Prognostic and Clinicopathological Significance of SERTAD1 in Various Types of Cancer Risk: A Systematic Review and Retrospective Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources and Search Strategy, Selection, Data Extraction and Quality Assessment
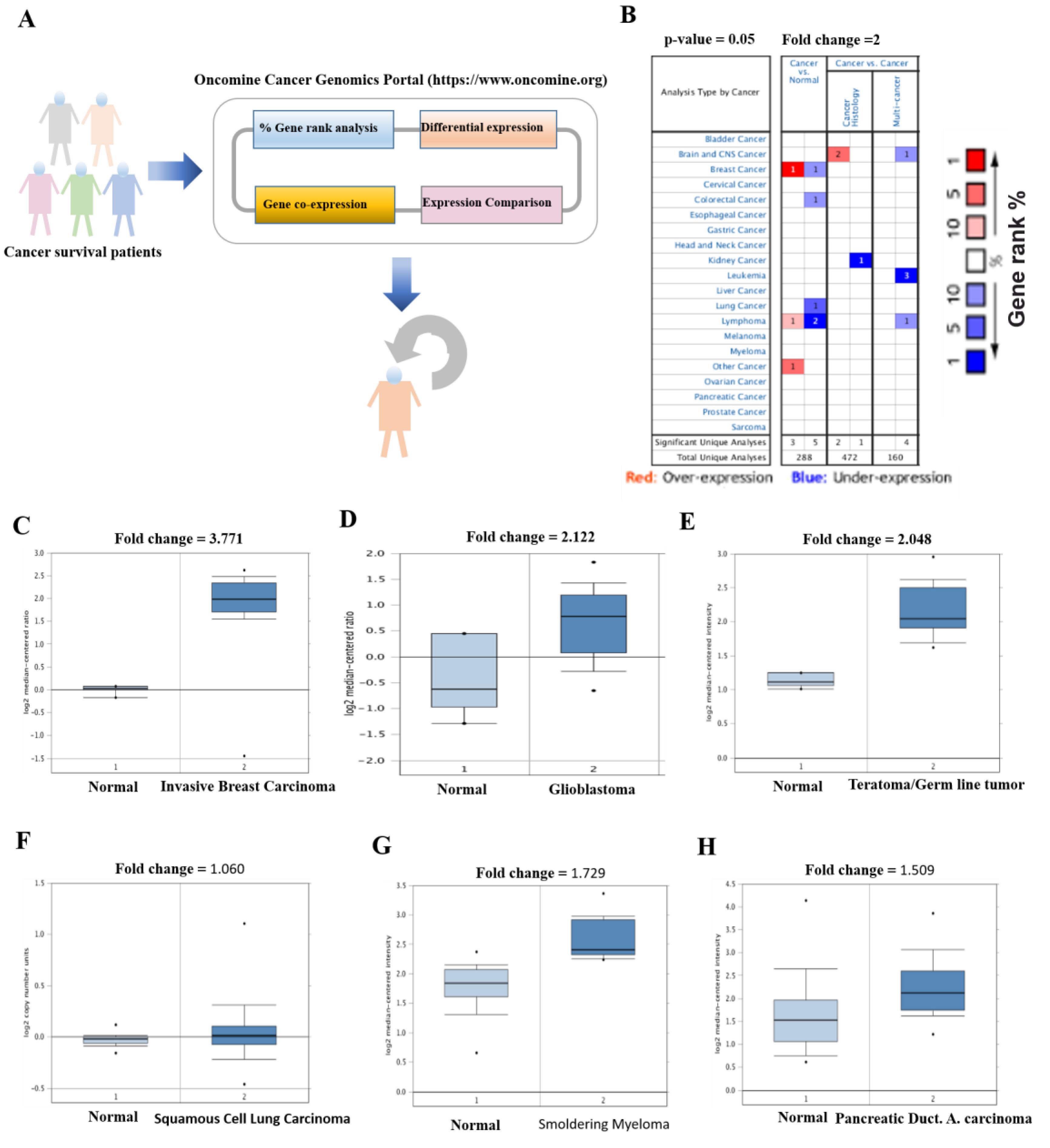
2.2. Transcriptomic and Differential Genes Expression Analysis
2.3. Patients Survival Prediction: Retrospective Analysis
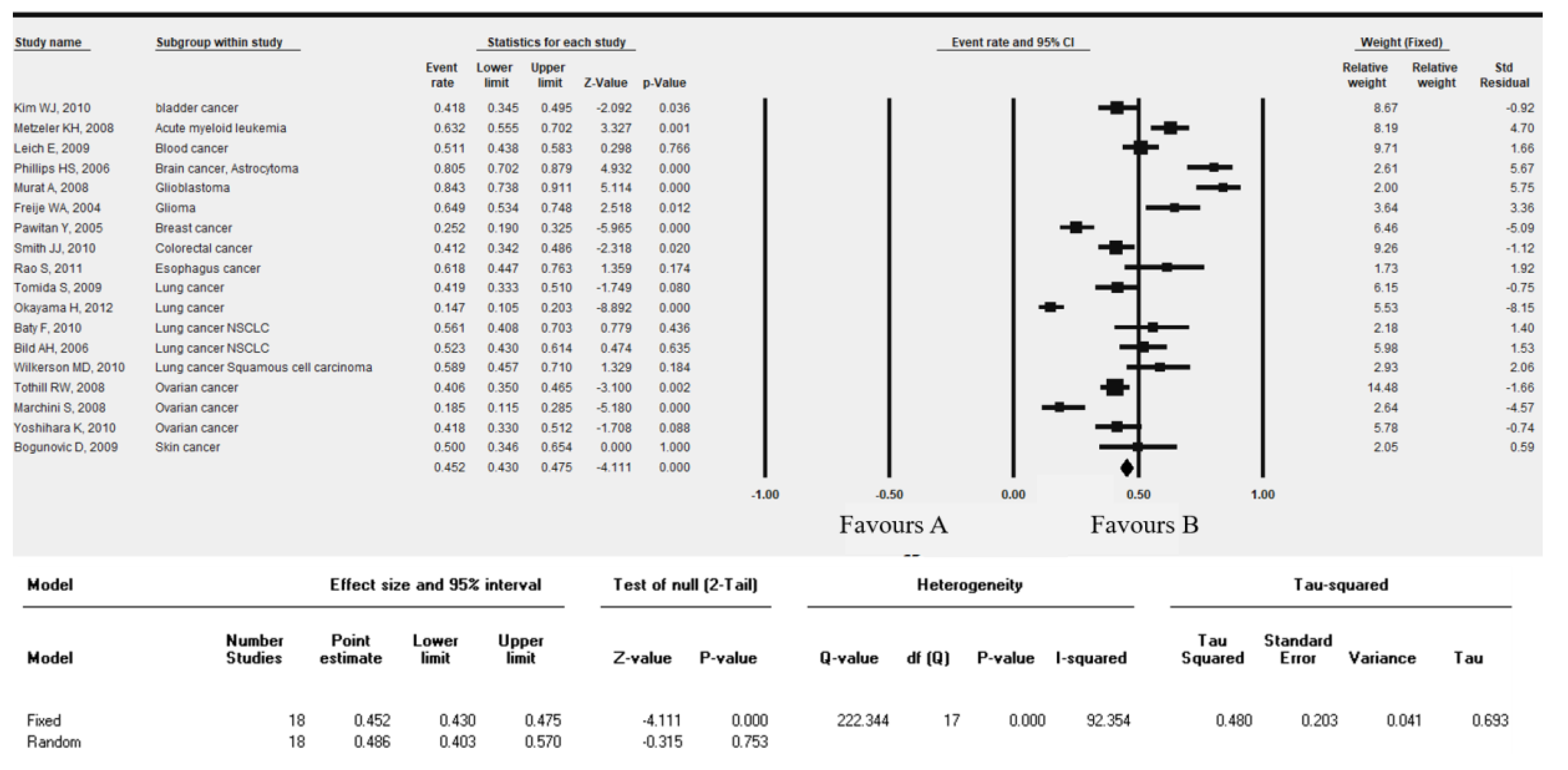
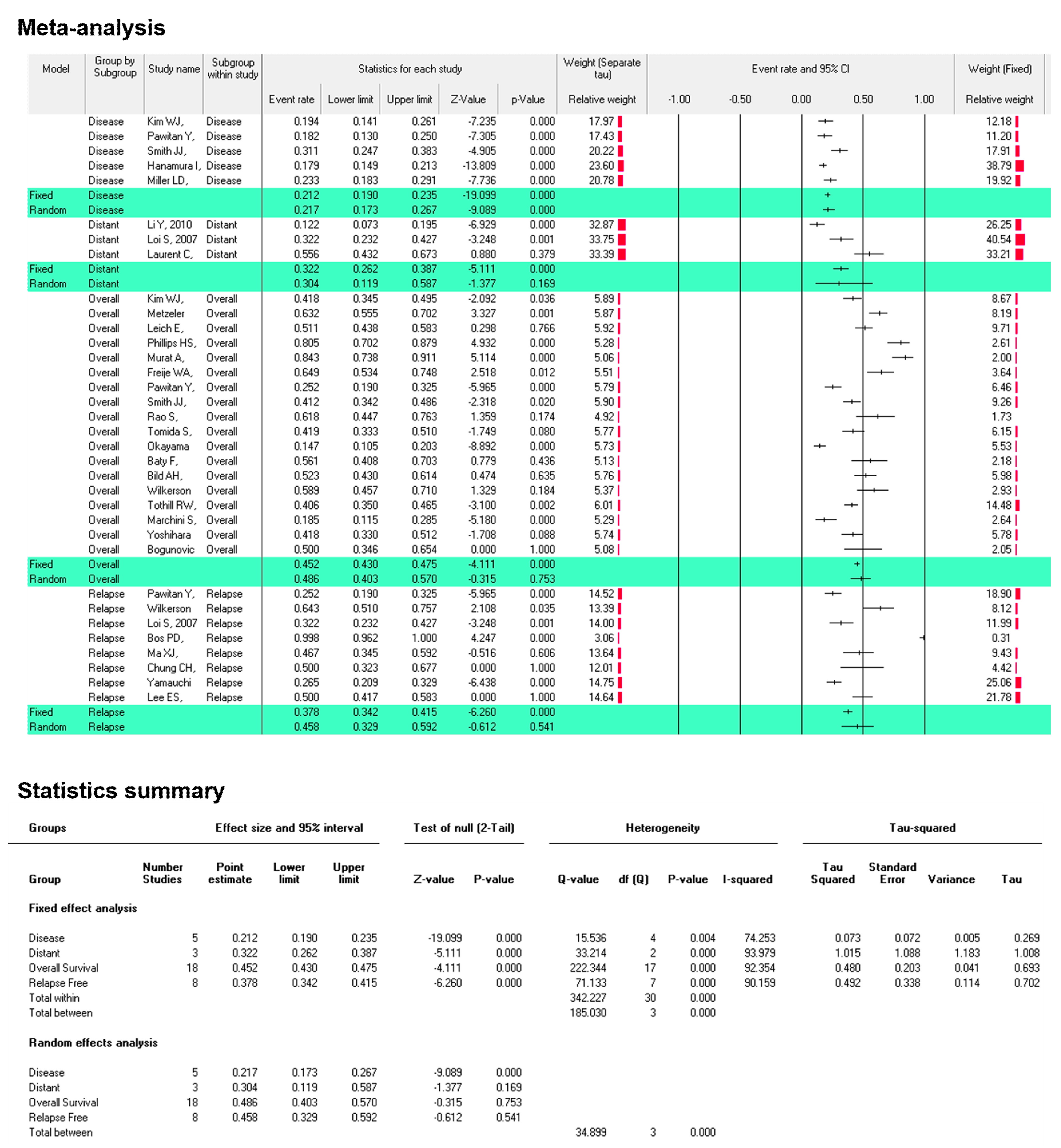
2.4. Genetic Alteration Study, Patient Prognosis and Clinical Outcome: Meta-Analysis
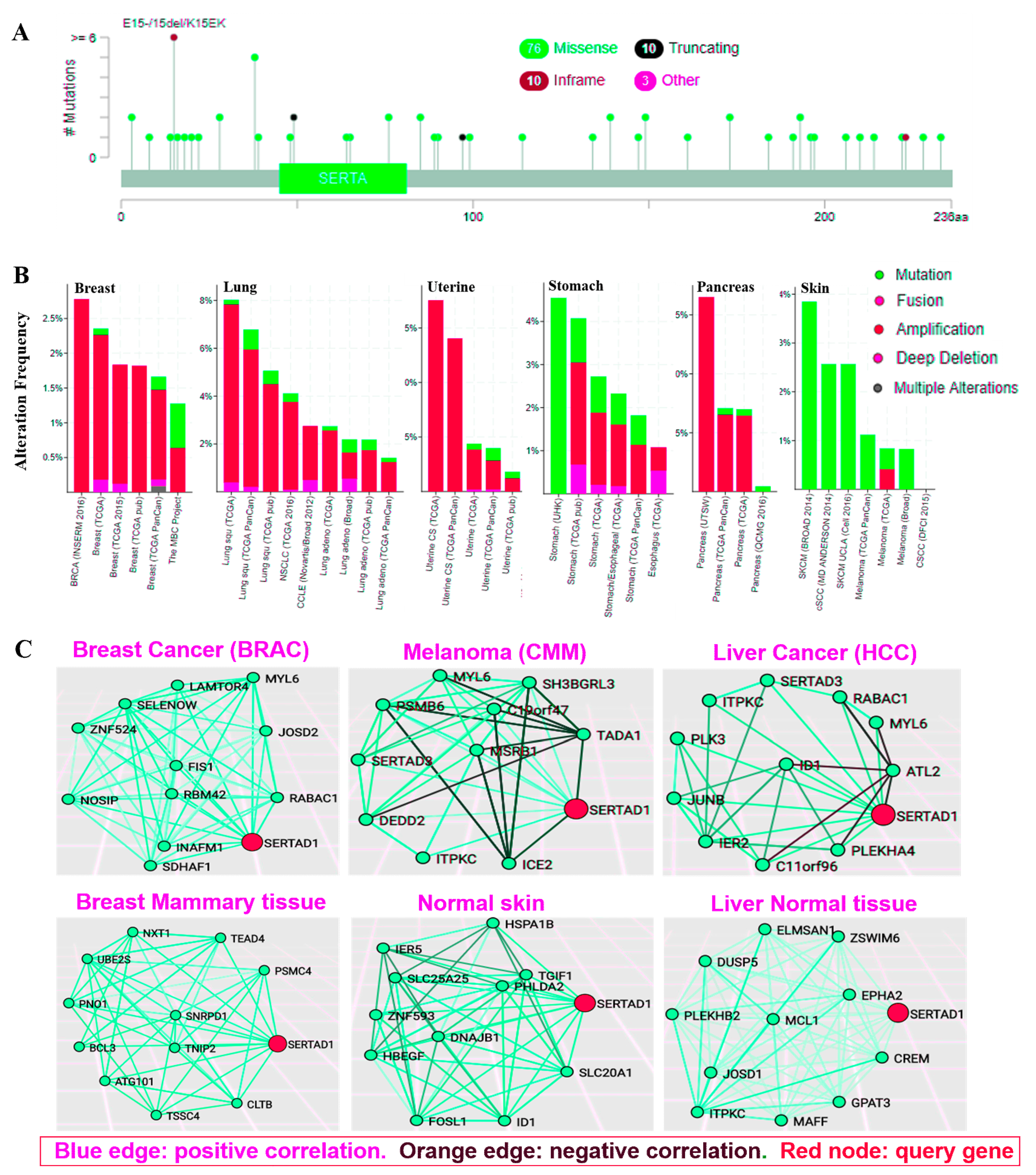
2.5. Tissue and Cancer Specific Biological Networks (TCSBN)
2.6. Protein-Protein Interaction, Gene Common Pathways and miRNAs Association with SERTAD1
2.7. Statistical Analysis
3. Results
3.1. Literature Search and Study Selection
3.2. Elevated Transcriptomic Levels of SERTAD1 Associated with Cancers
3.3. SERTAD1 Expression Define the Outcome of the Patient’s Survival in Cancers: A Meta-Analysis by KM-Plotter
3.4. SERTAD1 Expression Associated with Patient’s Survival: Meta-Analysis by ProgonoScan Database
3.5. Genetic Aberration in SERTAD1 Bestows More Invasive Cancers
3.6. The SERTAD1 Signature Prognosticate Better Outcome than Cases with Alteration: Meta-Analysis
3.7. SERTAD1 Cross Talks with the Certain Candidate Targets: As Bridge Avenue Model
4. Discussion
5. Concluding Remarks and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hsu, S.I.; Yang, C.M.; Sim, K.G.; Hentschel, D.M.; O’Leary, E.; Bonventre, J.V. TRIP-Br: A novel family of PHD zinc finger- and bromodomain-interacting proteins that regulate the transcriptional activity of E2F-1/DP-1. EMBO J. 2001, 20, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takhar, P.P.; Degenkolbe, R.; Koh, C.H.; Zimmermann, H.; Yang, C.M.; Guan Sim, K.; Hsu, S.I.; Bernard, H.U. The human papillomavirus type 11 and 16 E6 proteins modulate the cell-cycle regulator and transcription cofactor TRIP-Br1. Virology 2003, 317, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, Z.J.; Sim, K.G.; Cheong, J.K.; Yang, C.M.; Yap, C.S.; Hsu, S.I. Exploiting the TRIP-Br family of cell cycle regulatory proteins as chemotherapeutic drug targets in human cancer. Cancer Biol. Ther. 2007, 6, 712–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, Z.J.; Gunaratnam, L.; Cheong, J.K.; Lai, L.Y.; Hsiao, L.L.; O’Leary, E.; Sun, X.; Salto-Tellez, M.; Bonventre, J.V.; Hsu, S.I. Identification of PP2A as a novel interactor and regulator of TRIP-Br1. Cell Signal. 2009, 21, 34–42. [Google Scholar] [CrossRef]
- Jung, S.; Li, C.; Duan, J.; Lee, S.; Kim, K.; Park, Y.; Yang, Y.; Kim, K.I.; Lim, J.S.; Cheon, C.I.; et al. TRIP-Br1 oncoprotein inhibits autophagy, apoptosis, and necroptosis under nutrient/serum-deprived condition. Oncotarget 2015, 6, 29060–29075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockstaele, L.; Coulonval, K.; Kooken, H.; Paternot, S.; Roger, P.P. Regulation of CDK4. Cell Div. 2006, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Sim, K.G.; Cheong, J.K.; Hsu, S.I. The TRIP-Br family of transcriptional regulators is essential for the execution of cyclin E-mediated cell cycle progression. Cell Cycle 2006, 5, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Pantoja, C.; Gonzalez-Rodriguez, A.; Martin, N.; Flores, J.M.; Valverde, A.M.; Hara, E.; Serrano, M. Normal proliferation and tumorigenesis but impaired pancreatic function in mice lacking the cell cycle regulator sei1. PLoS ONE 2010, 5, e8744. [Google Scholar] [CrossRef]
- Darwish, H.; Cho, J.M.; Loignon, M.; Alaoui-Jamali, M.A. Overexpression of SERTAD3, a putative oncogene located within the 19q13 amplicon, induces E2F activity and promotes tumor growth. Oncogene 2007, 26, 4319–4328. [Google Scholar] [CrossRef] [Green Version]
- Thompson, F.H.; Nelson, M.A.; Trent, J.M.; Guan, X.Y.; Liu, Y.; Yang, J.M.; Emerson, J.; Adair, L.; Wymer, J.; Balfour, C.; et al. Amplification of 19q13.1-q13.2 sequences in ovarian cancer. G-band, FISH, and molecular studies. Cancer Genet. Cytogenet. 1996, 87, 55–62. [Google Scholar] [CrossRef]
- Tang, D.J.; Hu, L.; Xie, D.; Wu, Q.L.; Fang, Y.; Zeng, Y.; Sham, J.S.; Guan, X.Y. Oncogenic transformation by SEI-1 is associated with chromosomal instability. Cancer Res. 2005, 65, 6504–6508. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Shin, J.S.; Lee, Y.M.; Kim, D.G.; Lee, S.Y.; Yoon, D.H.; Jung, S.Y.; Hwang, J.J.; Lee, S.J.; Cho, D.H.; et al. p34 (SEI-1) inhibits ROS-induced cell death through suppression of ASK1. Cancer Biol. Ther. 2011, 12, 421–426. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Liu, J.; Bao, Y.; Wang, L.; Yu, Y.; Wang, L.; Wu, D.; Liu, C.; Wang, N.; Wang, F.; et al. SEI1 induces genomic instability by inhibiting DNA damage response in ovarian cancer. Cancer Lett. 2017, 385, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Kusano, S.; Shiimura, Y.; Eizuru, Y. I-mfa domain proteins specifically interact with SERTA domain proteins and repress their transactivating functions. Biochimie 2011, 93, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Eschenhagen, T.; Geisslinger, G.; Scholich, K. Capturing adenylyl cyclases as potential drug targets. Nat. Rev. Drug Discov. 2009, 8, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Yu, X.; Liu, Z.; Sun, Y.; Chen, X.; Yang, X.; Li, X.; Lam, W.K.; Duan, Y.; Cao, X.; et al. The complex of TRIP-Br1 and XIAP ubiquitinates and degrades multiple adenylyl cyclase isoforms. Elife 2017, 6, 28021. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Kim, C.J.; Park, W.S.; Shin, J.S.; Lee, S.D.; Ko, S.G.; Jung, S.I.; Park, I.C.; An, S.K.; Lee, W.K.; et al. p34SEI-1 inhibits apoptosis through the stabilization of the X-linked inhibitor of apoptosis protein: p34SEI-1 as a novel target for anti-breast cancer strategies. Cancer Res. 2009, 69, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Nakamura, T.; Ohtani, N.; Hampson, L.; Hampson, I.N.; Shimamoto, A.; Furuichi, Y.; Okumura, K.; Niwa, S.; Taya, Y.; Hara, E. Regulation of CDK4 activity by a novel CDK4-binding protein, SERTAD1. Genes Dev. 1999, 13, 3027–3033. [Google Scholar] [CrossRef]
- Li, J.; Muscarella, P.; Joo, S.H.; Knobloch, T.J.; Melvin, W.S.; Weghorst, C.M.; Tsai, M.D. Dissection of CDK4-binding and transactivation activities of p34(SEI-1) and comparison between functions of p34(SEI-1) and p16(INK4A). Biochemistry 2005, 44, 13246–13256. [Google Scholar] [CrossRef]
- Watanabe-Fukunaga, R.; Iida, S.; Shimizu, Y.; Nagata, S.; Fukunaga, R. SEI family of nuclear factors regulates p53-dependent transcriptional activation. Genes Cells 2005, 10, 851–860. [Google Scholar] [CrossRef]
- Semczuk, A.; Jakowicki, J.A. Alterations of pRb1-cyclin D1-cdk4/6-p16(INK4A) pathway in endometrial carcinogenesis. Cancer Lett. 2004, 203, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Liu, J.; You, J.; Wu, D.; Yu, Y.; Liu, C.; Wang, L.; Wang, F.; Xu, L.; Wang, L.; et al. Met promotes the formation of double minute chromosomes induced by Sei-1 in NIH-3T3 murine fibroblasts. Oncotarget 2016, 7, 56664–56675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Liu, C.; Wang, X.; Wang, F.; Wang, L.; Xu, L.; Ma, J.; Gao, Y.; Bao, Y.; Wang, F.; et al. Sei-1 promotes double minute chromosomes formation through activation of the PI3K/Akt/BRCA1-Abraxas pathway and induces double-strand breaks in NIH-3T3 fibroblasts. Cell Death Dis. 2018, 9, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Li, C.; Jung, S.; Yang, Y.; Kim, K.I.; Lim, J.S.; Cheon, C.I.; Lee, M.S. Inhibitory role of TRIP-Br1 oncoprotein in hypoxia-induced apoptosis in breast cancer cell lines. Int. J. Oncol. 2016, 48, 2639–2646. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Liu, J.; Lin, B.; Wang, Z.; Sun, J.; Huang, G. A Meta-Analysis of Interleukin-8 -251 Promoter Polymorphism Associated with Gastric Cancer Risk. PLoS ONE 2012, 7, e28083. [Google Scholar] [CrossRef] [PubMed]
- Stang, A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur. J. Epidemiol. 2010, 25, 603–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Zhang, C.; Arif, M.; Liu, Z.; Benfeitas, R.; Bidkhori, G.; Deshmukh, S.; Al Shobky, M.; Lovric, A.; Boren, J.; et al. TCSBN: A database of tissue and cancer specific biological networks. Nucleic Acids Res. 2018, 46, D595–D600. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Montojo, J.; Zuberi, K.; Rodriguez, H.; Bader, G.D.; Morris, Q. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Research 2014, 3, 153. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Gyorffy, A.; Tulassay, Z. The problem of multiple testing and solutions for genome-wide studies. Orv. Hetil. 2005, 146, 559–563. [Google Scholar]
- DerSimonian, R.; Laird, N. Meta-analysis in clinical trials. Control Clin. Trials 1986, 7, 177–188. [Google Scholar] [CrossRef]
- Hardy, R.J.; Thompson, S.G. Detecting and describing heterogeneity in meta-analysis. Stat. Med. 1998, 17, 841–856. [Google Scholar] [CrossRef]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Bredel, M.; Bredel, C.; Juric, D.; Harsh, G.R.; Vogel, H.; Recht, L.D.; Sikic, B.I. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 2005, 65, 8679–8689. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugières, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Korkola, J.E.; Houldsworth, J.; Chadalavada, R.S.; Olshen, A.B.; Dobrzynski, D.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006, 66, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Brune, V.; Tiacci, E.; Pfeil, I.; Döring, C.; Eckerle, S.; van Noesel, C.J.; Klapper, W.; Falini, B.; von Heydebreck, A.; Metzler, D.; et al. Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J. Exp. Med. 2008, 205, 2251–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haferlach, T.; Kohlmann, A.; Wieczorek, L.; Basso, G.; Kronnie, G.T.; Béné, M.C.; De Vos, J.; Hernández, J.M.; Hofmann, W.K.; Mills, K.I.; et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010, 28, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Aerts, J.; den Hamer, B.; van Ijcken, W.; den Bakker, M.; Riegman, P.; van der Leest, C.; van der Spek, P.; Foekens, J.A.; Hoogsteden, H.C.; et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007, 109, 1692–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef]
- Badea, L.; Herlea, V.; Dima, S.O.; Dumitrascu, T.; Popescu, I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 2008, 55, 2016–2027. [Google Scholar]
- Shyamsundar, R.; Kim, Y.H.; Higgins, J.P.; Montgomery, K.; Jorden, M.; Sethuraman, A.; van de Rijn, M.; Botstein, D.; Brown, P.O.; Pollack, J.R.; et al. A DNA microarray survey of gene expression in normal human tissues. Genome Biol. 2005, 6, R22. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H. Cancer Genome Atlas Research Network: Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Sim, K.G.; Zang, Z.; Yang, C.M.; Bonventre, J.V.; Hsu, S.I. TRIP-Br links E2F to novel functions in the regulation of cyclin E expression during cell cycle progression and the maintenance of genomic stability. Cell Cycle 2004, 3, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Li, C.; Jeong, D.; Lee, S.; Ohk, J.; Park, M.; Han, S.; Duan, J.; Kim, C.; Yang, Y.; et al. Oncogenic function of p34SEI1 via NEDD4-1-mediated PTEN ubiquitination/degradation and activation of the PI3K/AKT pathway. Int. J. Oncol. 2013, 43, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.C.; Zhang, Y.; Iyirhiaro, G.; Willett, R.T.; Rodriguez Gonzalez, Y.; Slack, R.S.; Park, D.S.; Greene, L.A. Sertad1 plays an essential role in developmental and pathological neuron death. J. Neurosci. 2010, 30, 3973–3982. [Google Scholar] [CrossRef]
- Li, Y.; Nie, C.J.; Hu, L.; Qin, Y.; Liu, H.B.; Zeng, T.T.; Chen, L.; Fu, L.; Deng, W.; Chen, S.P.; et al. Characterization of a novel mechanism of genomic instability involving the SEI1/SET/NM23H1 pathway in esophageal cancers. Cancer Res. 2010, 70, 5695–5705. [Google Scholar] [CrossRef]
- Glinsky, G.V.; Glinskii, A.B.; Stephenson, A.J.; Hoffman, R.M.; Gerald, W.L. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Investig. 2004, 113, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Lánczky, A.; Nagy, Á.; Bottai, G.; Munkácsy, G.; Szabó, A.; Santarpia, L.; Győrffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Meng, S.; Tripathy, D.; Shete, S.; Ashfaq, R.; Haley, B.; Perkins, S.; Beitsch, P.; Khan, A.; Euhus, D.; Osborne, C.; et al. HER-2 gene amplification can be acquired as breast cancer progresses. Proc. Natl. Acad. Sci. USA 2004, 101, 9393–9398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oba, K.; Paoletti, X.; Alberts, S.; Bang, Y.J.; Benedetti, J.; Bleiberg, H.; Catalano, P.; Lordick, F.; Michiels, S.; Morita, S.; et al. Disease-free survival as a surrogate for overall survival in adjuvant trials of gastric cancer: A meta-analysis. J. Natl. Cancer Inst. 2013, 105, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Calgaro, S.; Boube, M.; Cribbs, D.L.; Bourbon, H.M. The Drosophila gene taranis encodes a novel trithorax group member potentially linked to the cell cycle regulatory apparatus. Genetics 2002, 160, 547–560. [Google Scholar] [PubMed]
- Tiscornia, G.; Izpisúa Belmonte, J.C. MicroRNAs in embryonic stem cell function and fate. Genes Dev. 2010, 24, 2732–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Cancer Subtype | Fold Change | Rank % | Sample Size | Measured Genes | References |
|---|---|---|---|---|---|---|
| Breast cancer | Invasive breast carcinoma | 3.771 | 29 | 59 | 19,189 | [40] |
| Brain | Glioblastoma | 2.122 | 199 | 54 | 14,836 | [41] |
| Brain | Ganglioneuroma | 3.576 | 245 | 64 | 19,574 | [42] |
| Teratoma | Germ cell tumors | 2.048 | 584 | 107 | 17,779 | [43] |
| Blood | Lymphoma | 2.192 | 1213 | 67 | 19,574 | [44] |
| Blood | Leukemia | 1.512 | 3409 | 2,096 | 19,574 | [45] |
| Lung | Lung Adenocarcinoma | 1.951 | 270 | 156 | 19,574 | [46] |
| Blood | Smoldering Myeloma | 1.729 | 1486 | 78 | 19,574 | [47] |
| Lung | Squamous Cell Lung Carcinoma | 1.060 | 5041 | 291 | 18,823 | [48] |
| Pancreases | Pancreatic Ductal Adenocarcinoma | 1.509 | 5174 | 78 | 19,574 | [49] |
| Non-cancerous | Normal human tissues | 3.200 | 1926 | 123 | 14,430 | [50] |
| Dataset | Cancer Type | Endpoint | Probe ID | N | COX p-Value | HR (95%CI) |
|---|---|---|---|---|---|---|
| GSE13507 | Bladder cancer | Overall Survival | ILMN_1794017 | 165 | 0.251762 | 1.22 |
| GSE13507 | Bladder cancer | Disease Specific Survival | ILMN_1794017 | 165 | 0.242189 | 1.37 |
| GSE12417-GPL97 | Blood cancer | Overall Survival | 223394_at | 163 | 0.893883 | 1.03 |
| GSE12417-GPL570 | Blood cancer | Overall Survival | 223394_at | 79 | 0.668121 | 1.11 |
| GSE16131-GPL97 | Blood cancer | Overall Survival | 223394_at | 180 | 0.549863 | 1.21 |
| GSE2658 | Blood cancer | Disease Specific Survival | 223394_at | 559 | 0.185263 | 0.70 |
| GSE4271-GPL97 | Brain cancer | Overall Survival | 223394_at | 77 | 0.144382 | 1.39 |
| GSE7696 | Brain cancer | Overall Survival | 223394_at | 70 | 0.563036 | 0.84 |
| GSE4412-GPL97 | Brain cancer | Overall Survival | 223394_at | 74 | 0.149164 | 1.66 |
| GSE16581 | Brain cancer | Overall Survival | 223394_at | 67 | 0.223619 | 0.26 |
| GSE19615 | Breast cancer | Distant Metastasis Free Survival | 223394_at | 115 | 0.124646 | 0.22 |
| GSE12276 | Breast cancer | Relapse Free Survival | 223394_at | 204 | 0.171138 | 0.73 |
| GSE6532-GPL570 | Breast cancer | Relapse Free Survival | 223394_at | 87 | 0.494388 | 0.72 |
| GSE6532-GPL570 | Breast cancer | Distant Metastasis Free Survival | 223394_at | 87 | 0.494388 | 0.72 |
| GSE9195 | Breast cancer | Relapse Free Survival | 223394_at | 77 | 0.115978 | 0.33 |
| GSE9195 | Breast cancer | Distant Metastasis Free Survival | 223394_at | 77 | 0.029313 | 0.18 |
| GSE1378 | Breast cancer | Relapse Free Survival | 7818 | 60 | 0.980828 | 1.01 |
| GSE1379 | Breast cancer | Relapse Free Survival | 7818 | 60 | 0.400311 | 1.37 |
| GSE1456-GPL97 | Breast cancer | Disease Specific Survival | 223394_at | 159 | 0.864582 | 1.10 |
| GSE1456-GPL97 | Breast cancer | Overall Survival | 223394_at | 159 | 0.728309 | 0.84 |
| GSE1456-GPL97 | Breast cancer | Relapse Free Survival | 223394_at | 159 | 0.778333 | 1.15 |
| GSE3494-GPL97 | Breast cancer | Disease Specific Survival | 223394_at | 236 | 0.228813 | 1.91 |
| GSE4922-GPL97 | Breast cancer | Disease Free Survival | 223394_at | 249 | 0.276618 | 1.59 |
| GSE17536 | Colorectal cancer | Overall Survival | 223394_at | 177 | 0.861646 | 1.07 |
| GSE17536 | Colorectal cancer | Disease Specific Survival | 223394_at | 177 | 0.522633 | 1.31 |
| GSE17536 | Colorectal cancer | Disease Free Survival | 223394_at | 145 | 0.083306 | 2.36 |
| GSE14333 | Colorectal cancer | Disease Free Survival | 223394_at | 226 | 0.109716 | 1.51 |
| GSE17537 | Colorectal cancer | Overall Survival | 223394_at | 55 | 0.940023 | 1.04 |
| GSE17537 | Colorectal cancer | Disease Free Survival | 223394_at | 55 | 0.715296 | 0.80 |
| GSE17537 | Colorectal cancer | Disease Specific Survival | 223394_at | 49 | 0.781497 | 0.81 |
| GSE11595 | Esophagus cancer | Overall Survival | 756322 | 34 | 0.960091 | 1.02 |
| GSE22138 | Eye cancer | Distant Metastasis Free Survival | 223394_at | 63 | 0.743321 | 1.08 |
| GSE2837 | Head and neck cancer | Relapse Free Survival | g12803668_3p_at | 28 | 0.217278 | 1.60 |
| GSE13213 | Lung cancer | Overall Survival | A_23_P218463 | 117 | 0.598235 | 0.86 |
| GSE31210 | Lung cancer | Relapse Free Survival | 223394_at | 204 | 0.902867 | 1.05 |
| GSE31210 | Lung cancer | Overall Survival | 223394_at | 204 | 0.191555 | 1.89 |
| GSE11117 | Lung cancer | Overall Survival | H200004691 | 41 | 0.125025 | 1.49 |
| GSE3141 | Lung cancer | Overall Survival | 223394_at | 111 | 0.084274 | 1.48 |
| GSE8894 | Lung cancer | Relapse Free Survival | 223394_at | 138 | 0.214296 | 1.17 |
| GSE17710 | Lung cancer | Relapse Free Survival | 25284 | 56 | 0.804400 | 1.05 |
| GSE17710 | Lung cancer | Relapse Free Survival | 23819 | 56 | 0.892106 | 1.03 |
| GSE17710 | Lung cancer | Overall Survival | 25284 | 56 | 0.742781 | 1.08 |
| GSE17710 | Lung cancer | Overall Survival | 23819 | 56 | 0.797209 | 1.06 |
| GSE9891 | Ovarian cancer | Overall Survival | 223394_at | 278 | 0.097897 | 1.37 |
| GSE8841 | Ovarian cancer | Overall Survival | 12603 | 81 | 0.258771 | 1.69 |
| GSE17260 | Ovarian cancer | Progression Free Survival | A_23_P218463 | 110 | 0.419954 | 1.14 |
| GSE17260 | Ovarian cancer | Overall Survival | A_23_P218463 | 110 | 0.384906 | 1.19 |
| GSE19234 | Skin cancer | Overall Survival | 223394_at | 38 | 0.429824 | 1.53 |
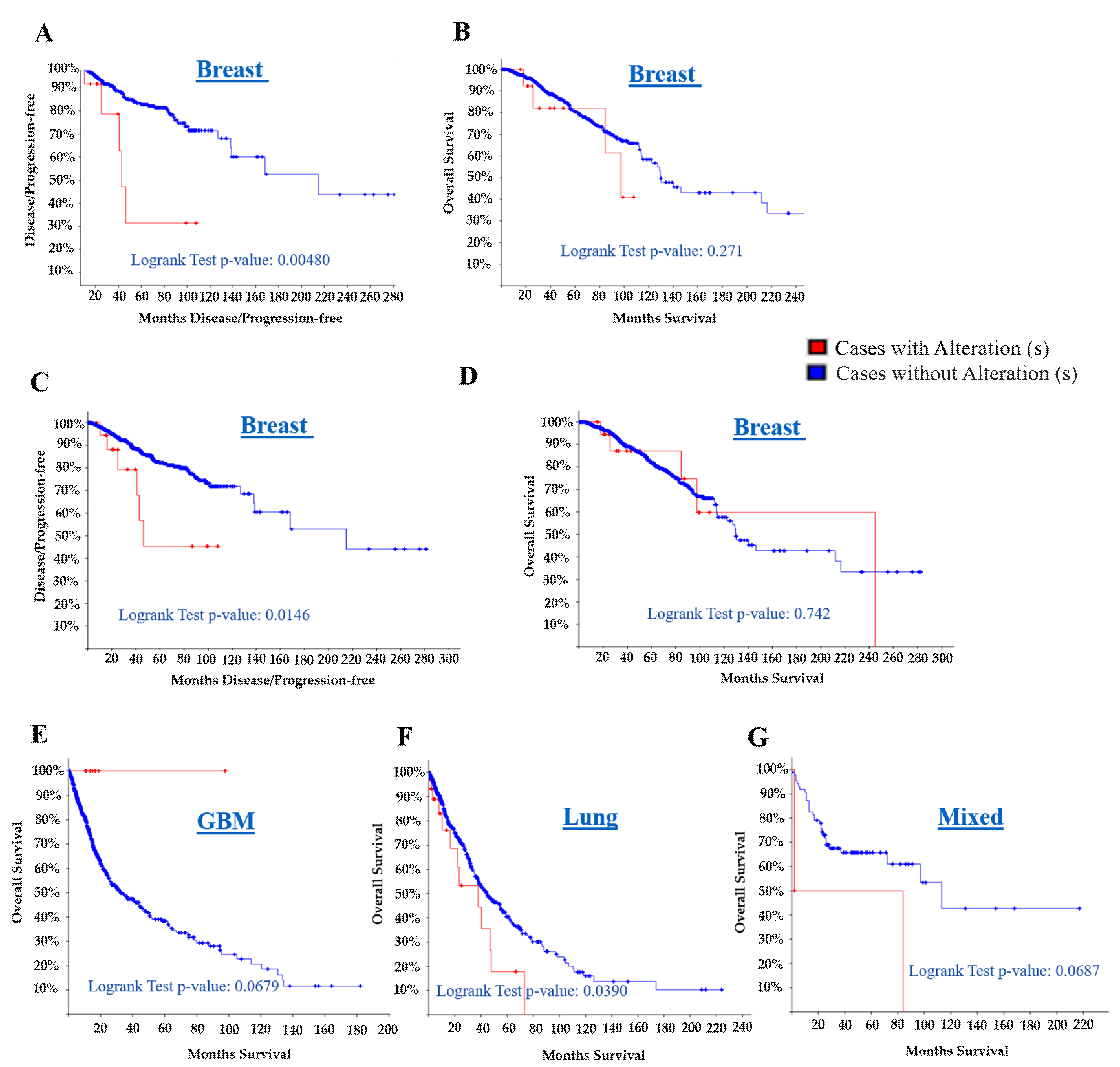
| Study | Overall Survival Kaplan-Meier Estimate | Disease/Progression-Free Kaplan-Meier Estimate | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log Rank Test p-Value | Altheration/Mutation | Total No. of Cases | No. of Cases with Deceased | Median Months Survival | % of Survival | Survival Months | Log Rank Test p-Value | Altheration/Mutation | Total No. of Cases | No. of Cases with Relapsed | Median Months Disease-Free | |
| A. | 0.271 | With | 15 | 4 | 97.4 | 41.03 | 107.85 | 0.00480 | With | 13 | 5 | 42.81 |
| Without | 799 | 114 | 129.6 | 65.94 | 234.10 | Without | 727 | 80 | 214.72 | |||
| B. | 0.742 | With | 23 | 5 | 244.91 | 59.78 | 244.91 | 0.0146 | With | 21 | 6 | 46.39 |
| Without | 938 | 130 | 129.6 | 65.93 | 282.69 | Without | 858 | 96 | 214.72 | |||
| C. | 0.0679 | With | 8 | 0 | NA | 100 | 97.80 | |||||
| Without | 721 | 263 | 32.4 | 24.64 | 182.20 | |||||||
| D. | 0.382 | With | 2 | 2 | 35 | 50 | 109 | |||||
| Without | 20 | 12 | 106 | 84.44 | 186 | |||||||
| E. | 0.442 | With | 13 | 5 | 86.85 | 34.92 | 60.84 | 0.177 | With | 11 | 4 | 32.62 |
| Without | 162 | 80 | 56.27 | 47.32 | 173.69 | Without | 110 | 39 | 61.6 | |||
| F. | 0.0687 | With | 2 | 2 | 2 | 50 | 84 | |||||
| Without | 86 | 31 | 113 | 97.67 | 217 | |||||||
| G. | 0.0390 | With | 40 | 13 | 37.83 | 17.77 | 73.16 | |||||
| Without | 914 | 259 | 44.21 | 36.50 | 224.10 | |||||||
- A
- Breast Invasive Carcinoma, TCGA, Cell 2015 [61], Tumor Samples with sequencing and CNA data (816 samples)/SERTAD1 Gene altered in 15 (1.8%) of queried samples;
- B
- Breast Invasive Carcinoma, TCGA, Provisional [30], Tumor Samples with sequencing and CNA data (963 samples)/SERTAD1 Gene altered in 23 (2.4%) of queried samples;
- C
- Merged Cohort of LGG and GBM, TCGA, Cell 2016 [62], Tumor Samples with sequencing and CNA data (794 samples)/SERTAD1 Gene altered in 8 (1%) of queried samples;
- D
- Low-Grade Gliomas, UCSF [30], Sequenced Tumors (61 samples)/SERTAD1 Gene altered in 2 (3.3%) of queried samples;
- E
- Lung Squamous Cell Carcinoma, TCGA, Provisional [30], Tumor Samples with sequencing and CNA data (178 samples)/SERTAD1 Gene altered in 13 (7.3%) of queried samples;
- F
- Mixed Tumors, PIP-Seq 2017 [30], Sequenced Tumors (103 samples)/SERTAD1 Gene altered in 3 (2.9%) of queried samples;
- G
- Pan-Lung Cancer, TCGA, Nat. Genet. 2016 [63], Tumor Samples with sequencing and CNA data (1144 samples)/SERTAD1 Gene altered in 47 (4.1%) of queried samples.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongre, R.K.; Jung, S.; Mishra, C.B.; Lee, B.S.; Kumari, S.; Lee, M.-S. Prognostic and Clinicopathological Significance of SERTAD1 in Various Types of Cancer Risk: A Systematic Review and Retrospective Analysis. Cancers 2019, 11, 337. https://doi.org/10.3390/cancers11030337
Mongre RK, Jung S, Mishra CB, Lee BS, Kumari S, Lee M-S. Prognostic and Clinicopathological Significance of SERTAD1 in Various Types of Cancer Risk: A Systematic Review and Retrospective Analysis. Cancers. 2019; 11(3):337. https://doi.org/10.3390/cancers11030337
Chicago/Turabian StyleMongre, Raj Kumar, Samil Jung, Chandra Bhushan Mishra, Beom Suk Lee, Shikha Kumari, and Myeong-Sok Lee. 2019. "Prognostic and Clinicopathological Significance of SERTAD1 in Various Types of Cancer Risk: A Systematic Review and Retrospective Analysis" Cancers 11, no. 3: 337. https://doi.org/10.3390/cancers11030337