Role of Calcium Signaling in GA101-Induced Cell Death in Malignant Human B Cells

 and
and
Abstract
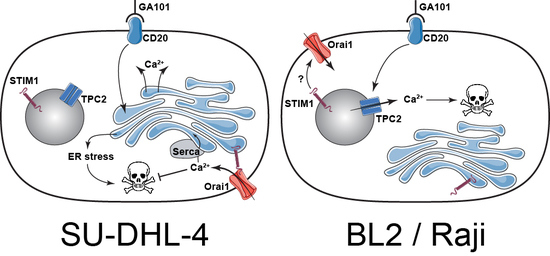
:
1. Introduction
2. Results
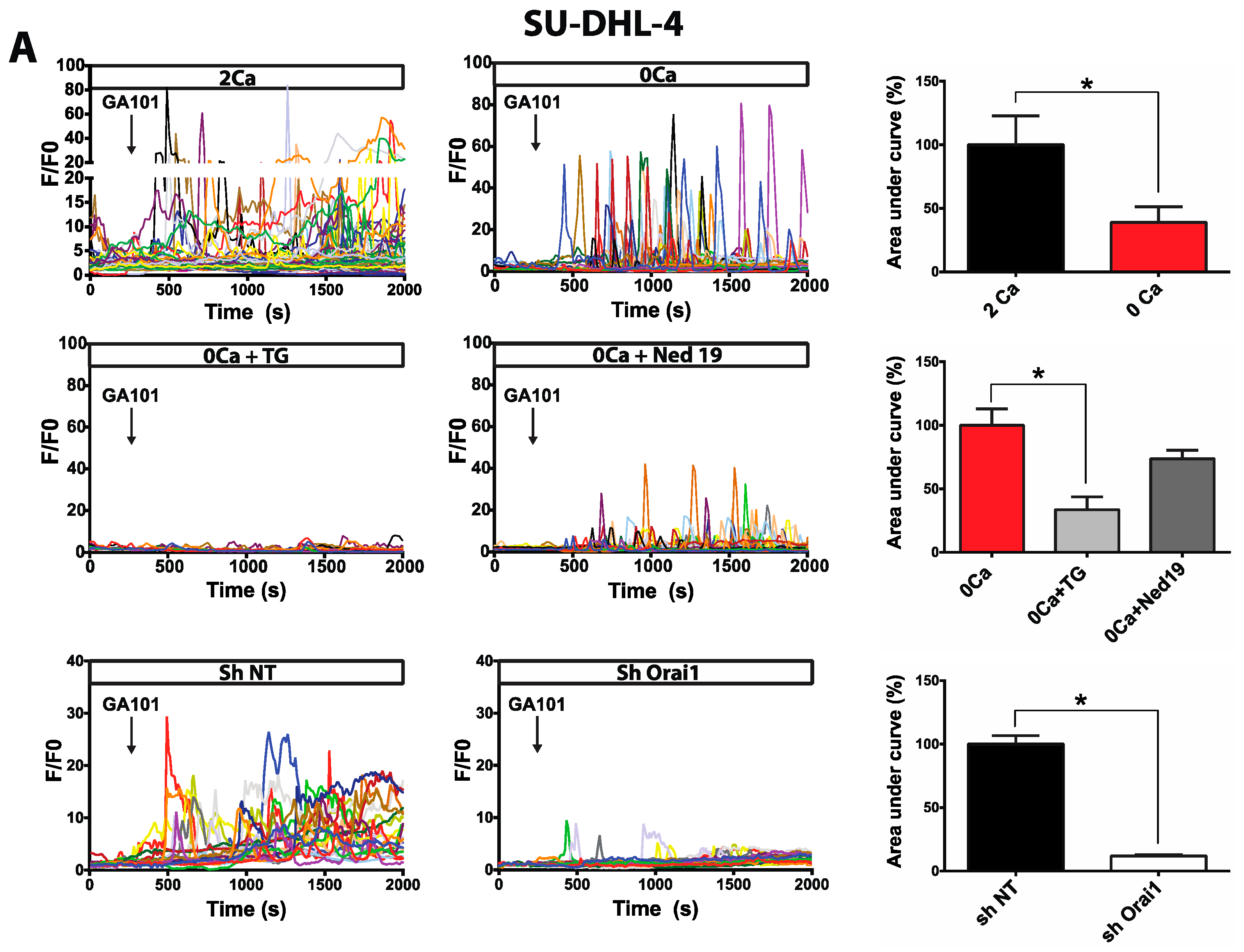
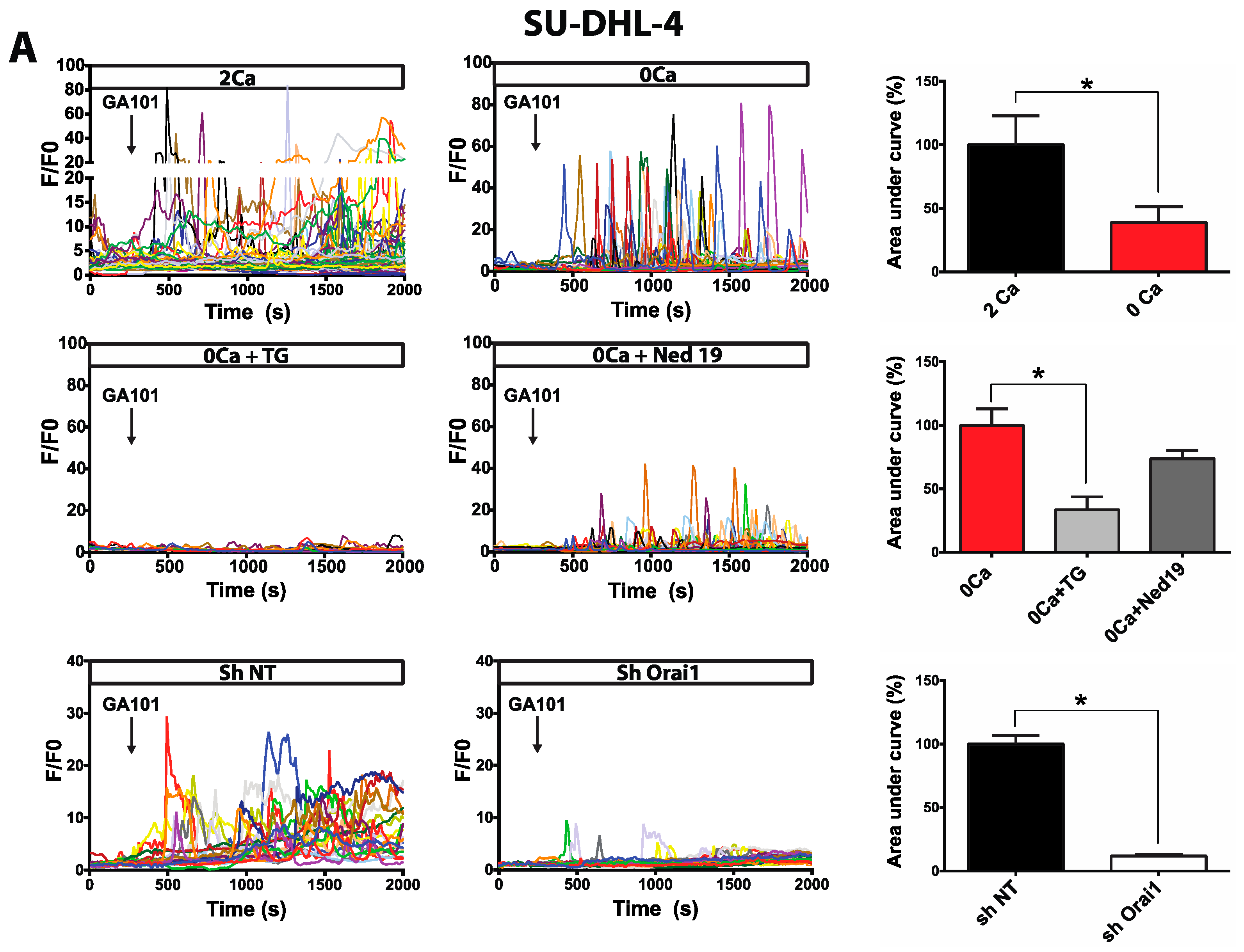
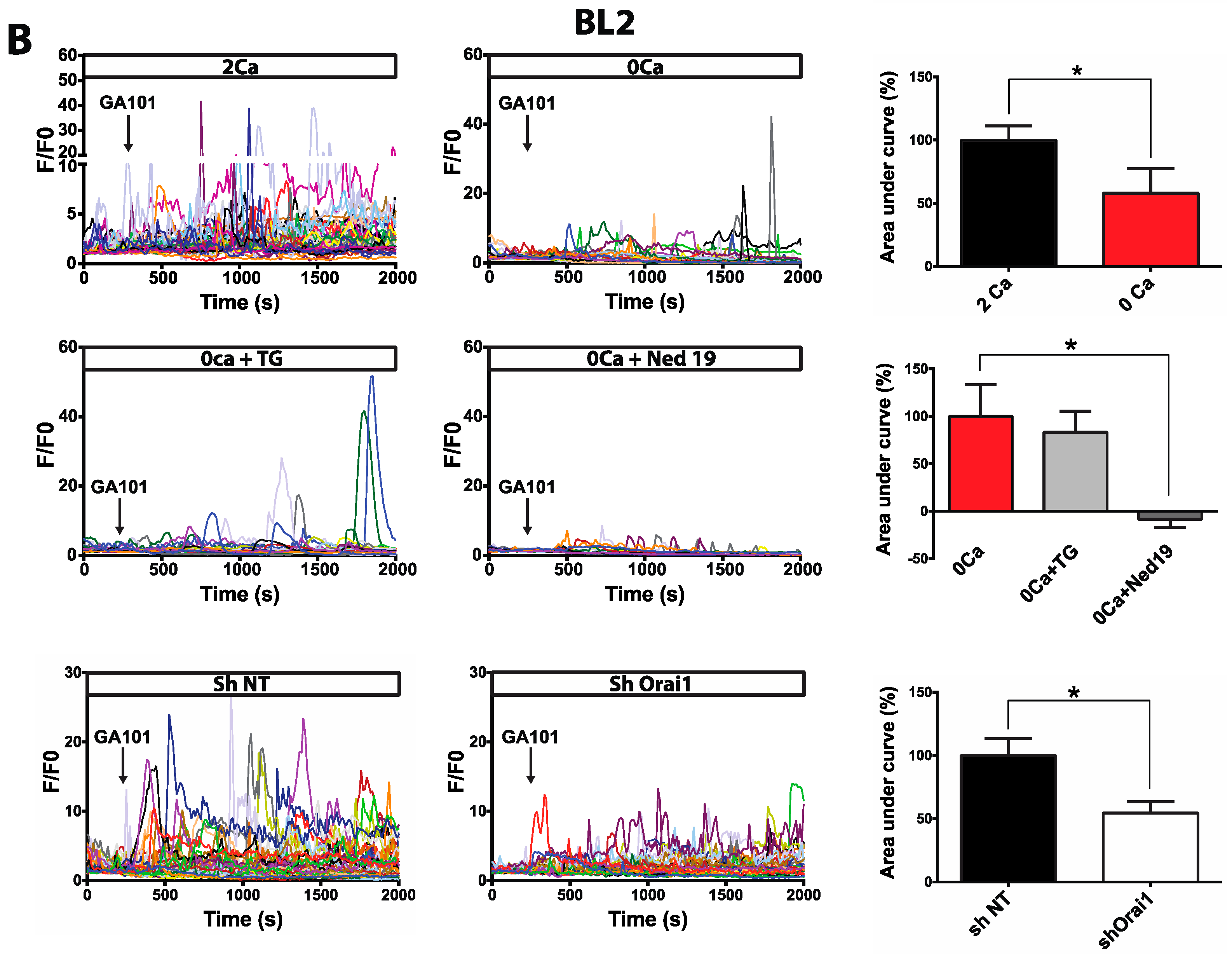
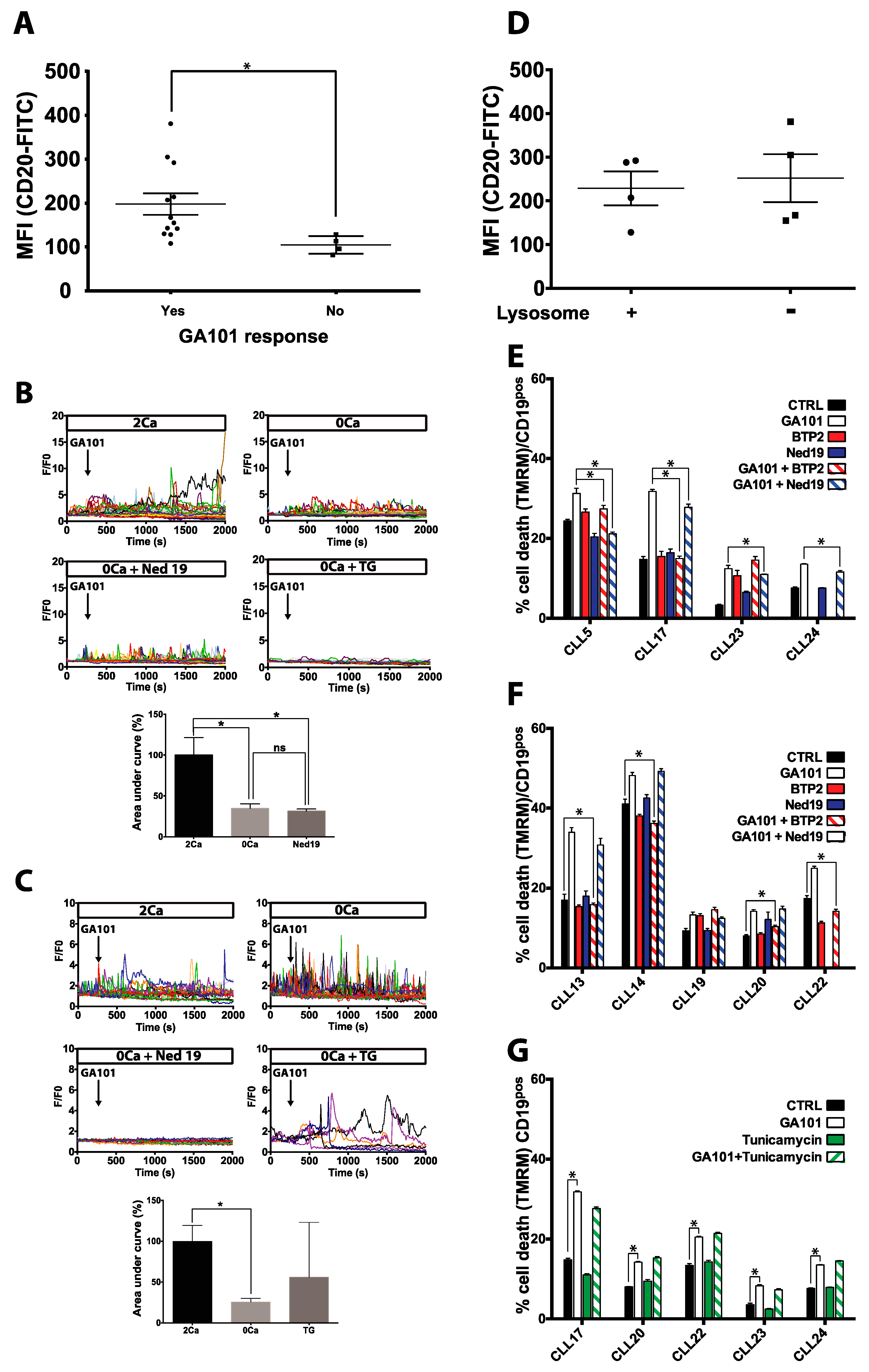
2.1. Calcium Responses Induced by GA101 in NHL Cell Lines
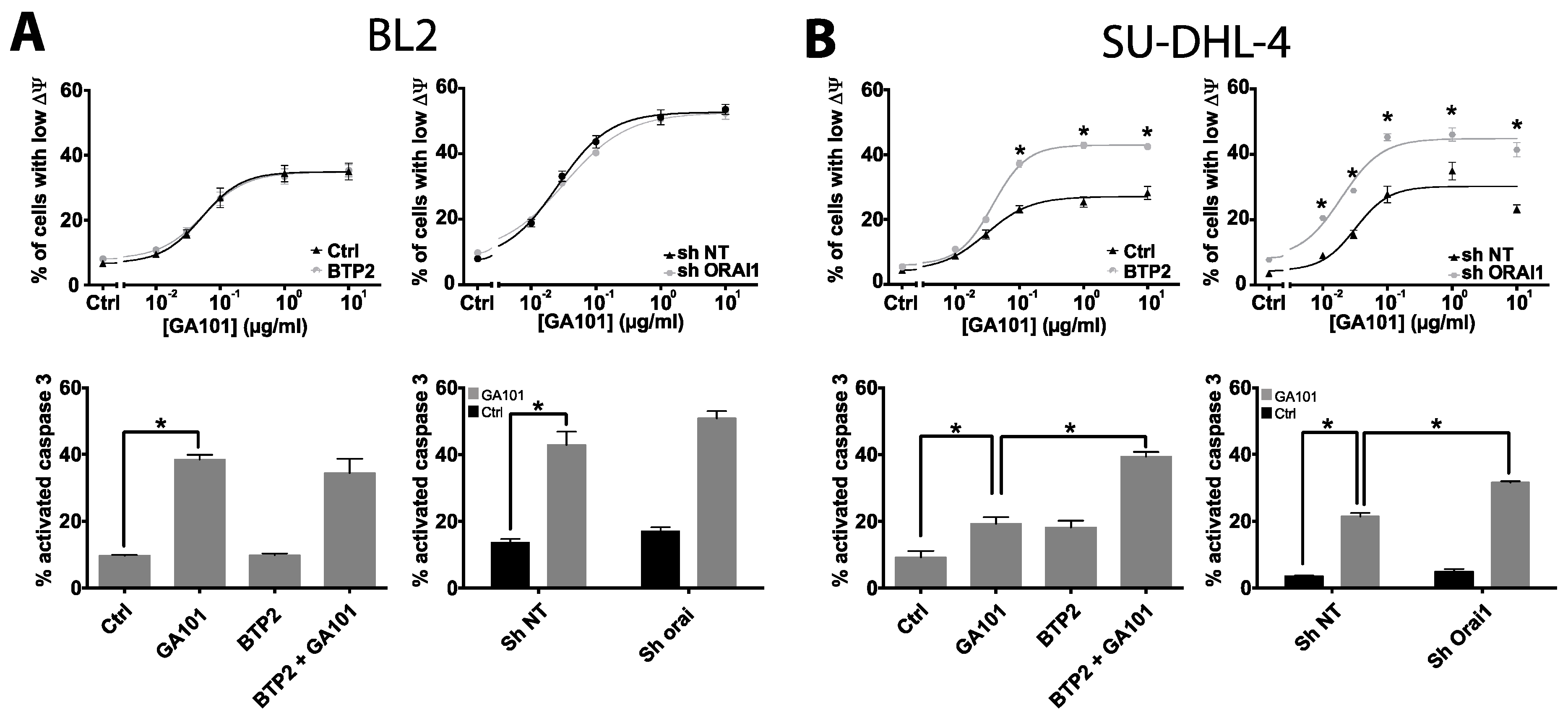
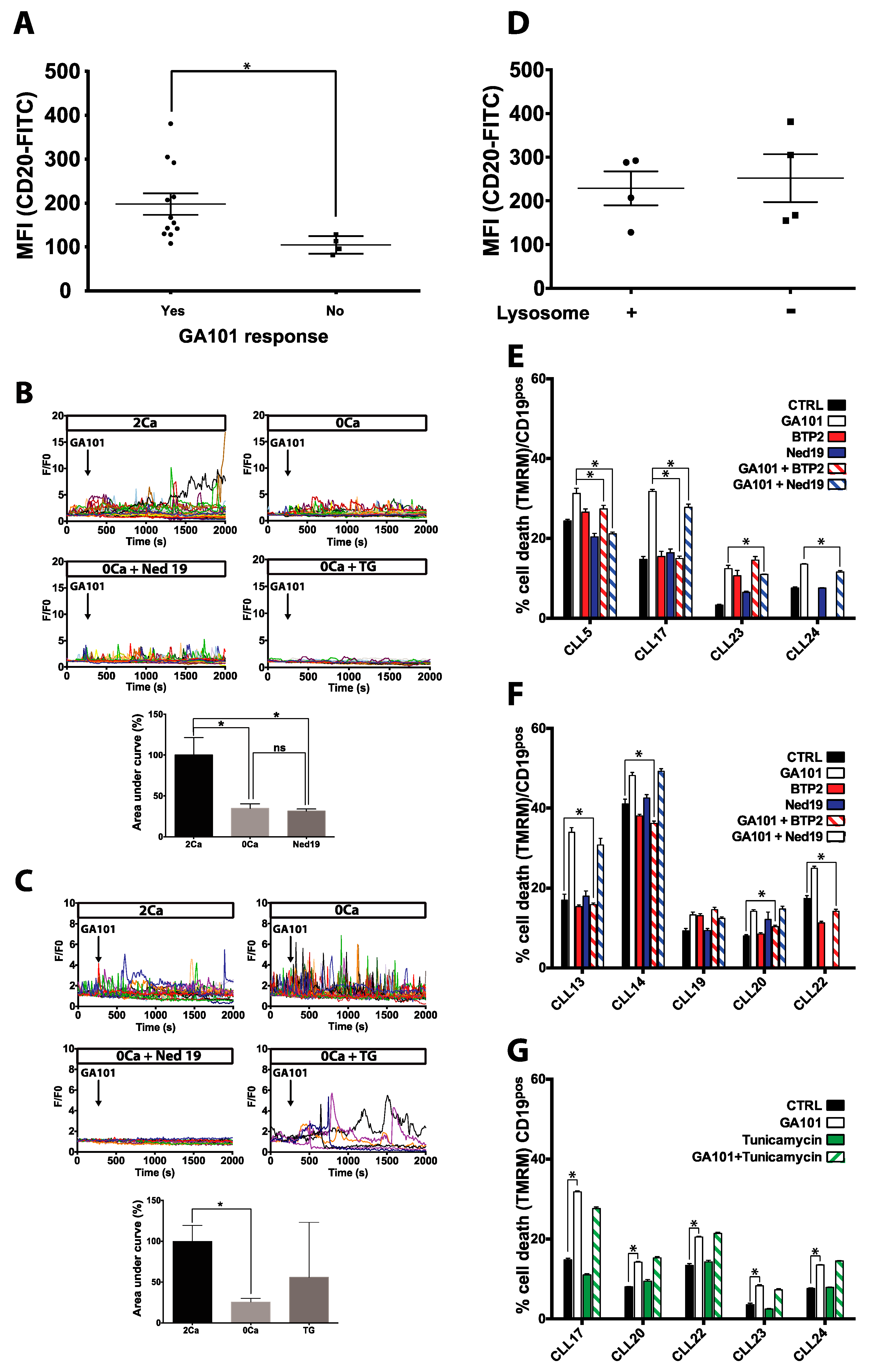
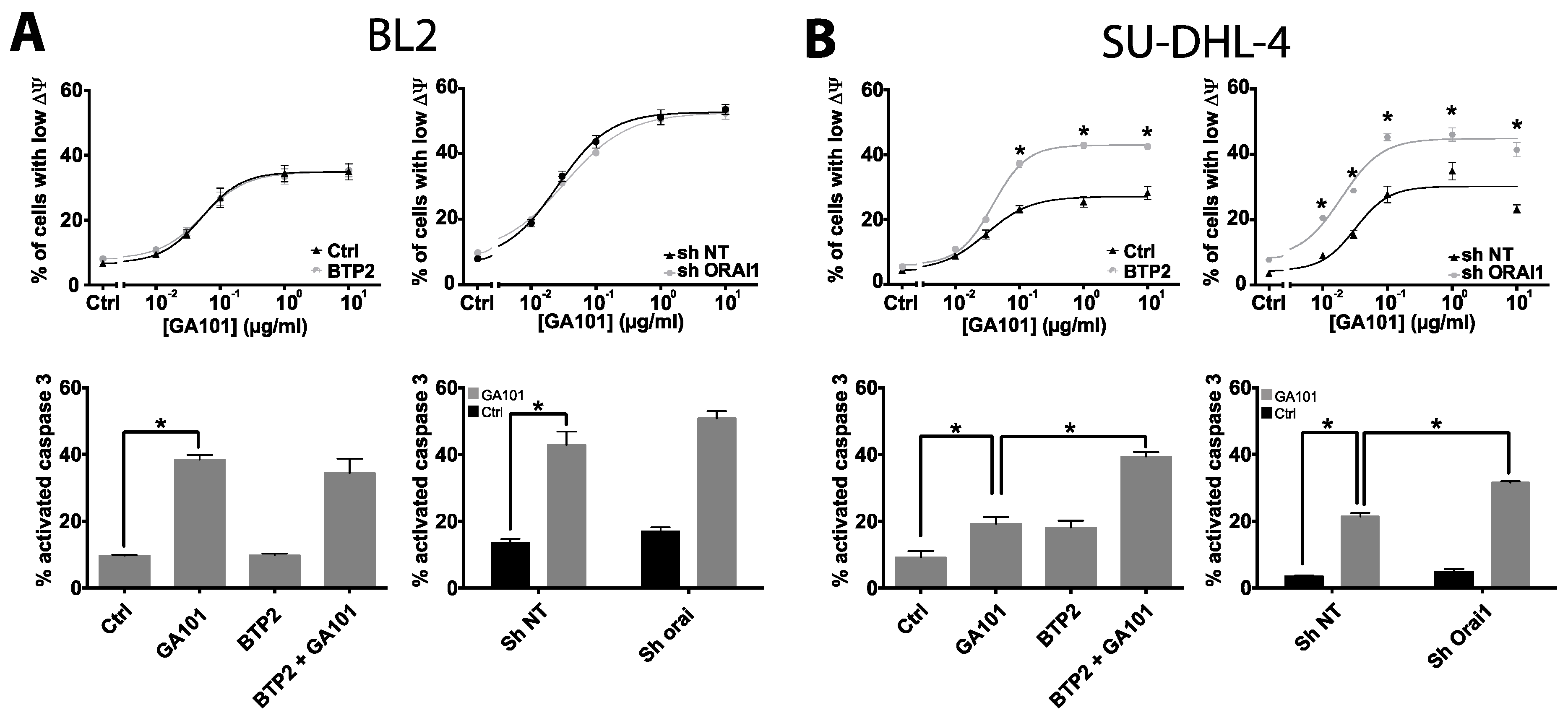
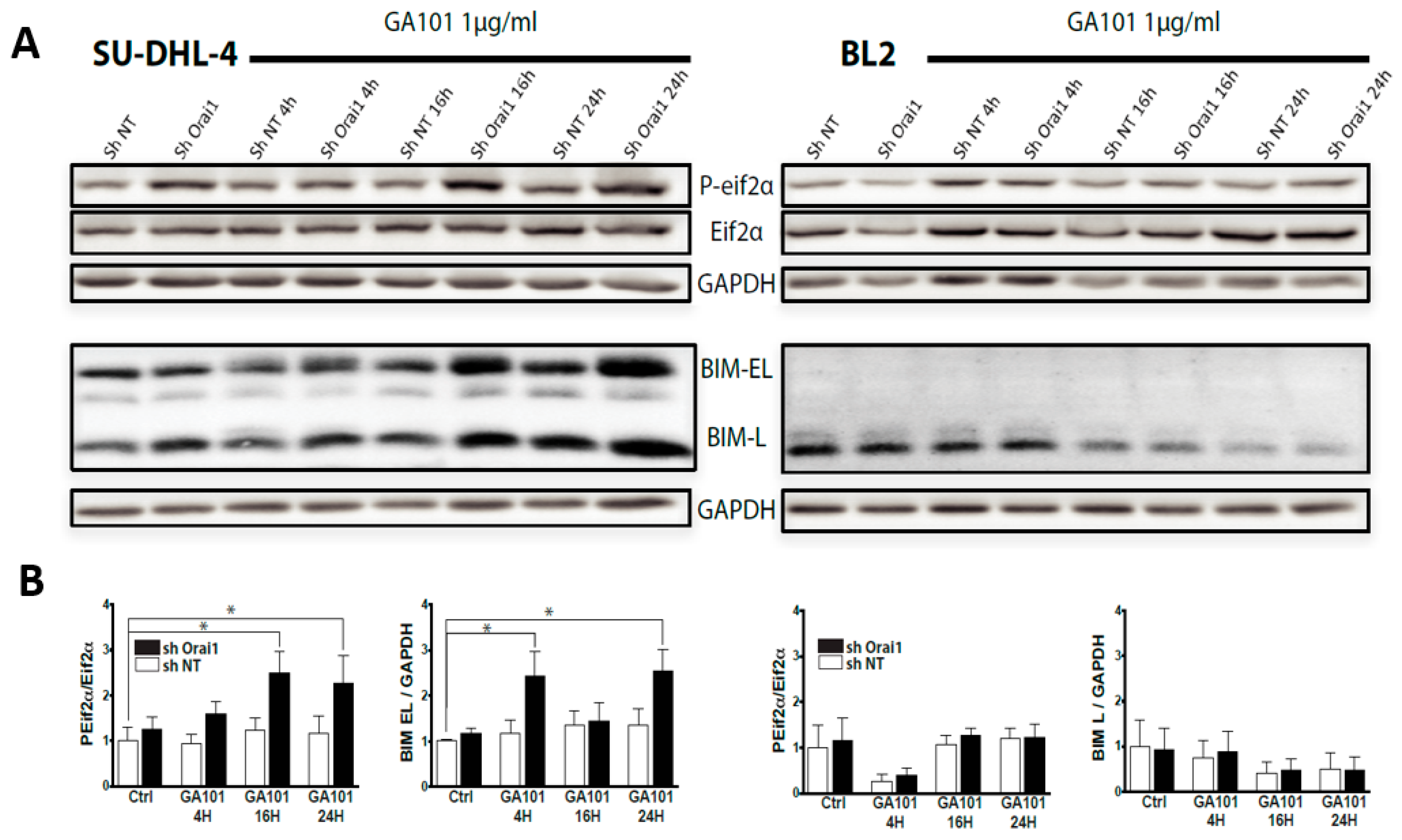
2.2. Role of Calcium Influx in GA101-Induced Cell Death
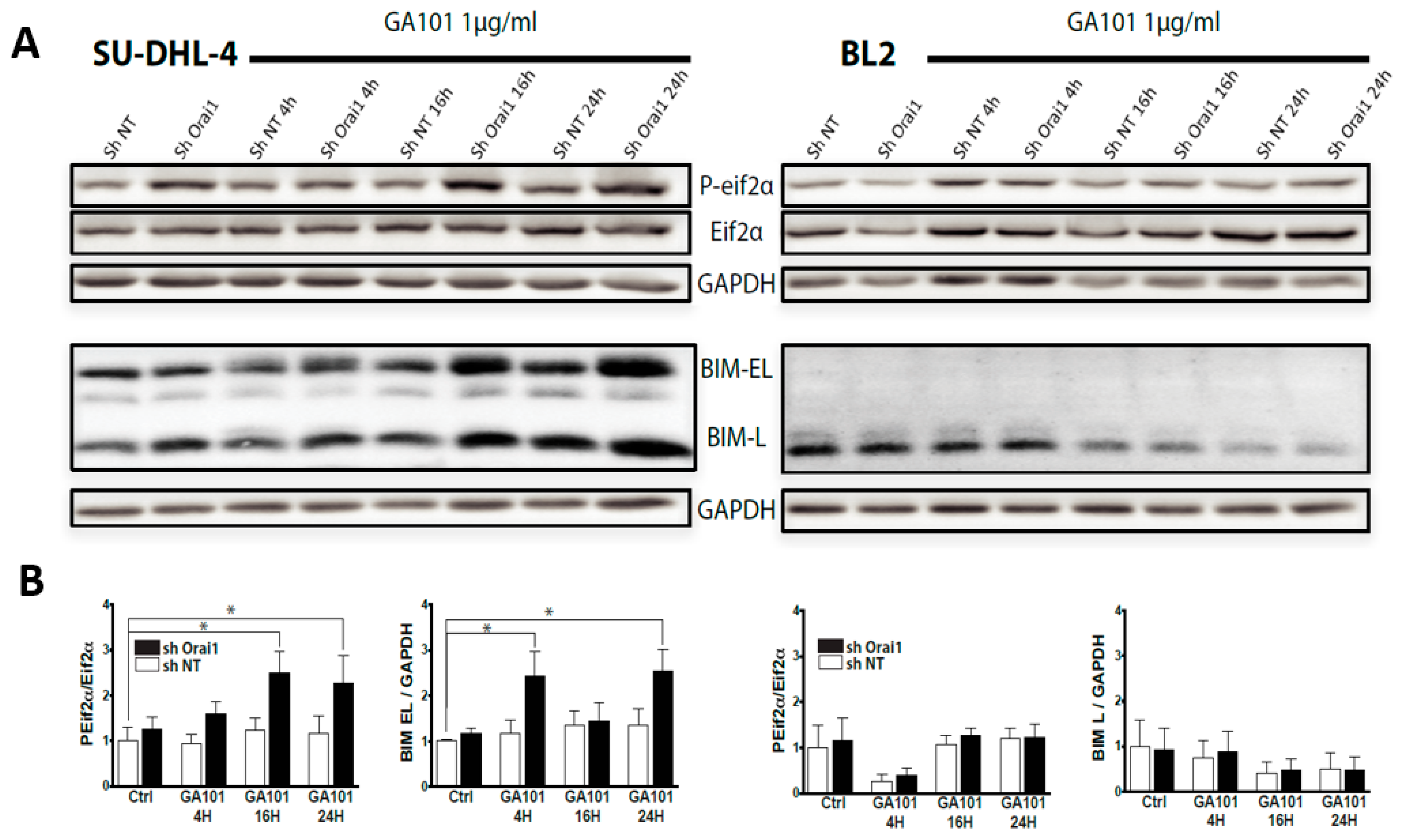
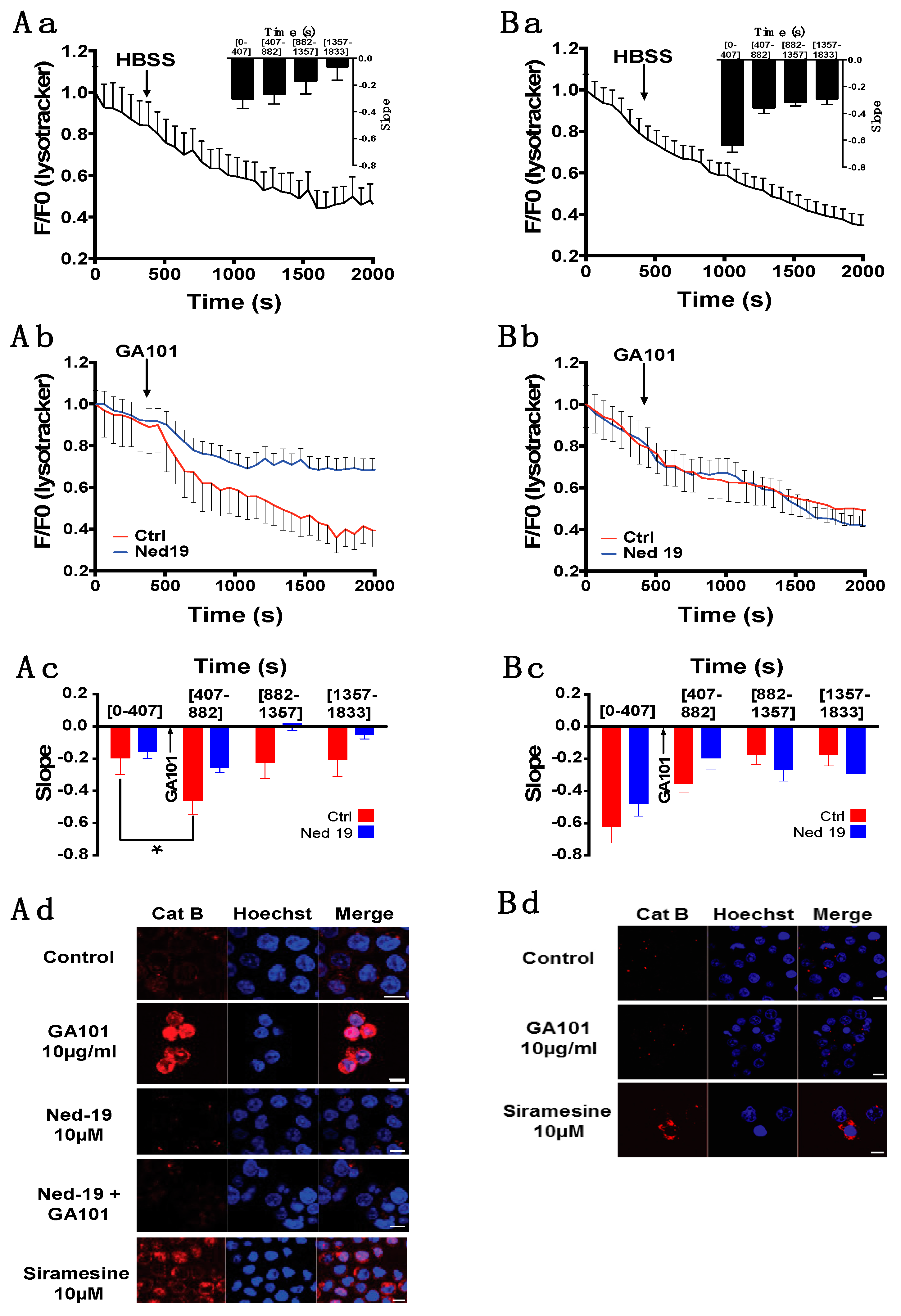
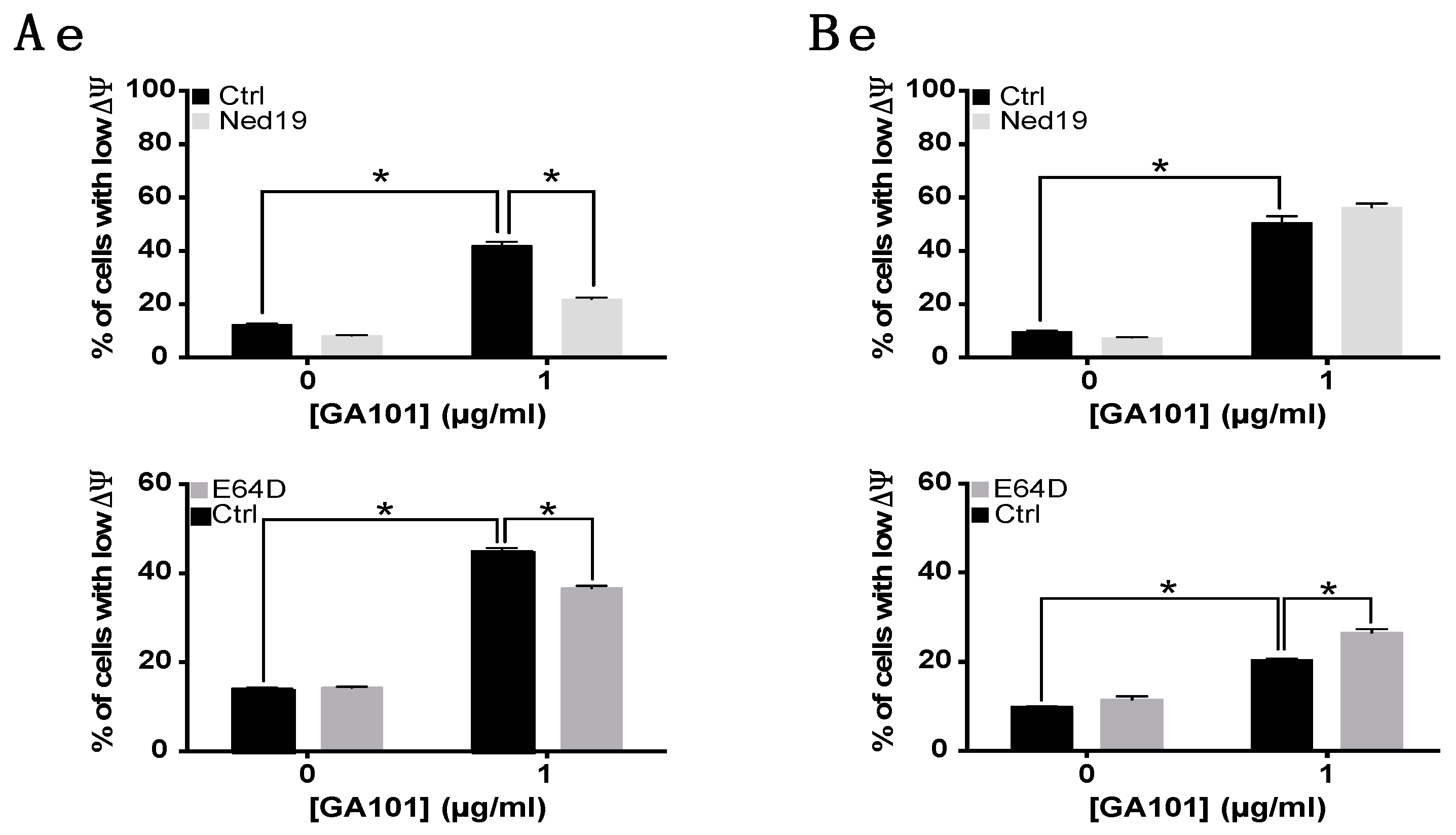
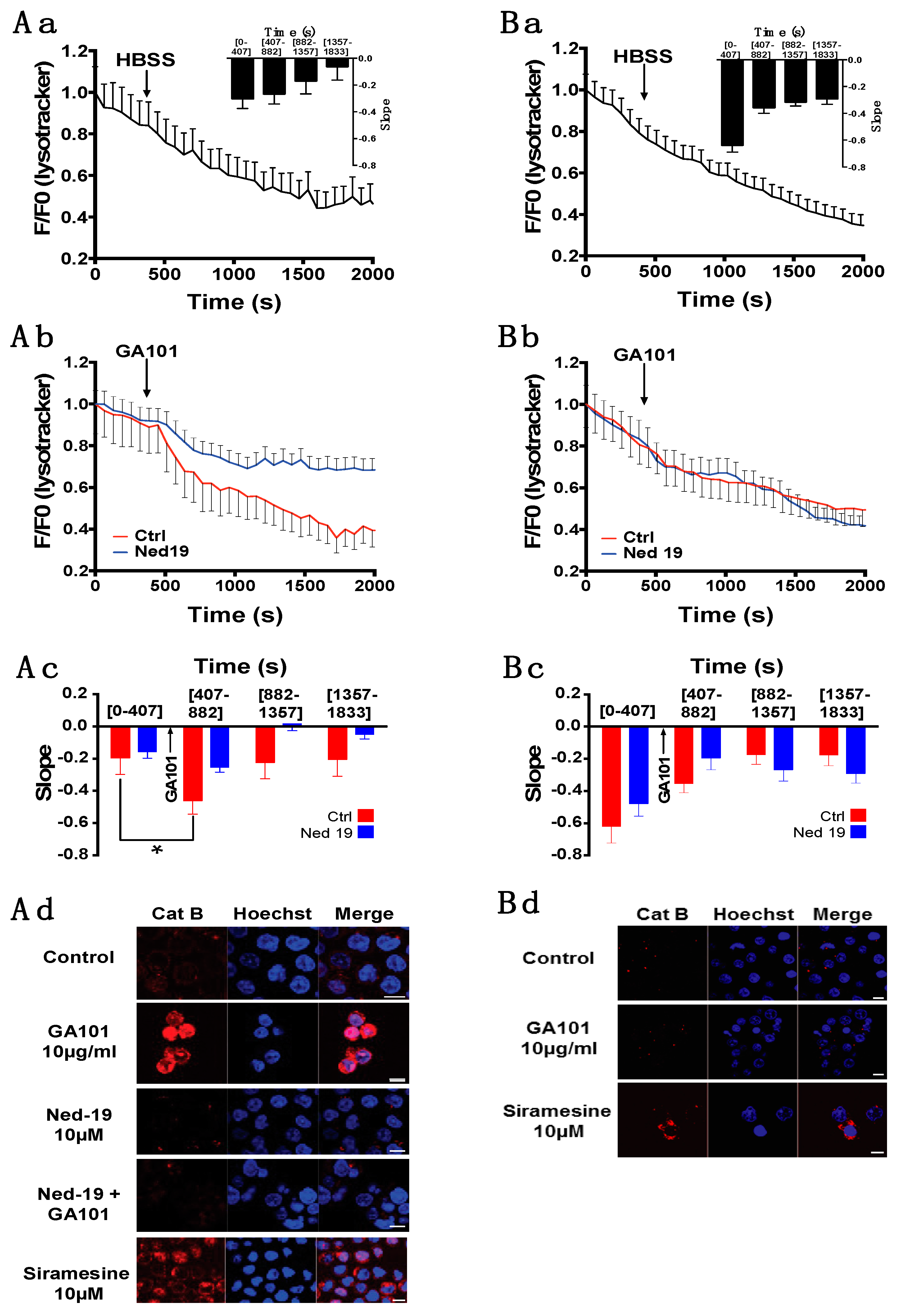
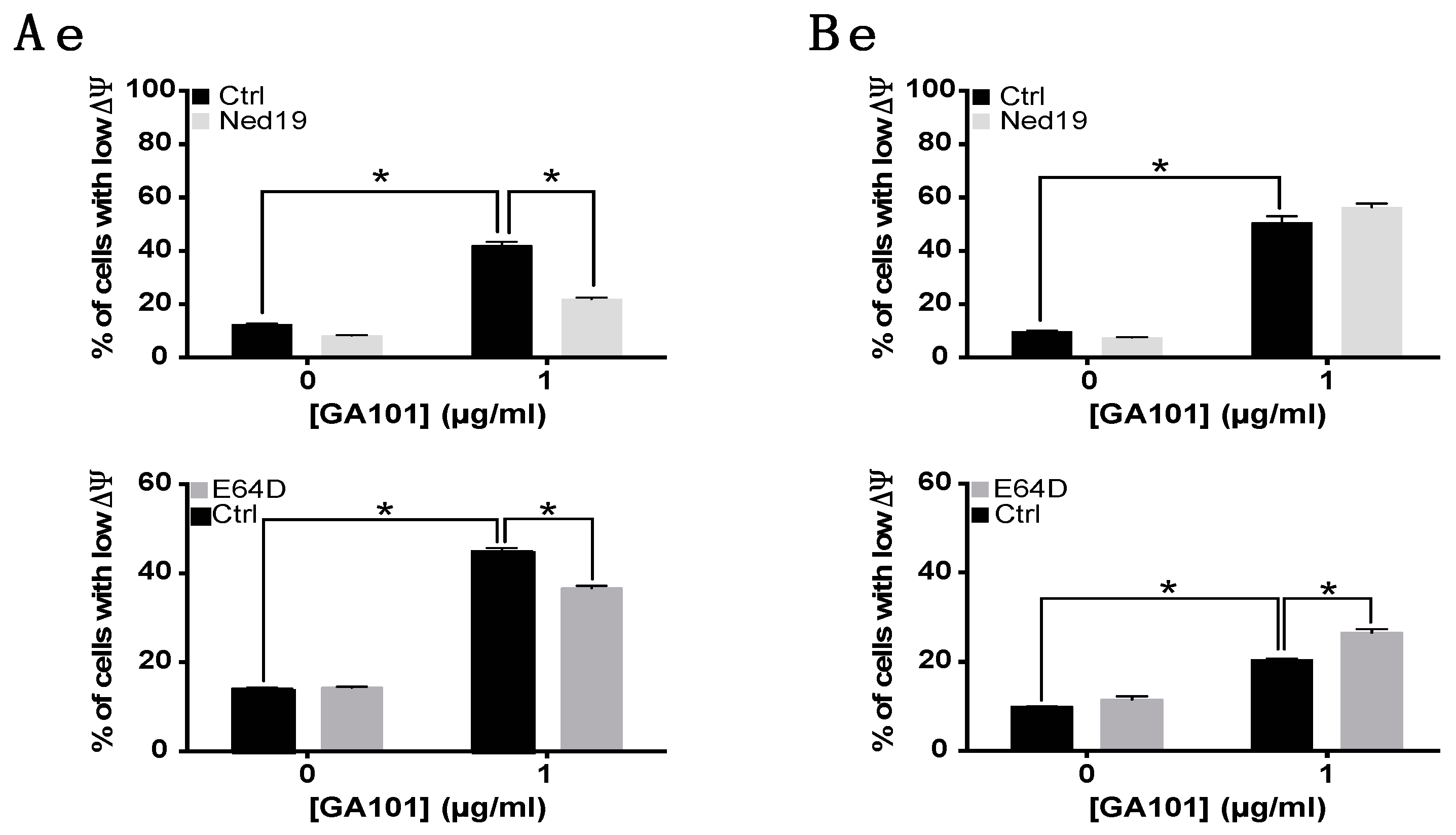
2.3. Role of Lysosomes in GA101-Induced Cell Death
2.4. Effect of GA101 on Primary B-CLL
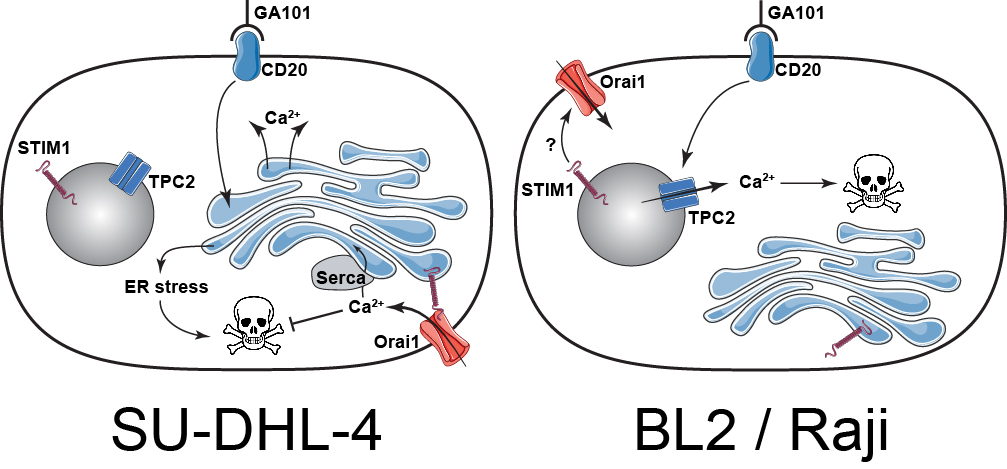
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Patient Samples and Cell Lines
4.3. Short Hairpin RNA Lentivirus Transduction
4.4. Apoptosis Assays
4.5. Intracellular Calcium Measurement
4.6. Assessement of Lysosomal Membrane Permeabilization
4.7. Western Blotting
4.8. RT-qPCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salles, G.; Seymour, J.F.; Offner, F.; López-Guillermo, A.; Belada, D.; Xerri, L.; Feugier, P.; Bouabdallah, R.; Catalano, J.V.; Brice, P.; et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): A phase 3, randomised controlled trial. Lancet Lond. Engl. 2011, 377, 42–51. [Google Scholar] [CrossRef]
- Pfreundschuh, M.; Trümper, L.; Österborg, A.; Pettengell, R.; Trneny, M.; Imrie, K.; Ma, D.; Gill, D.; Walewski, J.; Zinzani, P.-L.; et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: A randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 2006, 7, 379–391. [Google Scholar] [CrossRef]
- Hallek, M.; Fischer, K.; Fingerle-Rowson, G.; Fink, A.M.; Busch, R.; Mayer, J.; Hensel, M.; Hopfinger, G.; Hess, G.; von Grünhagen, U.; et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open-label, phase 3 trial. Lancet Lond. Engl. 2010, 376, 1164–1174. [Google Scholar] [CrossRef]
- Cartron, G.; Trappe, R.U.; Solal-Céligny, P.; Hallek, M. Interindividual variability of response to rituximab: From biological origins to individualized therapies. Clin. Cancer Res. 2011, 17, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell–mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Herting, F.; Mundigl, O.; Waldhauer, I.; Weinzierl, T.; Fauti, T.; Muth, G.; Ziegler-Landesberger, D.; Puijenbroek, E.V.; Lang, S.; et al. Preclinical Activity of the Type II CD20 Antibody GA101 (Obinutuzumab) Compared with Rituximab and Ofatumumab In Vitro and in Xenograft Models. Mol. Cancer Ther. 2013, 12, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Herting, F.; Friess, T.; Bader, S.; Muth, G.; Hölzlwimmer, G.; Rieder, N.; Umana, P.; Klein, C. Enhanced anti-tumor activity of the glycoengineered type II CD20 antibody obinutuzumab (GA101) in combination with chemotherapy in xenograft models of human lymphoma. Leuk. Lymphoma 2014, 55, 2151–5160. [Google Scholar] [CrossRef] [PubMed]
- Patz, M.; Isaeva, P.; Forcob, N.; Müller, B.; Frenzel, L.P.; Wendtner, C.-M.; Klein, C.; Umana, P.; Hallek, M.; Krause, G. Comparison of the in vitro effects of the anti-CD20 antibodies rituximab and GA101 on chronic lymphocytic leukaemia cells. Br. J. Haematol. 2011, 152, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Alduaij, W.; Ivanov, A.; Honeychurch, J.; Cheadle, E.J.; Potluri, S.; Lim, S.H.; Shimada, K.; Chan, C.H.T.; Tutt, A.; Beers, S.A.; et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 2011, 117, 4519–4529. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, J.; Alduaij, W.; Azizyan, M.; Cheadle, E.J.; Pelicano, H.; Ivanov, A.; Huang, P.; Cragg, M.S.; Illidge, T.M. Antibody-induced nonapoptotic cell death in human lymphoma and leukemia cells is mediated through a novel reactive oxygen species-dependent pathway. Blood 2012, 119, 3523–3533. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; O’Brien, S.; Kingsley, C.D.; Eradat, H.; Pagel, J.M.; Lymp, J.; Hirata, J.; Kipps, T.J. Obinutuzumab plus fludarabine/cyclophosphamide or bendamustine in the initial therapy of CLL patients: The phase 1b GALTON trial. Blood 2015, 125, 2779–2785. [Google Scholar] [CrossRef] [PubMed]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Goy, A.; Offner, F.C.; Martinelli, G.; Caballero, M.D.; Gadeberg, O.; Baetz, T.; Zelenetz, A.D.; Gaidano, G.; Fayad, L.E.; et al. Randomized Phase II Trial Comparing Obinutuzumab (GA101) With Rituximab in Patients With Relapsed CD20+ Indolent B-Cell Non-Hodgkin Lymphoma: Final Analysis of the GAUSS Study. J. Clin. Oncol. 2015, 33, 3467–3474. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Woodard, G.E.; Rosado, J.A. Orais and STIMs: Physiological mechanisms and disease. J. Cell. Mol. Med. 2012, 16, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, M.D.; Zhang, S.L.; Yeromin, A.V.; Ohlsen, K.; Roos, J.; Stauderman, K.A. Molecular basis of the CRAC channel. Cell Calcium 2007, 42, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Agellon, L.B.; Michalak, M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu. Rev. Physiol. 2013, 75, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Bologna, L.; André, P.-A.; Buchegger, F.; Mach, J.P.; Boumsell, L.; Introna, M. Possible misinterpretation of the mode of action of therapeutic antibodies in vitro: Homotypic adhesion and flow cytometry result in artefactual direct cell death. Blood 2010, 116, 3372–3373. [Google Scholar] [CrossRef] [PubMed]
- Dalle, S.; Reslan, L.; de Horts, T.B.; Herveau, S.; Herting, F.; Plesa, A.; Friess, T.; Umana, P.; Klein, C.; Dumontet, C. Preclinical Studies on the Mechanism of Action and the Anti-Lymphoma Activity of the Novel Anti-CD20 Antibody GA101. Mol. Cancer Ther. 2011, 10, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Vacher, P.; Vacher, A.-M.; Pineau, R.; Latour, S.; Soubeyran, I.; Pangault, C.; Tarte, K.; Soubeyran, P.; Ducret, T.; Bresson-Bepoldin, L. Localized Store-Operated Calcium Influx Represses CD95-Dependent Apoptotic Effects of Rituximab in Non-Hodgkin B Lymphomas. J. Immunol. 2015, 195, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Decaup, E.; Jean, C.; Laurent, C.; Gravelle, P.; Fruchon, S.; Capilla, F.; Marrot, A.; Saati, T.A.; Frenois, F.-X.; Laurent, G.; et al. Anti-tumor activity of obinutuzumab and rituximab in a follicular lymphoma 3D model. Blood Cancer J. 2013, 3, e131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shu, L.; Wu, J. Ceramide participates in lysosome-mediated cell death induced by type II anti-CD20 monoclonal antibodies. Leuk. Lymphoma 2015, 56, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.-T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596. [Google Scholar] [CrossRef] [PubMed]
- Galione, A.; Evans, A.M.; Ma, J.; Parrington, J.; Arredouani, A.; Cheng, X.; Zhu, M.X. The acid test: The discovery of two-pore channels (TPCs) as NAADP-gated endolysosomal Ca(2+) release channels. Pflug. Arch. 2009, 458, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Patel, S. Function and dysfunction of two-pore channels. Sci. Signal. 2015, 8, re7. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [PubMed]
- Walshe, C.A.; Beers, S.A.; French, R.R.; Chan, C.H.T.; Johnson, P.W.; Packham, G.K.; Glennie, M.J.; Cragg, M.S. Induction of cytosolic calcium flux by CD20 is dependent upon B Cell antigen receptor signaling. J. Biol. Chem. 2008, 283, 16971–16984. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, J.K.; Cooney, D.; Coggeshall, K.M. Clustered CD20 Induced Apoptosis: Src-Family Kinase, the Proximal Regulator of Tyrosine Phosphorylation, Calcium Influx, and Caspase 3-Dependent Apoptosis. Blood Cells Mol. Dis. 2000, 26, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lammens, A.; Schäfer, W.; Georges, G.; Schwaiger, M.; Mössner, E.; Hopfner, K.-P.; Umaña, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. mAbs 2013, 5, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Packham, G.; Krysov, S.; Allen, A.; Savelyeva, N.; Steele, A.J.; Forconi, F.; Stevenson, F.K. The outcome of B-cell receptor signaling in chronic lymphocytic leukemia: Proliferation or anergy. Haematologica 2014, 99, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Stoka, V. Protease signalling in cell death: Caspases versus cysteine cathepsins. FEBS Lett. 2007, 581, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Khadra, N.; Penna, A.; Chaigne-Delalande, B.; Segui, B.; Levade, T.; Vacher, A.-M.; Reiffers, J.; Ducret, T.; Moreau, J.-F.; Cahalan, M.D.; et al. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) 2, and prevents death-inducing signaling complex formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19072–19077. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of Bim in Health and Disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef] [PubMed]
- Gottardi, D.; Alfarano, A.; De Leo, A.M.; Stacchini, A.; Aragno, M.; Rigo, A.; Veneri, D.; Zanotti, R.; Pizzolo, G.; Caligaris-Cappio, F. In leukaemic CD5+ B cells the expression of BCL-2 gene family is shifted toward protection from apoptosis. Br. J. Haematol. 1996, 94, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Kitada, S.; Andersen, J.; Akar, S.; Zapata, J.M.; Takayama, S.; Krajewski, S.; Wang, H.G.; Zhang, X.; Bullrich, F.; Croce, C.M.; et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: Correlations with In vitro and In vivo chemoresponses. Blood 1998, 91, 3379–3389. [Google Scholar] [PubMed]
- Parys, J.B. The IP3 Receptor as a Hub for Bcl-2 Family Proteins in Cell Death Control and Beyond. Sci. Signal. 2014, 7, pe4. [Google Scholar] [CrossRef] [PubMed]
- Abdoul-Azize, S.; Buquet, C.; Vannier, J.-P.; Dubus, I. Pyr3, a TRPC3 channel blocker, potentiates dexamethasone sensitivity and apoptosis in acute lymphoblastic leukemia cells by disturbing Ca2+ signaling, mitochondrial membrane potential changes and reactive oxygen species production. Eur. J. Pharmacol. 2016, 784, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Pottosin, I.; Delgado-Enciso, I.; Bonales-Alatorre, E.; Nieto-Pescador, M.G.; Moreno-Galindo, E.G.; Dobrovinskaya, O. Mechanosensitive Ca2+-permeable channels in human leukemic cells: Pharmacological and molecular evidence for TRPV2. Biochim. Biophys. Acta BBA-Biomembr. 2015, 1848, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Geuna, M. A flow cytometry assay simultaneously detects independent apoptotic parameters. Cytometry 2001, 45, 151–157. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | GA101-Induced Cell Death in CD19pos Cells (% of Control) | |||||
|---|---|---|---|---|---|---|
| Patient ID | Age/Gender | Stage | TP53 (del17p) | % WBC | % CD19pos | |
| CLL1 | 79/F | A | Positive | 78 | 91.2 | 9.7 * |
| CLL2 | 73/F | C | Positive | 69 | 82.1 | 2 |
| CLL3 | 77/F | A | Negative | 55 | 82 | −15.7 |
| CLL4 | 60/F | A | Negative | 35 | 66 | 22 * |
| CLL5 | 79/F | C | Negative | 56 | 76.8 | 6.9 * |
| CLL6 | 75/M | A | Negative | 24 | 56.7 | 8.2 * |
| CLL7 | 79/F | A | Negative | 45 | 23 | 0.2 |
| CLL8 | 66/F | A | Negative | 34 | 50.4 | 5.2 * |
| CLL10 | 62/F | A | Negative | 39 | 66 | −2 |
| CLL11 | 86/F | A | Negative | 86 | 92 | 1.8 |
| CLL12 | 70/F | A | Negative | 33 | 68 | 10.7 * |
| CLL13 | 66/F | A | Negative | 36 | 66 | 15.8 * |
| CLL14 | 73/F | A | Negative | 38 | 63 | 7 * |
| CLL15 | 56/M | A | Negative | 23 | 51 | −1.5 |
| CLL16 | 67/M | A | Negative | 58 | 76 | 1 |
| CLL17 | 43/F | A | Negative | 36 | 69 | 16.9 * |
| CLL18 | 73/F | A | Negative | 41 | 81 | −1.2 |
| CLL19 | 54/M | A | Negative | 55 | 73 | 4 * |
| CLL20 | 51/F | A | Negative | 24 | 47 | 6.2 * |
| CLL21 | 74/F | B | Postive | 69 | 92 | 6.8 * |
| CLL22 | 70/M | A | Negative | 69 | 85 | 7.5 * |
| CLL23 | 49/M | B | Negative | 52 | 87 | 9.1 * |
| CLL24 | 75/M | B | Negative | 62 | 86 | 5.9 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latour, S.; Zanese, M.; Le Morvan, V.; Vacher, A.-M.; Menard, N.; Bijou, F.; Durrieu, F.; Soubeyran, P.; Savina, A.; Vacher, P.; et al. Role of Calcium Signaling in GA101-Induced Cell Death in Malignant Human B Cells. Cancers 2019, 11, 291. https://doi.org/10.3390/cancers11030291
Latour S, Zanese M, Le Morvan V, Vacher A-M, Menard N, Bijou F, Durrieu F, Soubeyran P, Savina A, Vacher P, et al. Role of Calcium Signaling in GA101-Induced Cell Death in Malignant Human B Cells. Cancers. 2019; 11(3):291. https://doi.org/10.3390/cancers11030291
Chicago/Turabian StyleLatour, Simon, Marion Zanese, Valérie Le Morvan, Anne-Marie Vacher, Nelly Menard, Fontanet Bijou, Francoise Durrieu, Pierre Soubeyran, Ariel Savina, Pierre Vacher, and et al. 2019. "Role of Calcium Signaling in GA101-Induced Cell Death in Malignant Human B Cells" Cancers 11, no. 3: 291. https://doi.org/10.3390/cancers11030291