) | 17 (81) | |
| Smoker | ||||
| Former | 20 (26) | 12 (21) | 8 (38) | <0.001 |
| Never | 7 (9) | 1 (2) | 6 (29) | |
| Unknown | 2 (3) | 2 (4) | 0 (0) | |
| Yes | 48 (62) | 41 (73) | 7 (33) | |
| Alcoholic consumption | ||||
| Former alcoholic | 13 (17) | 13 (23) | 0 (0) | 0.005 |
| Never | 10 (13) | 4 (7) | 6 (29) | |
| Unknown | 2 (3) | 2 (4) | 0 (0) | |
| Yes | 52 (68) | 37 (66) | 15 (71) | |
| Median age at diagnosis (range) | 58 (27–78) | 58.5 (37–78) | 57 (27–77) | 0.175 |
| Median follow-up (95% CI) | 8.2 (6.7–10.5) | 9.3 (7.8–NA) | 6.6 (5.9–10.2) | 0.113 |
| Number of PFS events (%) | 47 (61) | 43 (77) | 4 (19) | <0.001 |
| Median PFS (95% CI) | 5.8 (4.5–8.9) | 4.5 (1.8–6.2) | NA | <0.001 |
| Gene | Mutation a | Prevalence | Survival Analysis b | ||||
|---|---|---|---|---|---|---|---|
| All Patients | HPV-Negative | HPV-Positive | p-Value | PFS | |||
| N = 77 | N = 56 | N = 21 | |||||
| N (%) | N (%) | N (%) | HR | p-Value | |||
| TP53 | SPM | 48 (62) | 47 (84) | 1 (5) | <0.001 | 0.87 | 0.753 |
| CCND1 | A | 39 (51) | 39 (70) | 0 (0) | <0.001 | 2.1 | 0.079 |
| CDKN2A | SPM or D | 23 (30) | 23 (41) | 0 (0) | <0.001 | 1.17 | 0.663 |
| PIK3CA | SPM | 16 (21) | 10 (18) | 6 (29) | 0.35 | 1.16 | 0.732 |
| Variable | Prevalence | Survival Analysis c | ||||
|---|---|---|---|---|---|---|
| All Patients | HPV-Negative | HPV-Positive | p-Value | PFS | ||
| N = 77 | N = 56 | N = 21 | ||||
| N (%) | N (%) | N (%) | HR | p-Value | ||
| Mutational burdena | ||||||
| Per tumor | 5 (0–16) | 5 (1–14) | 2 (0–16) | 0.006 | 1.08 | 0.197 |
| Mutational burden groupsb | ||||||
| High | 31 (40) | 26 (46) | 5 (24) | 0.116 | 1.87 | 0.089 |
| Low | 46 (60) | 30 (54) | 16 (76) | |||
| CINb | ||||||
| Positive | 32 (42) | 30 (54) | 2 (10) | <0.001 | 2.3 | 0.023 |
| Negative | 45 (58) | 26 (46) | 19 (90) | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vossen, D.M.; Verhagen, C.V.M.; van der Heijden, M.; Essers, P.B.M.; Bartelink, H.; Verheij, M.; Wessels, L.F.A.; van den Brekel, M.W.M.; Vens, C. Genetic Factors Associated with a Poor Outcome in Head and Neck Cancer Patients Receiving Definitive Chemoradiotherapy. Cancers 2019, 11, 445. https://doi.org/10.3390/cancers11040445
Vossen DM, Verhagen CVM, van der Heijden M, Essers PBM, Bartelink H, Verheij M, Wessels LFA, van den Brekel MWM, Vens C. Genetic Factors Associated with a Poor Outcome in Head and Neck Cancer Patients Receiving Definitive Chemoradiotherapy. Cancers. 2019; 11(4):445. https://doi.org/10.3390/cancers11040445
Chicago/Turabian StyleVossen, David M., Caroline V.M. Verhagen, Martijn van der Heijden, Paul B.M. Essers, Harry Bartelink, Marcel Verheij, Lodewyk F.A. Wessels, Michiel W.M. van den Brekel, and Conchita Vens. 2019. "Genetic Factors Associated with a Poor Outcome in Head and Neck Cancer Patients Receiving Definitive Chemoradiotherapy" Cancers 11, no. 4: 445. https://doi.org/10.3390/cancers11040445