Compositional and Functional Differences between Microbiota and Cervical Carcinogenesis as Identified by Shotgun Metagenomic Sequencing

Abstract
:1. Introduction
2. Results
2.1. Characteristics of Subjects
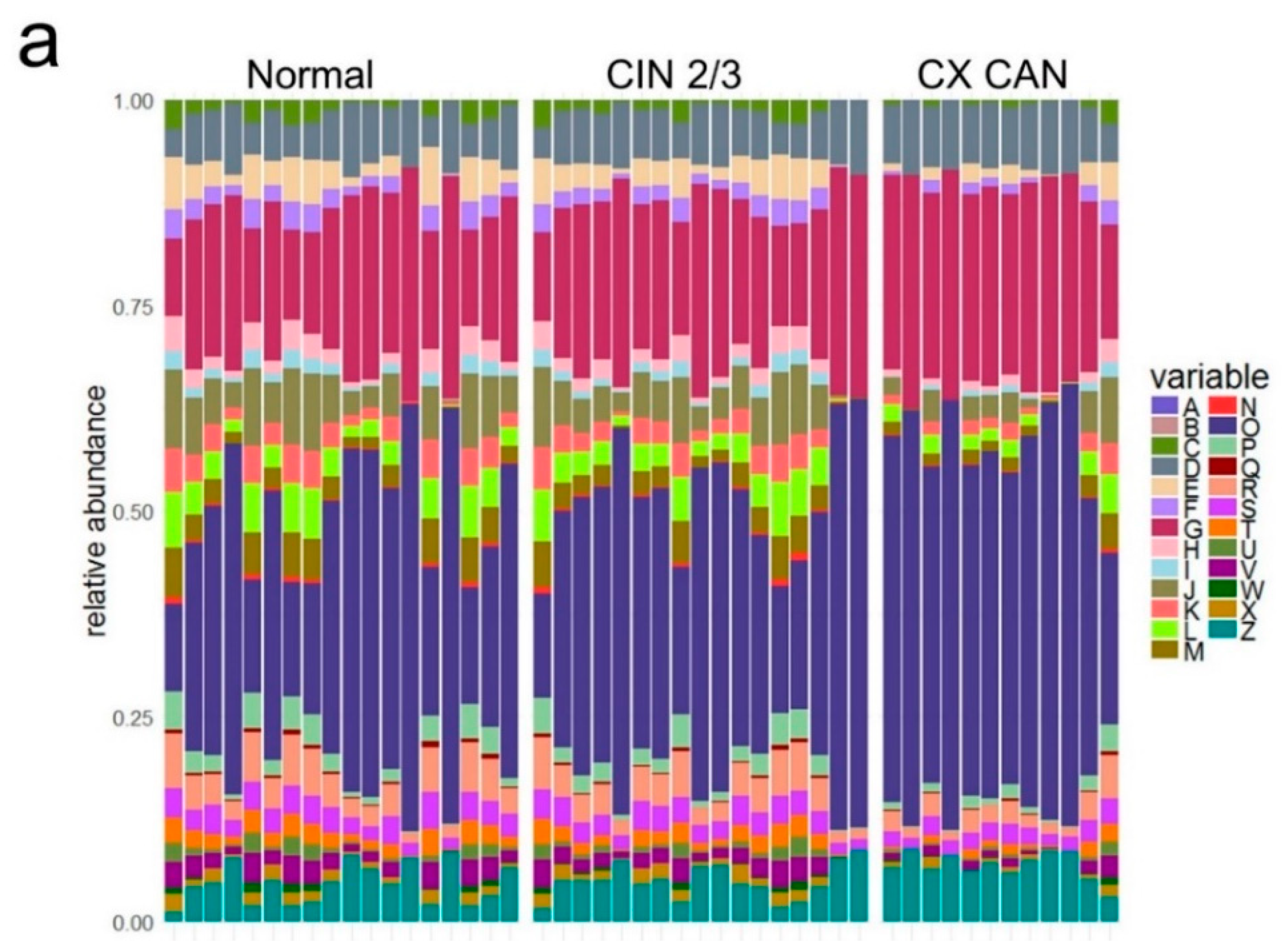
2.2. Taxonomic Characterization of Cervical Microbiome
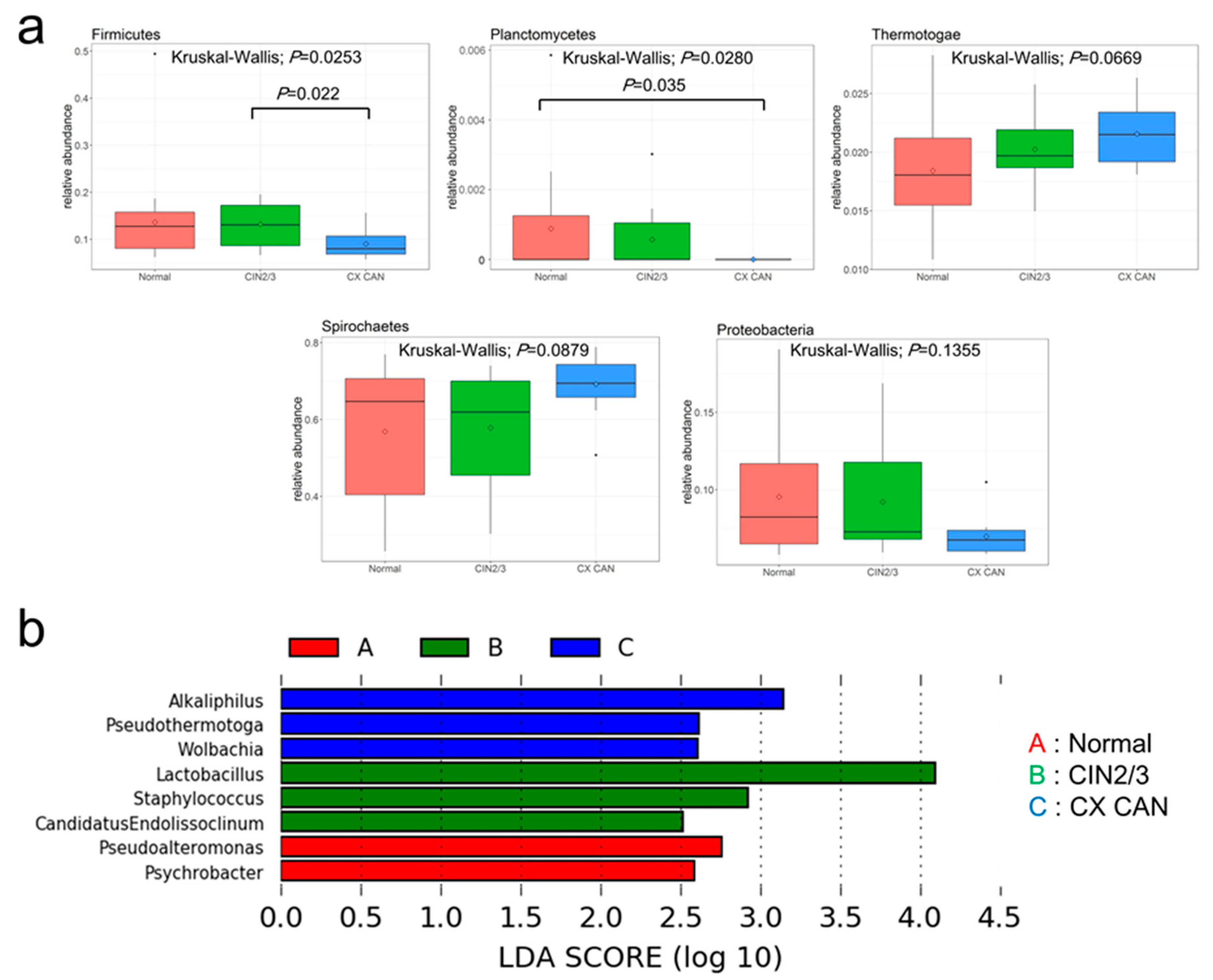
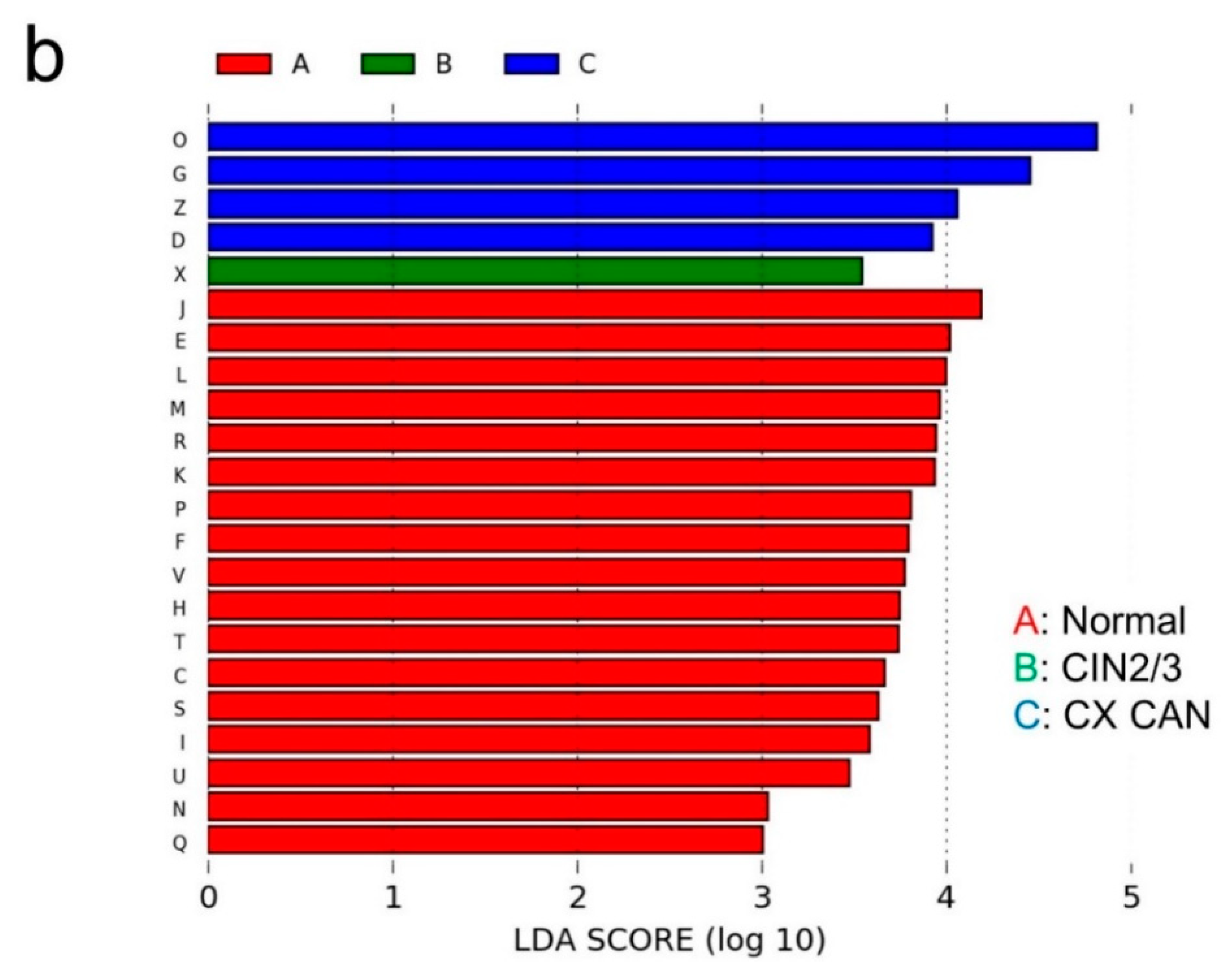
2.3. Microbial Composition among Normal, Cervical Intraepithelial Neoplasia 2/3, and Cervical Cancer
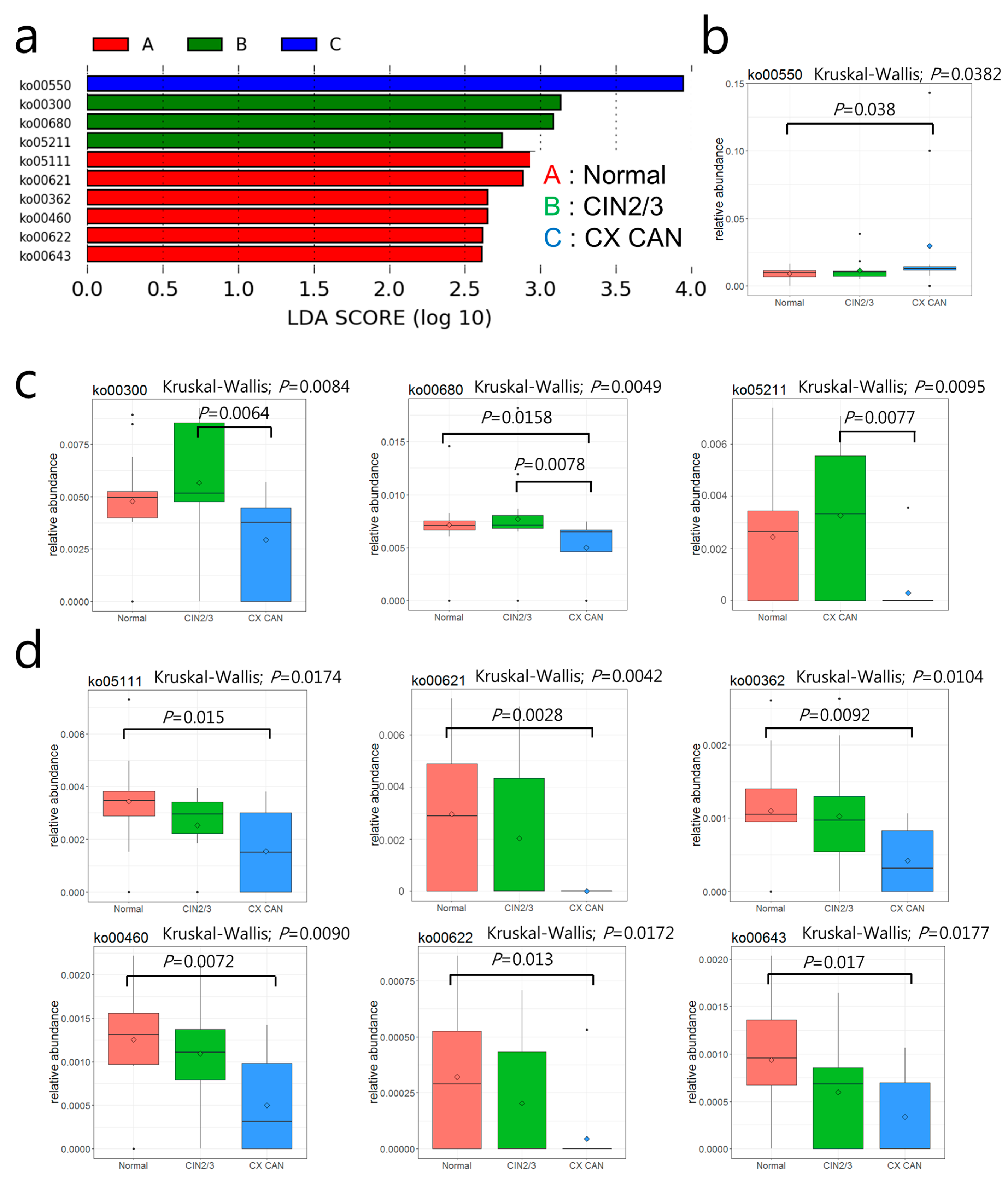
2.4. Distribution and Differences in Relative Abundances of COG Categories
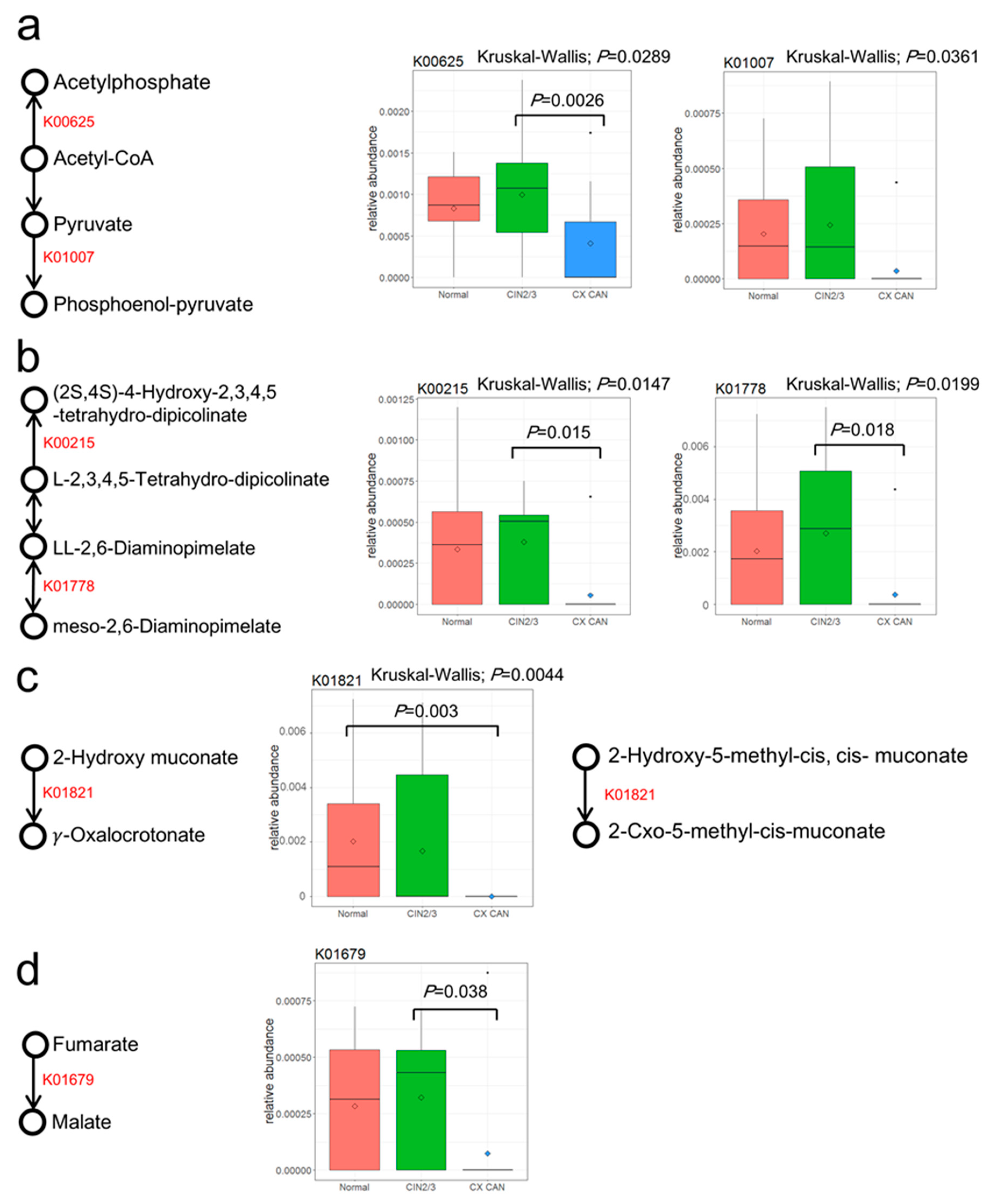
2.5. Metabolic Functions of Cervical Microbiota
3. Discussion
4. Materials and Methods
4.1. Study Design and Subjects
4.2. DNA Extraction, Sequencing, and Quality Check
4.3. Taxonomical Analysis
4.4. De Novo Assembly and Gene Prediction
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CIN | cervical intraepithelial neoplasia |
| LEfSe | Linear discriminant analysis (LDA) effect size |
| COG | Cluster of Orthologous Groups |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PCoA | principal coordinates analysis |
| PERMANOVA | Permutational multivariate analysis of variance |
| Kos | KEGG orthologies |
| LLETZ | Large Loop Excision of the Transformation Zone |
References
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Barsotti, C.; Cosio, S.; Domenici, L.; Riccardo Genazzani, A. Smoking habit, immune suppression, oral contraceptive use, and hormone replacement therapy use and cervical carcinogenesis: A review of the literature. Gynecol. Endocrinol. 2011, 27, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Kyrgiou, M.; Mitra, A.; Moscicki, A.B. Does the vaginal microbiota play a role in the development of cervical cancer? Transl. Res. 2017, 179, 168–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; MacIntyre, D.A.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Lehne, B.; Bhatia, R.; Lyons, D.; Paraskevaidis, E.; Li, J.V.; et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 2015, 5, 16865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, Y.; Gao, W.; Pan, Y.; Gao, Y.; Shen, J.; Xiong, H. The direct and indirect association of cervical microbiota with the risk of cervical intraepithelial neoplasia. Cancer Med. 2018, 7, 2172–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviles-Jimenez, F.; Yu, G.; Torres-Poveda, K.; Madrid-Marina, V.; Torres, J. On the Search to Elucidate the Role of Microbiota in the Genesis of Cancer: The Cases of Gastrointestinal and Cervical Cancer. Arch. Med. Res. 2017, 48, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Antonio, M.A.; Hawes, S.E.; Hillier, S.L. The identification of vaginal Lactobacillus species and the demographic and microbiologic characteristics of women colonized by these species. J. Infect. Dis. 1999, 180, 1950–1956. [Google Scholar] [CrossRef] [PubMed]
- Gillet, E.; Meys, J.F.; Verstraelen, H.; Verhelst, R.; de Sutter, P.; Temmerman, M.; Vanden Broeck, D. Association between bacterial vaginosis and cervical intraepithelial neoplasia: Systematic review and meta-analysis. PLoS ONE 2012, 7, e45201. [Google Scholar] [CrossRef] [PubMed]
- Livengood, C.H. Bacterial Vaginosis: An Overview for 2009. Rev Obstet. Gynecol. 2009, 2, 28–37. [Google Scholar] [PubMed]
- Oh, H.Y.; Kim, B.S.; Seo, S.S.; Kong, J.S.; Lee, J.K.; Park, S.Y.; Hong, K.M.; Kim, H.K.; Kim, M.K. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin. Microbiol. Infect. 2015, 21, 674.e1–674.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.S.; Oh, H.Y.; Lee, J.K.; Kong, J.S.; Lee, D.O.; Kim, M.K. Combined effect of diet and cervical microbiome on the risk of cervical intraepithelial neoplasia. Clin. Nutr. 2016, 35, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.Y.; Seo, S.S.; Kong, J.S.; Lee, J.K.; Kim, M.K. Association between Obesity and Cervical Microflora Dominated by Lactobacillus iners in Korean Women. J. Clin. Microbiol. 2015, 53, 3304–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Tang, H.; Doak, T.G.; Ye, Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Pac. Symp. Biocomput. 2011, 16, 165–176. [Google Scholar]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, S.; Dogan, I.; Dogruman-Al, F.; Nalbantoglu, U.; Ustek, D.; Sarzhanov, F.; Yildirim, S. Association of Enteric Protist Blastocystis spp. and Gut Microbiota with Hepatic Encephalopathy. J. Gastrointestin. Liver Dis. 2016, 25, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Simón, F.; Prieto, G.; Morchón, R.; Bazzocchi, C.; Bandi, C.; Genchi, C. Immunoglobulin G Antibodies against the Endosymbionts of Filarial Nematodes (Wolbachia) in Patients with Pulmonary Dirofilariasis. Clin. Diagn. Lab. Immunol. 2003, 10, 180–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piyathilake, C.J.; Ollberding, N.J.; Kumar, R.; Macaluso, M.; Alvarez, R.D.; Morrow, C.D. Cervical Microbiota Associated with Higher Grade Cervical Intraepithelial Neoplasia in Women Infected with High-Risk Human Papillomaviruses. Cancer Prev. Res. 2016, 9, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, N.; Fukuzawa, M.; Wajima, T.; Yokose, K.; Suzuki, M.; Nakaminami, H.; Kawai, T.; Moriyasu, F.; Sasatsu, M. Specific clones of Staphylococcus lugdunensis may be associated with colon carcinoma. J. Infect. Public Health 2017, 11, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, M.; Genillier, P.L.; Dunais, B.; Kaphan, R.; Saudes, L.; Duval, Y.; Rolland, F.; Jullien, V.; Weiss, N.; Blanchouin, E.; et al. Short-course daptomycin lock and systemic therapy for catheter-related bloodstream infections: A retrospective cohort study in cancer patients with surgically implanted devices. J. Chemother 2017, 29, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Barca, E.; Carratala, J.; Mykietiuk, A.; Fernandez-Sevilla, A.; Gudiol, F. Predisposing factors and outcome of Staphylococcus aureus bacteremia in neutropenic patients with cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2001, 20, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhou, L.; Gong, Y.; Song, Z.; He, L.; Lin, S.; Zhang, J. Non-pylori Helicobacters (NHPHs) Induce Shifts in Gastric Microbiota in Helicobacter pylori—Infected Patients. Front. Microbiol. 2017, 8, 1038. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Brotman, R.M. Vaginal microbiome and sexually transmitted infections: An epidemiologic perspective. J. Clin. Investig. 2011, 121, 4610–4617. [Google Scholar] [CrossRef] [PubMed]
- Vogtmann, E.; Hua, X.; Zeller, G.; Sunagawa, S.; Voigt, A.Y.; Hercog, R.; Goedert, J.J.; Shi, J.; Bork, P.; Sinha, R. Colorectal Cancer and the Human Gut Microbiome: Reproducibility with Whole-Genome Shotgun Sequencing. PLoS ONE 2016, 11, e0155362. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, H.J.; Perkins, H.R.; Ward, J.B. Biosynthesis of Peptidoglycan. In Microbial Cell Walls and Membranes; Rogers, H.J., Ed.; Chapman and Hall: London, UK, 1980; pp. 239–297. [Google Scholar]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Umbarger, H.E. Amino acid biosynthesis and its regulation. Annu. Rev. Biochem. 1978, 47, 533–606. [Google Scholar] [CrossRef] [PubMed]
- Zamani, Z.; Arjmand, M.; Vahabi, F.; Eshaq Hosseini, S.M.; Fazeli, S.M.; Iravani, A.; Bayat, P.; Oghalayee, A.; Mehrabanfar, M.; Haj Hosseini, R.; et al. A metabolic study on colon cancer using (1)h nuclear magnetic resonance spectroscopy. Biochem. Res. Int. 2014, 2014, 348712. [Google Scholar] [CrossRef] [PubMed]
- Holma, R.; Korpela, R.; Sairanen, U.; Blom, M.; Rautio, M.; Poussa, T.; Saxelin, M.; Osterlund, P. Colonic methane production modifies gastrointestinal toxicity associated with adjuvant 5-fluorouracil chemotherapy for colorectal cancer. J. Clin. Gastroenterol. 2013, 47, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Haines, A.; Dilawari, J.; Metz, G.; Blendis, L.; Wiggins, H. Breath-methane in patients with cancer of the large bowel. Lancet 1977, 2, 481–483. [Google Scholar] [CrossRef]
- Piqué, J.M.; Pallarés, M.; Cusó, E.; Vilar-Bonet, J.; Gassull, M.A. Methane production and colon cancer. Gastroenterology 1984, 87, 601–605. [Google Scholar] [PubMed]
- Zhang, B.; Xia, C.; Lin, Q.; Huang, J. Identification of key pathways and transcription factors related to Parkinson disease in genome wide. Mol. Biol. Rep. 2012, 39, 10881–10887. [Google Scholar] [CrossRef] [PubMed]
- Jansson, B.; Jankovic, J. Low cancer rates among patients with Parkinson’s disease. Ann. Neurol. 1985, 17, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Manor, O.; Borenstein, E. Revised computational metagenomic processing uncovers hidden and biologically meaningful functional variation in the human microbiome. Microbiome 2017, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Wu, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Dong, H.; Sun, J. Lack of Vitamin D Receptor Causes Dysbiosis and Changes the Functions of the Murine Intestinal Microbiome. Clin. Ther. 2015, 37, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Mohandhas, B.; Vennila, J.J.; Singh, S. Drug target identification in Helicobacter pylori associated with Gastric Cancer: An insilico approach. J. Pharm. Res. 2012, 5, 4303–4306. [Google Scholar]
- Wu, J.; Peters, B.A.; Dominianni, C.; Zhang, Y.; Pei, Z.; Yang, L.; Ma, Y.; Purdue, M.P.; Jacobs, E.J.; Gapstur, S.M.; et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 2016, 10, 2435–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgman, A.; Perfetti, T.A. The Chemical Components of Tobacco and Tobacco Smoke, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Zhao, H.; Shi, Z.-Z.; Jiang, R.; Zhao, D.-B.; Zhou, H.-T.; Liang, J.-W.; Bi, X.-Y.; Zhao, J.-J.; Li, Z.-Y.; Zhou, J-G. Metastasis associated genomic aberrations in stage II rectal cancer. Genes Genom. 2016, 38, 1085–1094. [Google Scholar] [CrossRef]
- Oh, H.Y.; Seo, S.S.; Kim, M.K.; Lee, D.O.; Chung, Y.K.; Lim, M.C.; Kim, J.Y.; Lee, C.W.; Park, S.Y. Synergistic effect of viral load and alcohol consumption on the risk of persistent high-risk human papillomavirus infection. PLoS ONE 2014, 9, e104374. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Lee, J.K.; Kim, T.J.; Kim, M.K. The association between fruit and vegetable consumption and HPV viral load in high-risk HPV-positive women with cervical intraepithelial neoplasia. Cancer Causes Control 2010, 21, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality-Control Tool for High-Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 18 December 2018).
- Huttenhower Lab. KneadData. Available online: http://huttenhower.sph.harvard.edu/kneaddata (accessed on 18 December 2018).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; MacDonald, N.J.; Beiko, R.G. Classifying short genomic fragments from novel lineages using composition and homology. BMC Bioinform. 2011, 12, 328. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; Stevens, M.; Wagner, H. vegan: Community Ecology Package, Version 2.4-1. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 18 December 2018).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Pohlert, T. The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR). R Package. 2014. Available online: http://CRAN.R-project.org/package=PMCMR (accessed on 18 December 2018).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Normal (Group A, n = 18) | CIN 2/3 (Group B, n = 17) | Cervical Cancer (Group C, n = 12) | pa |
|---|---|---|---|---|
| Age (year) b | 45.6 (7.72) | 41.1 (6.98) | 55.9 (10) | 0.0004 |
| BMI (kg/m2) b | 21.3 (2.66) | 21.3 (2.56) | 22.9 (4.03) | 0.4852 |
| Energy intake (kcal/day) b | 1892 (367) | 1839 (435) | 1953 (452) | 0.8232 |
| HPV c | 11 (61.1) | 12 (80) | 8 (72.7) | 0.5435 |
| Post-menopausal c | 7 (41.2) | 3 (18.7) | 7 (63.6) | 0.0604 |
| Oral contraceptive use (Yes&ever) c | 3 (16.7) | 5 (29.4) | 4 (33.3) | 0.5343 |
| Hormone treatment (Yes&ever) c | 4 (23.6) | 0 (0) | 1 (9.1) | 0.1552 |
| Current smoker c | 2 (11.8) | 2 (12.5) | 1 (9.1) | 1.0 |
| Passive smoker c | 10 (58.8) | 13 (81.3) | 7 (63.6) | 0.3587 |
| Alcohol drinker c | 12 (70.6) | 14 (87.5) | 6 (54.5) | 0.0615 |
| KEGG Pathway | Pathway Name | Normal | CIN2/3 | Cervical Cancer | pa | KEGG Orthology | KO Name | Normal | CIN2/3 | Cervical Cancer | pa |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ko00550 | Peptidoglycan biosynthesis | 0.8881 | 1.1465 | 2.9481 | 0.0382 | ||||||
| ko00300 | Lysine biosynthesis | 0.4785 | 0.5669 | 0.2940 | 0.0084 | K00215 | 4-Hydroxy-tetrahydrodipicolinate reductase | 0.0334 | 0.0380 | 0.0055 | 0.0146 |
| K01778 | Diaminopimelate epimerase | 0.0202 | 0.0271 | 0.0036 | 0.0199 | ||||||
| ko00680 | Methane metabolism | 0.7161 | 0.7716 | 0.4996 | 0.0049 | K00625 | Phosphate acetyltransferase | 0.0831 | 0.0997 | 0.0407 | 0.0289 |
| K01007 | Pyruvate, water dikinase | 0.0203 | 0.0243 | 0.0036 | 0.0361 | ||||||
| ko05211 | Renal cell carcinoma | 0.0244 | 0.0327 | 0.0030 | 0.0095 | K01679 | Fumarate hydratase, class II | 0.0284 | 0.0322 | 0.0073 | 0.0318 |
| ko05111 | Biofilm formation in Vibrio cholerae | 0.3439 | 0.2532 | 0.1552 | 0.0174 | ||||||
| ko00621 | Dioxin degradation | 0.0296 | 0.0204 | 0 | 0.0042 | K01821 | 4-Oxalocrotonate tautomerase | 0.0202 | 0.0167 | 0 | 0.0044 |
| ko00362 | Benzoate degradation | 0.1100 | 0.1027 | 0.0421 | 0.0104 | K01821 | 4-Oxalocrotonate tautomerase | 0.0202 | 0.0167 | 0 | 0.0044 |
| ko00460 | Cyanoamino acid metabolism | 0.1251 | 0.1093 | 0.0502 | 0.0090 | ||||||
| ko00622 | Xylene degradation | 0.0320 | 0.0204 | 0.0044 | 0.0172 | K01821 | 4-Oxalocrotonate tautomerase | 0.0202 | 0.0167 | 0 | 0.0044 |
| ko00643 | Styrene degradation | 0.0939 | 0.0596 | 0.0339 | 0.0177 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, M.; Seo, S.-S.; Kim, M.K.; Lee, D.O.; Lim, M.C. Compositional and Functional Differences between Microbiota and Cervical Carcinogenesis as Identified by Shotgun Metagenomic Sequencing. Cancers 2019, 11, 309. https://doi.org/10.3390/cancers11030309
Kwon M, Seo S-S, Kim MK, Lee DO, Lim MC. Compositional and Functional Differences between Microbiota and Cervical Carcinogenesis as Identified by Shotgun Metagenomic Sequencing. Cancers. 2019; 11(3):309. https://doi.org/10.3390/cancers11030309
Chicago/Turabian StyleKwon, Minji, Sang-Soo Seo, Mi Kyung Kim, Dong Ock Lee, and Myoung Cheol Lim. 2019. "Compositional and Functional Differences between Microbiota and Cervical Carcinogenesis as Identified by Shotgun Metagenomic Sequencing" Cancers 11, no. 3: 309. https://doi.org/10.3390/cancers11030309