A Developmental Perspective on Paragangliar Tumorigenesis

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Intersections between Tumorigenesis, Histogenesis, and Tissue Regeneration
1.2. Paragangliomas and Pheochromocytomas
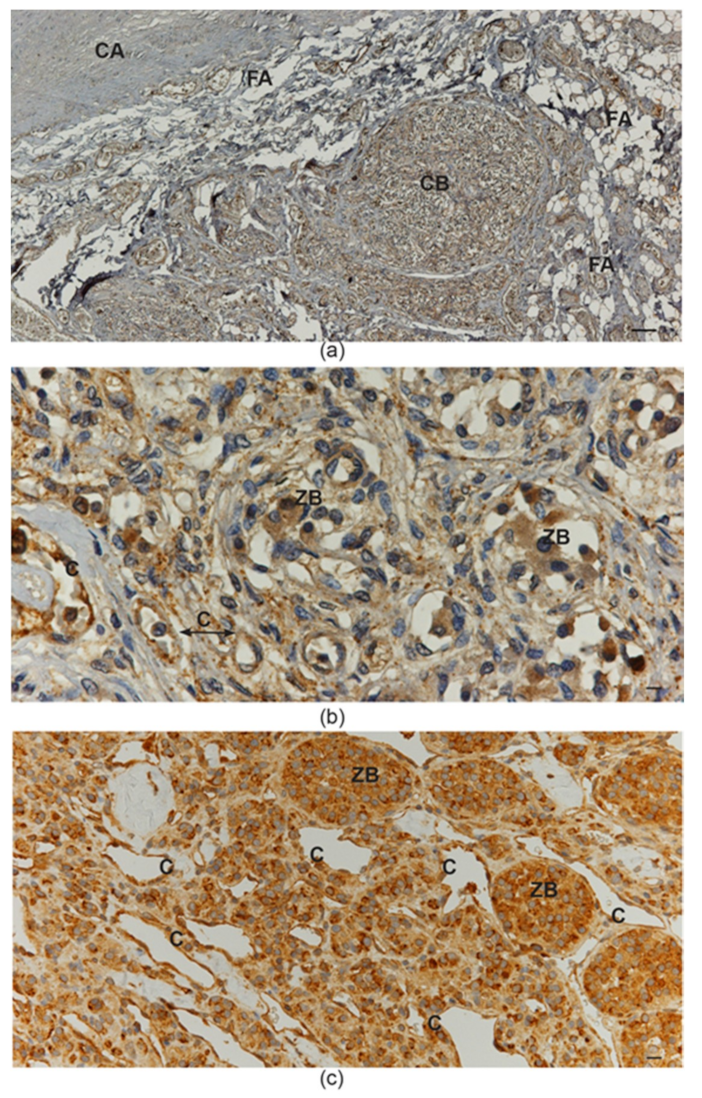
2. The Physiological Model of Carotid Body Hyperplasia Under Chronic Hypoxia May Illuminate Paraganglioma Development
3. Molecular Heterogeneities Do Not Exclude a Developmental Model of Paragangliar Tumorigenesis
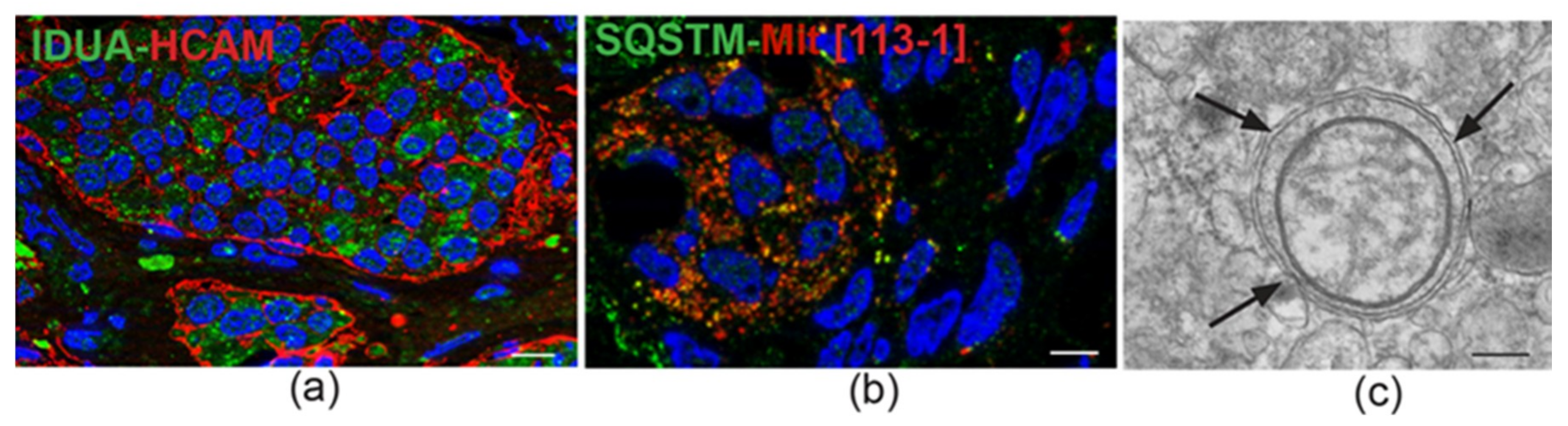
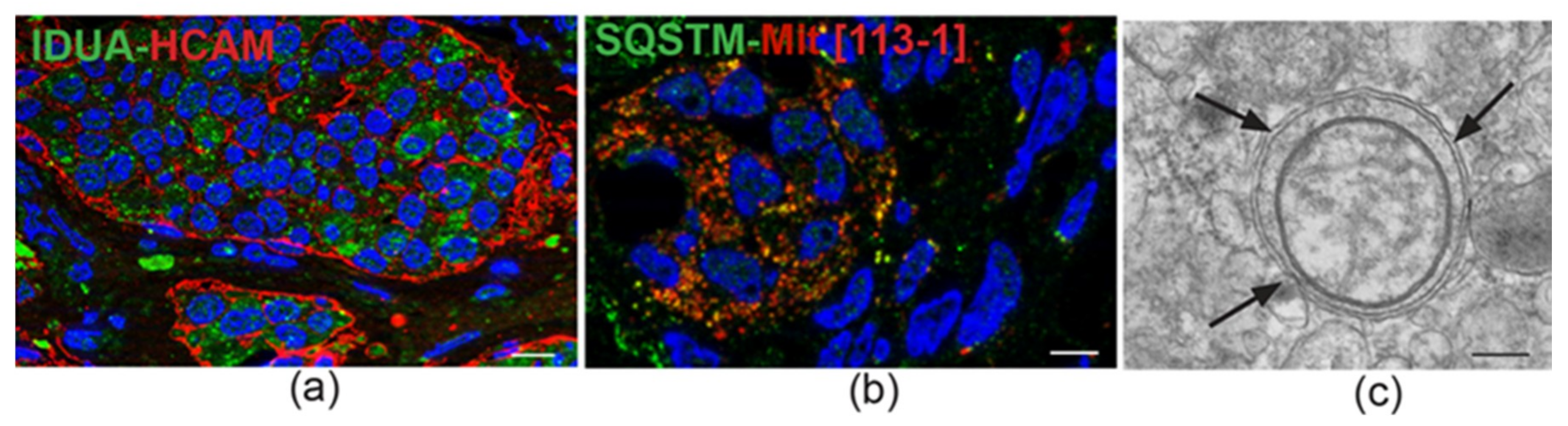
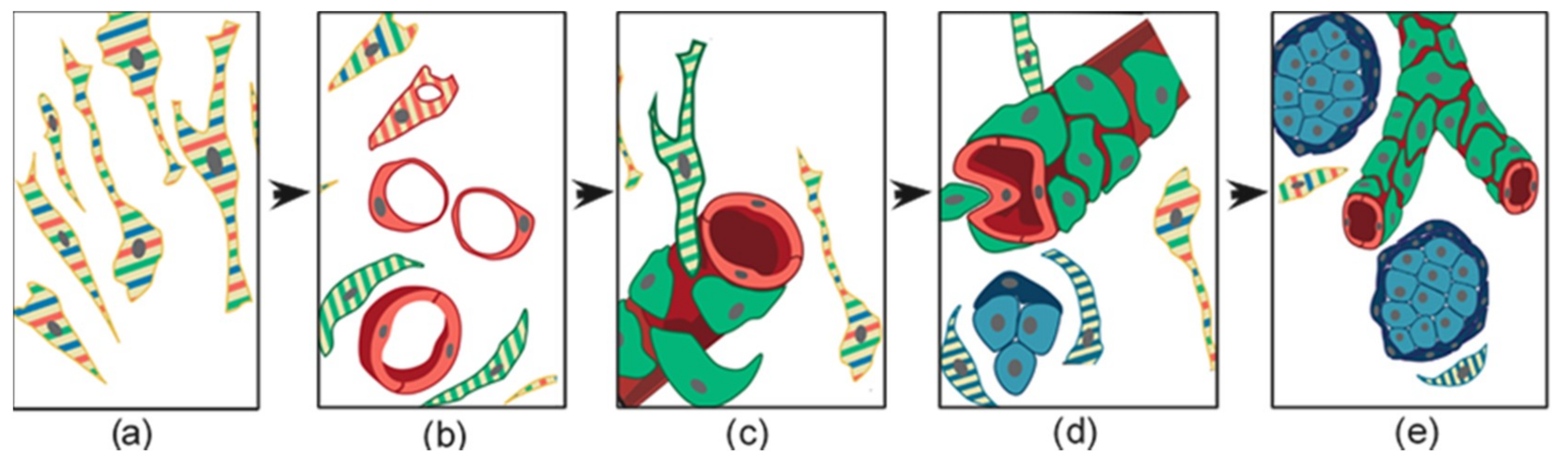
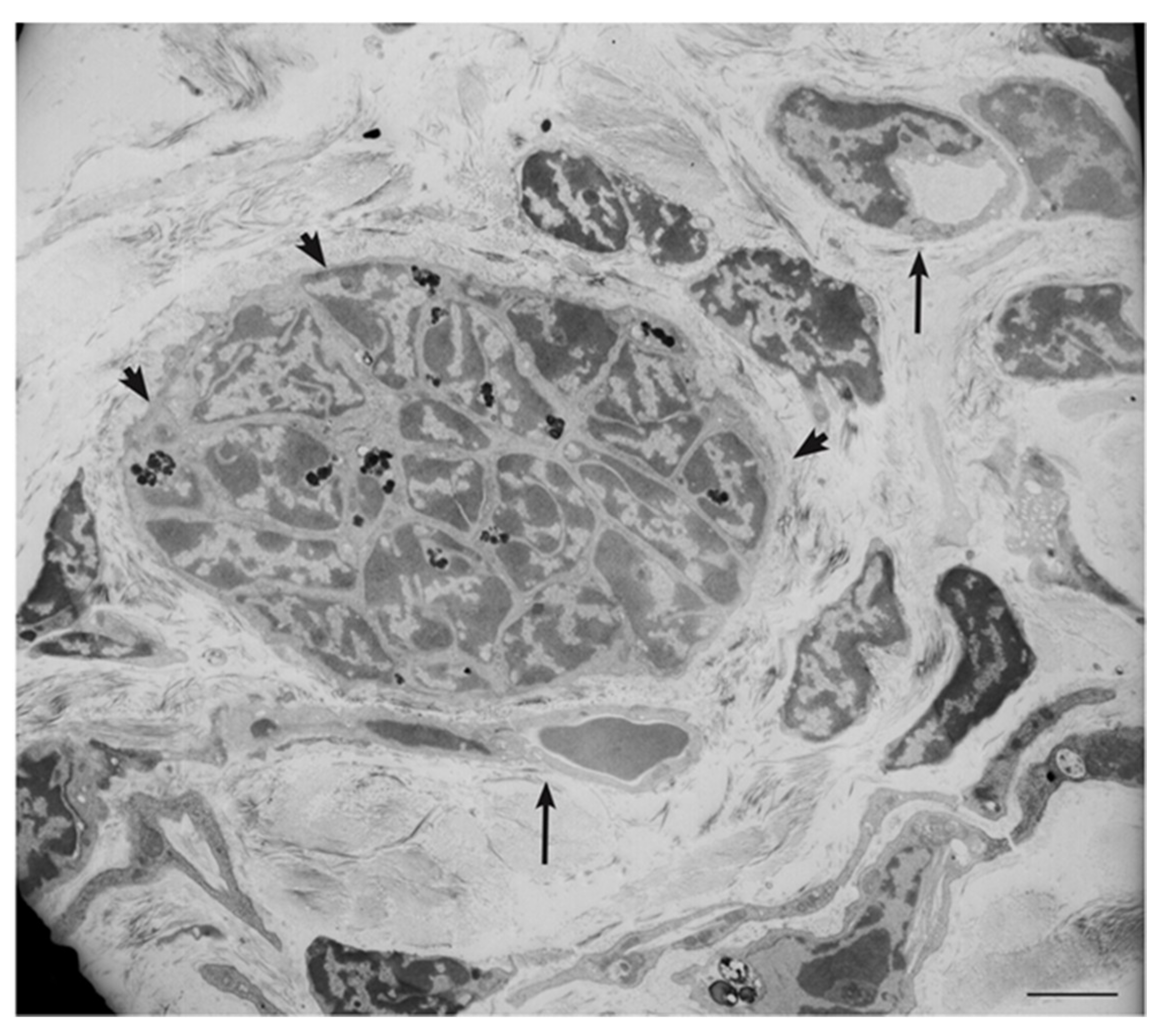
4. Ultrastructural and Immunomorphological Relationships Between the Vascular and Neural Compartments of Head and Neck Paragangliomas
5. Our Approach to the Study of Genes and Pathways Shared Among Head and Neck Paragangliomas
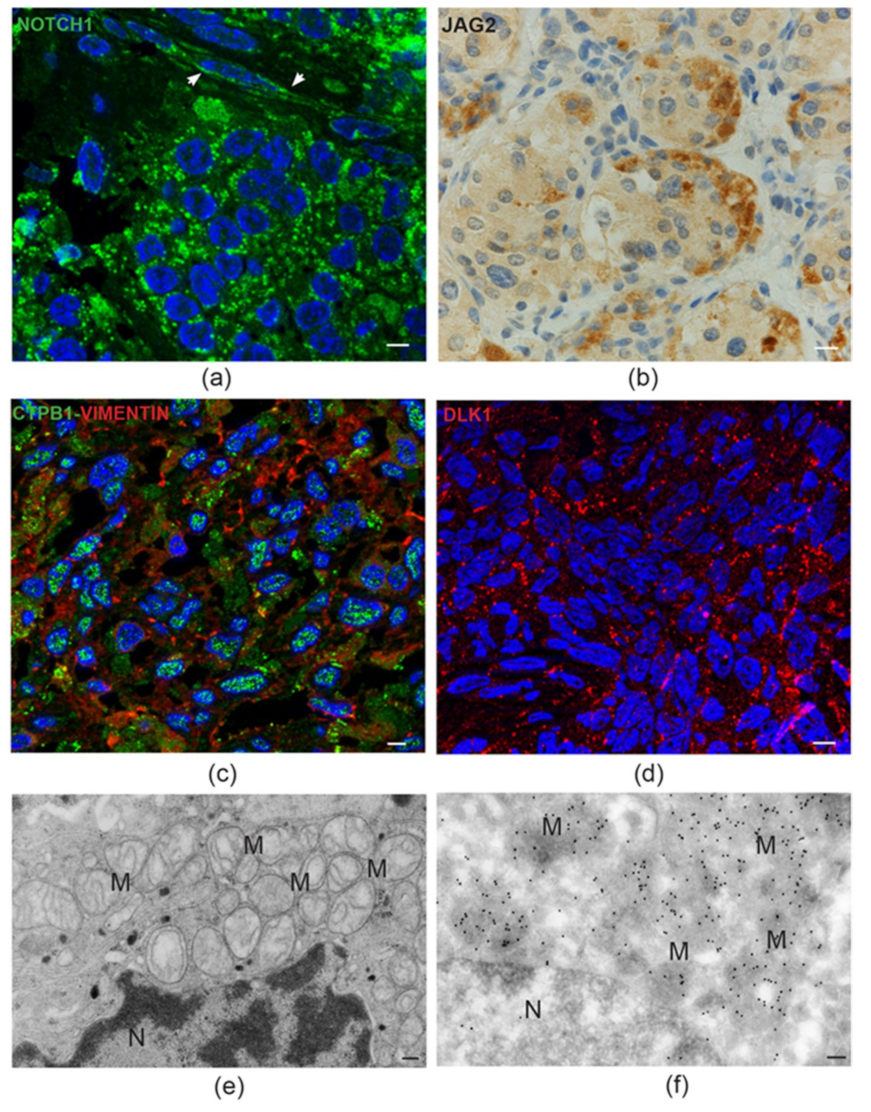
6. Constitutive Notch Signaling in Head and Neck Paraganglioma
7. Patient-Derived Head and Neck Paraganglioma Cultures Exhibit a Multipotent Mesenchymal-Like Phenotype
8. The microRNA-200s and -34s Modulate NOTCH1, ZEB1, and PDGFRA Levels in Paraganglioma
9. The Lesson of the Xenograft Models
10. Imatinib Blocks HNPGL Cell Growth and Inhibits Xenograft Formation
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kho, A.T.; Zhao, Q.; Cai, Z.; Butte, A.J.; Kim, J.Y.; Pomeroy, S.L.; Rowitch, D.H.; Kohane, I.S. Conserved mechanisms across development and tumorigenesis revealed by a mouse development perspective of human cancers. Genes Dev. 2004, 18, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna Priya, S.; Nagare, R.P.; Sneha, V.S.; Sidhanth, C.; Bindhya, S.; Manasa, P.; Ganesan, T.S. Tumour angiogenesis-Origin of blood vessels. Int. J. Cancer 2016, 139, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, M. Morphogenetic fields in embryogenesis, regeneration, and cancer: Non-local control of complex patterning. Biosystems 2012, 109, 243–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Maguire, L.H.; Thomas, A.R.; Goldstein, A.M. Tumors of the neural crest: Common themes in development and cancer. Dev. Dyn. 2015, 244, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Dupin, E.; Sommer, L. Neural crest progenitors and stem cells: From early development to adulthood. Dev. Biol. 2010, 366, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Wolsky, A. Regeneration and cancer. Growth 1978, 42, 425–426. [Google Scholar] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Bianco, P. “Mesenchymal” stem cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Remoli, C.; Giannicola, G.; Kogler, G.; Liedtke, S.; Cossu, G.; Serafini, M.; Sampaolesi, M.; Tagliafico, E.; et al. No Identical “Mesenchymal Stem Cells” at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem Cell Rep. 2016, 14, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Martucci, V.L.; Pacak, K. Pheochromocytoma and paraganglioma: Diagnosis, genetics, management, and treatment. Curr. Probl. Cancer 2014, 38, 7–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantorovich, V.; Eisenhofer, G.; Pacak, K. Pheochromocytoma: An endocrine stress mimicking disorder. Ann. N. Y. Acad. Sci. 2008, 1148, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Harari, A.; Inabnet, W.B., 3rd. Malignant pheochromocytoma: A review. Am. J. Surg. 2011, 201, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Kim, J.H.; Hong, A.R.; Seong, M.W.; Lee, K.E.; Kim, S.J.; Kim, S.W.; Shin, C.S.; Kim, S.Y. Disentangling of Malignancy from Benign Pheochromocytomas/Paragangliomas. PLoS ONE 2016, 11, e0168413. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takekoshi, K.; Naruse, M. Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine. J. Clin. Med. 2018, 7, 242. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D.; Young, W.F.; Kawashima, A.; Komminoth, P. Malignant adrenal phaeochromocytoma. In World Health Organization Classification of Tumours Pathology & Genetics, Tumours of Endocrine Organs, 3rd ed.; DeLellis, R.A., Lloyd, R.V., Eds.; IARC: Lyon, France, 2004; pp. 147–150. [Google Scholar]
- Kimura, N.; Capella, C. Extraadrenal paraganglioma. In WHO Classification of Tumors of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Eds.; IARC Press: Lyons, France, 2017; pp. 190–195. [Google Scholar]
- Tischler, A.S.; de Krijger, R.R. Phaeochromocytoma. In WHO Classification of Tumors of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Eds.; IARC Press: Lyons, France, 2017; pp. 183–189. [Google Scholar]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Cama, A.; Verginelli, F.; Lotti, L.V.; Napolitano, F.; Morgano, A.; D’Orazio, A.; Vacca, M.; Perconti, S.; Pepe, F.; Romani, F.; et al. Integrative genetic, epigenetic and pathological analysis of paraganglioma reveals complex dysregulation of NOTCH signaling. Acta Neuropathol. 2013, 126, 575–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Zhikrivetskaya, S.O.; Snezhkina, A.V.; Zaretsky, A.R.; Alekseev, B.Y.; Pokrovsky, A.V.; Golovyuk, A.L.; Melnikova, N.V.; Stepanov, O.A.; Kalinin, D.V.; Moskalev, A.A.; et al. Molecular markers of paragangliomas/pheochromocytomas. Oncotarget 2017, 8, 25756–25782. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.-P.; Oldenburg, R.A.; Nuk, J.; Hoekstra, A.S.; van der Meer, C.A.; Korpershoek, E.; McGillivray, B.; Corssmit, E.P.M.; Dinjens, W.N.M.; de Krijger, R.R.; et al. Paraganglioma and pheochromocytoma upon maternal transmission of SDHD mutations. BMC Med. Genet. 2014, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Zhu, Y.; Andrews, K.A.; Wilding, M.; Duncan, E.L.; Dwight, T.; Tothill, R.W.; Burgess, J.; Crook, A.; Gill, A.J.; et al. Bayesian approach to determining penetrance of pathogenic SDH variants. J. Med. Genet. 2018, 55, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Piruat, J.I.; Millán-Uclés, Á. Genetically Modeled Mice with Mutations in Mitochondrial Metabolic Enzymes for the Study of Cancer. Front. Oncol. 2014, 4, 200. [Google Scholar] [CrossRef] [PubMed]
- Saldana, M.J.; Salem, L.E.; Travezan, R. High altitude hypoxia and chemodectomas. Hum. Pathol. 1973, 4, 251–263. [Google Scholar] [CrossRef]
- Chedid, A.; Jao, W. Hereditary tumors of the carotid bodies and chronic obstructive pulmonary disease. Cancer 1974, 33, 1635–1641. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, J.H.; Killien, F.C.; Troupin, R.H. Bilateral carotid body tumors and cyanotic heart disease. AJR Am. J. Roentgenol. 1980, 134, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Cerecer-Gil, N.Y.; Figuera, L.E.; Llamas, F.J.; Lara, M.; Escamilla, J.G.; Ramos, R.; Estrada, G.; Hussain, A.K.; Gaal, J.; Korpershoek, E.; et al. Mutation of SDHB is a Cause of Hypoxia-Related High-Altitude Paraganglioma. Clin. Cancer Res. 2010, 16, 4148–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M.S.; Sajan, M.P.; Farese, R.V.; Freeman, R.S.; Carter, B.D.; Kaelin, W.G., Jr.; et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005, 8, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieneke, J.A.; Smith, A. Paraganglioma: Carotid body tumor. Head Neck Pathol. 2009, 3, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Pardal, R.; Ortega-Sáenz, P.; Durán, R.; López-Barneo, J. Glia-like stem cells sustain physiologic neurogenesis in the adult mammalian carotid body. Cell 2007, 131, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Annese, V.; Navarro-Guerrero, E.; Rodríguez-Prieto, I.; Pardal, R. Physiological Plasticity of Neural-Crest-Derived Stem Cells in the Adult Mammalian Carotid Body. Cell Rep. 2017, 19, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Sobrino, V.; Annese, V.; Navarro-Guerrero, E.; Platero-Luengo, A.; Pardal, R. The carotid body: A physiologically relevant germinal niche in the adult peripheral nervous system. Cell. Mol. Life Sci. 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Kubo, S.; Nakano-Doi, A.; Sakuma, R.; Lu, S.; Narita, A.; Kawahara, M.; Taguchi, A.; Matsuyama, T. Brain vascular pericytes following ischemia have multipotential stem cell activity to differentiate into neural and vascular lineage cells. Stem Cells 2015, 33, 1962–1974. [Google Scholar] [CrossRef] [PubMed]
- Farahani, R.M.; Rezaei-Lotfi, S.; Simonian, M.; Xaymardan, M.; Hunter, N. Neural microvascular pericytes contribute to human adult neurogenesis. J. Comp. Neurol. 2019, 527, 780–796. [Google Scholar] [CrossRef] [PubMed]
- Salman, S.; Buttigieg, J.; Nurse, C.A. Ontogeny of O2 and CO2//H+ chemosensitivity in adrenal chromaffin cells: Role of innervation. J. Exp. Biol. 2014, 217 Pt 5, 673–681. [Google Scholar] [CrossRef]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T.; Akkuratova, N.; Usoskin, D.; Kamenev, D.; et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 2017, 357, eaal3753. [Google Scholar] [CrossRef] [PubMed]
- Hockman, D.; Adameyko, I.; Kaucka, M.; Barraud, P.; Otani, T.; Hunt, A.; Hartwig, A.C.; Sock, E.; Waithe, D.; Franck, M.C.M.; et al. Striking parallels between carotid body glomus cell and adrenal chromaffin cell development. Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Devilee, P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr. Opin. Genet. Dev. 2010, 20, 324–329. [Google Scholar] [CrossRef] [PubMed]
- van Schothorst, E.M.; Beekman, M.; Torremans, P.; Kuipers-Dijkshoorn, N.J.; Wessels, H.W.; Bardoel, A.F.; van der Mey, A.G.; van der Vijver, M.J.; van Ommen, G.J.; Devilee, P.; et al. Paragangliomas of the head and neck region show complete loss of heterozygosity at 11q22-q23 in chief cells and the flowsorted DNA aneuploid fraction. Hum. Pathol. 1998, 29, 1045–1049. [Google Scholar] [CrossRef]
- Douwes Dekker, P.B.; Corver, W.E.; Hogendoorn, P.C.; van der Mey, A.G.; Cornelisse, C.J. Multiparameter DNA flow-sorting demonstrates diploidy and SDHD wild-type gene retention in the sustentacular cell compartment of head and neck paragangliomas: Chief cells are the only neoplastic component. J. Pathol. 2004, 202, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.C.; Seftor, E.A.; Hess, A.R.; Seftor, R.E.B. Vasculogenic mimicry and tumour-cell plasticity: Lessons from melanoma. Nat. Rev. Cancer 2003, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Kuratani, S.; Kusakabe, R.; Hirasawa, T. The neural crest and evolution of the head/trunk interface in vertebrates. Dev. Biol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2016, 8, 30502–30510. [Google Scholar] [CrossRef] [PubMed]
- Turkova, H.; Prodanov, T.; Maly, M.; Martucci, V.; Adams, K.; Widimsky, J., Jr.; Chen, C.C.; Ling, A.; Kebebew, E.; Stratakis, C.A.; et al. Characteristics and outcomes of metastatic SDHB and sporadic pheochromcytoma/paraganglioma: An National Institutes of Halth Study. Endocr. Pract. 2016, 22, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Comino-Méndez, I.; Tejera, Á.M.; Currás-Freixes, M.; Remacha, L.; Gonzalvo, P.; Tonda, R.; Letón, R.; Blasco, M.A.; Robledo, M.; Cascón, A. ATRX driver mutation in a composite malignant pheochromocytoma. Cancer Genet. 2016, 209, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.L.; Terzic, A. Metabolic determinants of embryonic development and stem cell fate. Reprod. Fertil. Dev. 2014, 27, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Chisolm, D.A.; Weinmann, A.S. Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annu. Rev. Immunol. 2018, 36, 221–246. [Google Scholar] [CrossRef] [PubMed]
- Janke, R.; Dodson, A.E.; Rine, J. Metabolism and Epigenetics. Annu. Rev. Cell Dev. Biol. 2015, 31, 473–496. [Google Scholar] [CrossRef] [PubMed]
- Verginelli, F.; Perconti, S.; Vespa, S.; Schiavi, F.; Prasad, S.C.; Lanuti, P.; Cama, A.; Tramontana, L.; Esposito, D.L.; Guarnieri, S.; et al. Paragangliomas arise through an autonomous vasculo-angio-neurogenic program inhibited by imatinib. Acta Neuropathol. 2018, 135, 779–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Serpa, J. Metabolic cooperation between cancer and non-cancerous stromal cells is pivotal in cancer progression. Tumour Biol. 2018, 40. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Kaliski, A.; Boedeker, C.C.; Martucci, V.; Fojo, T.; Adler, J.R.; Pacak, K. Current Approaches and Recent Developments in the Management of Head and Neck Paragangliomas. Endocr. Rev. 2014, 35, 795–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise review: Evidence for CD34 as a common marker for diverse progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dong, M.; Wang, X.G. The Implication and Significance of Beta 2 Microglobulin: A Conservative Multifunctional Regulator. Chin. Med. J. 2016, 129, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A. Mucopolysaccharidosis Type I. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1162/ (accessed on 12 December 2018).
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavao, M.; Tzanakakis, G.M.; Karamanos, N.K. Glycosaminoglycans: Key players in cancer cell biology and treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Christianson, H.C.; Belting, M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Huang, M.; Sun, S.; Wu, Y.; Lin, X. Epithelial heparan sulfate regulates Sonic Hedgehog signaling in lung development. PLoS Genet. 2017, 13, e1006992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Mata, M.; Cotán, D.; Villanueva-Paz, M.; de Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Suárez-Rivero, J.M.; Tiscornia, G.; Oropesa-Ávila, M. Mitochondrial Dysfunction in Lysosomal Storage Disorders. Diseases 2016, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Kotiadis, V.N.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 2014, 1840, 1254–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caolo, V.; Molin, D.G.; Post, M.J. Notch regulation of hematopoiesis, endothelial precursor cells, and blood vessel formation: Orchestrating the vasculature. Stem Cells Int. 2012, 2012, 805602. [Google Scholar] [CrossRef] [PubMed]
- Kofler, N.M.; Shawber, C.J.; Kangsamaksin, T.; Reed, H.O.; Galatioto, J.; Kitajewski, J. Notch signaling in developmental and tumor angiogenesis. Genes Cancer 2011, 2, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Kageyama, R. The role of Notch signaling in adult neurogenesis. Mol. Neurobiol. 2011, 44, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; von Stedingk, K.; Lindgren, D.; Påhlman, S.; Axelson, H. JAG2 induction in hypoxic tumor cells alters Notch signaling and enhances endothelial cell tube formation. Mol. Cancer Res. 2011, 9, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Hitoshi, S.; Ishino, Y.; Kumar, A.; Jasmine, S.; Tanaka, K.F.; Kondo, T.; Kato, S.; Hosoya, T.; Hotta, Y.; Ikenaka, K. Mammalian Gcm genes induce Hes5 expression by active DNA demethylation and induce neural stem cells. Nat. Neurosci. 2011, 14, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chen, Y.G. Dishevelled: The hub of Wnt signaling. Cell. Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Blevins, M.A.; Huang, M.; Zhaoa, R. The role of CtBP1 in oncogenic processes and its potential as a therapeutic target. Mol. Cancer Ther. 2017, 16, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, H.; Yin, X.; Liu, H.; Liu, J.; Guo, D.; Shi, Z. C-Terminal Binding Protein 1 Modulates Cellular Redox via Feedback Regulation of MPC1 and MPC2 in Melanoma Cells. Med. Sci. Monit. 2018, 24, 7614–7624. [Google Scholar] [CrossRef] [PubMed]
- Lorendeau, D.; Rinaldi, G.; Boon, R.; Spincemaille, P.; Metzger, K.; Jäger, C.; Christen, S.; Dong, X.; Kuenen, S.; Voordeckers, K.; et al. Dual loss of succinate dehydrogenase (SDH) and complex I activity is necessary to recapitulate the metabolic phenotype of SDH mutant tumors. Metab. Eng. 2017, 43, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NAD+/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef] [PubMed]
- Perumalsamy, L.R.; Nagala, M.; Sarin, A. Notch-activated signaling cascade interacts with mitochondrial remodeling proteins to regulate cell survival. Proc. Natl. Acad. Sci. USA 2010, 107, 6882–6887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, N.P.; Roy, A.; Banerjee, S. Alteration of mitochondrial proteome due to activation of Notch1 signaling pathway. J. Biol. Chem. 2014, 289, 7320–7334. [Google Scholar] [CrossRef] [PubMed]
- Mutvei, A.P.; Landor, S.K.-J.; Fox, R.; Braune, E.-B.; Tsoi, Y.L.; Phoon, Y.P.; Sahlgren, C.; Hartman, J.; Bergh, J.; Jin, S.; et al. Notch signaling promotes a HIF2α-driven hypoxic response in multiple tumor cell types. Oncogene 2018, 37, 6083–6095. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Agüera, M.C.; Gao, L.; González-Rodríguez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grego-Bessa, J.; Díez, J.; Timmerman, L.; de la Pompa, J.L. Notch and epithelial-mesenchyme transition in development and tumor progression: Another turn of the screw. Cell Cycle 2004, 3, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/miR-34 axis in development and disease. J. Mol. Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farahani, R.M.; Xaymardan, M. Platelet-Derived Growth Factor Receptor Alpha as a Marker of Mesenchymal Stem Cells in Development and Stem Cell Biology. Stem Cells Int. 2015, 2015, 362753. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Smith, S.C.; Faber, A.C.; Trent, J.; Grossman, S.R.; Stratakis, C.A.; Boikos, S.A. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer 2018, 4, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Kamei, M.; Saunders, W.B.; Bayless, K.J.; Dye, L.; Davis, G.E.; Weinstein, B.M. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 2006, 442, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Iruela-Arispe, M.L.; Beitel, G.J. Tubulogenesis. Development 2013, 140, 2851–2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D.; Meissner, N.; Kleff, V.; Jastrow, H.; Yamaguchi, M.; Ergün, S.; Jendrossek, V. Nestin(+) tissue-resident multipotent stem cells contribute to tumor progression by differentiating into pericytes and smooth muscle cells resulting in blood vessel remodeling. Front. Oncol. 2014, 4, 169. [Google Scholar] [CrossRef] [PubMed]
- Mentzer, S.J.; Konerding, M.A. Intussusceptive angiogenesis: Expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lin, Q.; Zelterman, D.; Yun, Z. Hypoxia-regulated delta-like 1 homologue enhances cancer cell stemness and tumorigenicity. Cancer Res. 2009, 69, 9271–9280. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.S.; Huang, P.H. The pathobiology of collagens in glioma. Mol. Cancer Res. 2013, 11, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, J.; Miosge, N. Basement membrane components are key players in specialized extracellular matrices. Cell. Mol. Life Sci. 2010, 67, 2879–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, K.E.; Joy, S.; Delhove, J.M.K.M.; Kotiadis, V.N.; Fernandez, E.; Fitzpatrick, L.M.; Whiteford, J.R.; King, P.J.; Bolanos, J.P.; Duchen, M.R.; et al. NRF2 Orchestrates the Metabolic Shift during Induced Pluripotent Stem Cell Reprogramming. Cell Rep. 2016, 14, 1883–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, S.K.; Kusumbe, A.P.; Adams, R.H. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol. 2015, 25, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertmer, A.; Huber, V.; Gilch, S.; Yoshimori, T.; Erfle, V.; Duyster, J.; Elsässer, H.P.; Schätzl, H.M. The anticancer drug imatinib induces cellular autophagy. Leukemia 2007, 21, 936–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, A.; Zent, R. Extracellular matrix receptors in branched organs. Curr. Opin. Cell Biol. 2011, 23, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannezo, E.; Scheele, C.L.G.J.; Moad, M.; Drogo, N.; Heer, R.; Sampogna, R.V.; van Rheenen, J.; Simons, B.D. A Unifying Theory of Branching Morphogenesis. Cell 2017, 171, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Casaletto, J.B.; McClatchey, A.I. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 2012, 12, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnenschein, C.; Soto, A.M.; Rangarajan, A.; Kulkarni, P. Competing views on cancer. J. Biosci. 2014, 39, 281–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecot, C.V.; Rupaimoole, R.; Yang, D.; Akbani, R.; Ivan, C.; Lu, C.; Wu, S.; Han, H.D.; Shah, M.Y.; Rodriguez-Aguayo, C.; et al. Tumour angiogenesis regulation by the miR-200 family. Nat. Commun. 2013, 4, 2427. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, J.; Chen, A.F. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E110–E116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C. Treatment for Patients with Malignant Pheochromocytomas and Paragangliomas: A Perspective from the Hallmarks of Cancer. Front. Endocrinol. 2018, 9, 277. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.C.; Mimoune, H.A.; D’Orazio, F.; Medina, M.; Bacciu, A.; Mariani-Costantini, R.; Piazza, P.; Sanna, M. The role of wait-and-scan and the efficacy of radiotherapy in the treatment of temporal bone paragangliomas. Otol. Neurotol. 2014, 35, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Briere, J.J.; Burnichon, N.; Riviere, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; De Reynies, A.; Bertherat, J.; Badoual, C.; et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lotti, L.V.; Vespa, S.; Pantalone, M.R.; Perconti, S.; Esposito, D.L.; Visone, R.; Veronese, A.; Paties, C.T.; Sanna, M.; Verginelli, F.; et al. A Developmental Perspective on Paragangliar Tumorigenesis. Cancers 2019, 11, 273. https://doi.org/10.3390/cancers11030273
Lotti LV, Vespa S, Pantalone MR, Perconti S, Esposito DL, Visone R, Veronese A, Paties CT, Sanna M, Verginelli F, et al. A Developmental Perspective on Paragangliar Tumorigenesis. Cancers. 2019; 11(3):273. https://doi.org/10.3390/cancers11030273
Chicago/Turabian StyleLotti, Lavinia Vittoria, Simone Vespa, Mattia Russel Pantalone, Silvia Perconti, Diana Liberata Esposito, Rosa Visone, Angelo Veronese, Carlo Terenzio Paties, Mario Sanna, Fabio Verginelli, and et al. 2019. "A Developmental Perspective on Paragangliar Tumorigenesis" Cancers 11, no. 3: 273. https://doi.org/10.3390/cancers11030273