Reprogramming of Energy Metabolism in Response to Radiotherapy in Head and Neck Squamous Cell Carcinoma

 ,
,  and
and 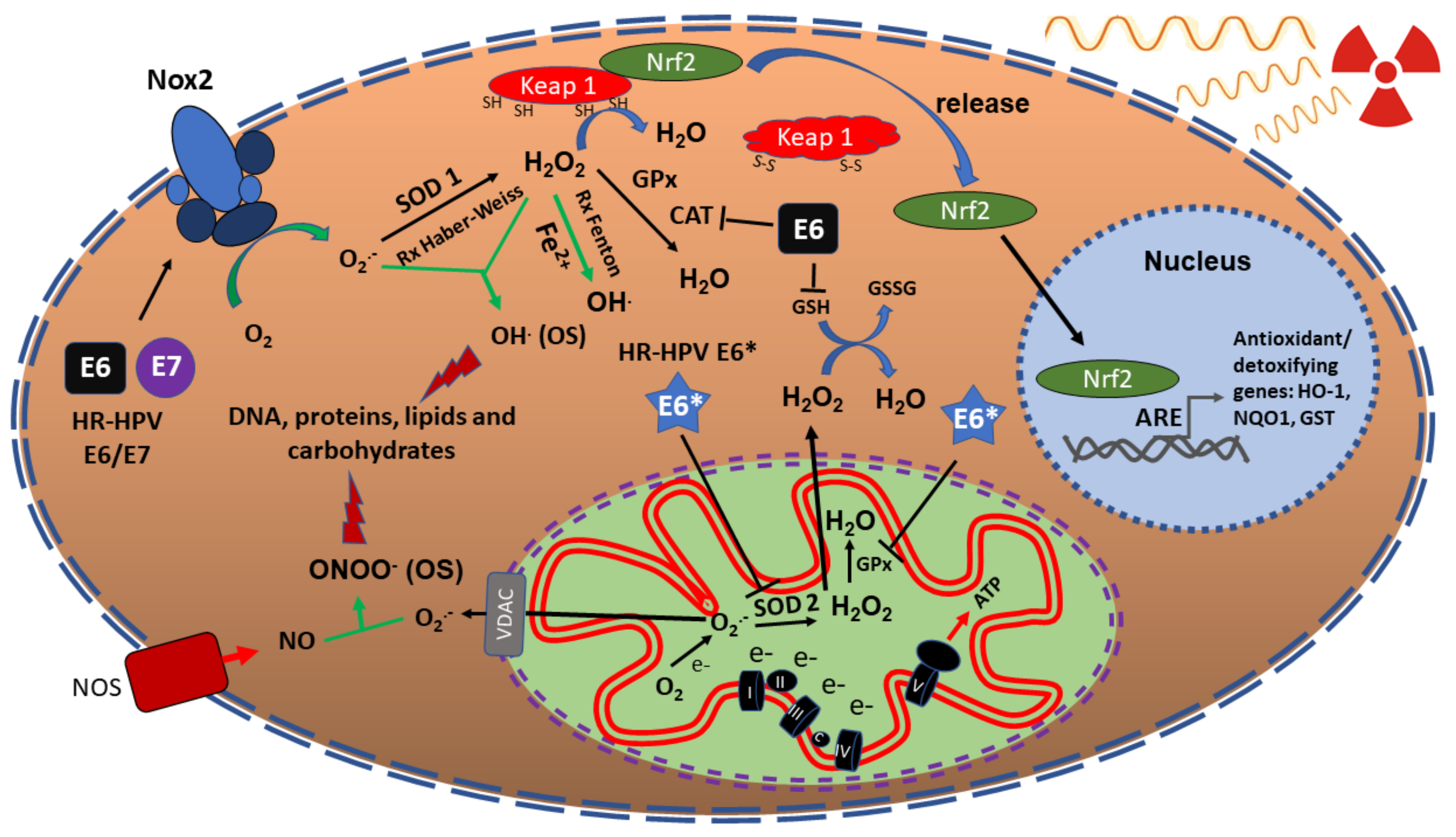{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genetic Alterations in HPV-Positive and HPV-Negative HNSCC
3. Radiosensitivity in HPV-Positive HNSCC
4. Nuclear Factor (Erythoid-Derived 2)-Like 2 (Nrf2) Induces Radioresistance in HNSCC
5. Radioresistance and Glucose Metabolism in HNSCC
6. Role of Mitochondria in Irradiated HNSCC
7. Hypoxia and ROS in Response to Radiotherapy in HNSCC
8. Lipid Metabolism in HNSCC Radioresistance
9. Autophagy in HNSCC Radioresistance
10. Metabolic Therapy for HNSCC
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Mirghani, H.; Amen, F.; Tao, Y.; Deutsch, E.; Levy, A. Increased radiosensitivity of HPV-positive head and neck cancers: Molecular basis and therapeutic perspectives. Cancer Treat. Rev. 2015, 41, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Spence, T.; Bruce, J.; Yip, K.W.; Liu, F.-F. HPV Associated Head and Neck Cancer. Cancers 2016, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 2010, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pinatti, L.M.; Walline, H.M.; Carey, T.E. Human Papillomavirus Genome Integration and Head and Neck Cancer. J. Dent. Res. 2018, 97, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.C.; Myers, S.L.; Persing, D.H.; Sarkar, G.; McGovern, R.M.; Gostout, B.S.; Smith, D.I. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res. 2000, 60, 5916–5921. [Google Scholar] [PubMed]
- Winder, D.M.; Pett, M.R.; Foster, N.; Shivji, M.K.K.; Herdman, M.T.; Stanley, M.A.; Venkitaraman, A.R.; Coleman, N. An increase in DNA double-strand breaks, induced by Ku70 depletion, is associated with human papillomavirus 16 episome loss and de novo viral integration events. J. Pathol. 2007, 213, 27–34. [Google Scholar] [CrossRef]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef]
- Chen Wongworawat, Y.; Filippova, M.; Williams, V.M.; Filippov, V.; Duerksen-Hughes, P.J. Chronic oxidative stress increases the integration frequency of foreign DNA and human papillomavirus 16 in human keratinocytes. Am. J. Cancer Res. 2016, 6, 764–780. [Google Scholar]
- Bernier, J.; Hall, E.J.; Giaccia, A. Radiation oncology: A century of achievements. Nat. Rev. Cancer 2004, 4, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Nijkamp, M.M.; Span, P.N.; Bussink, J.; Kaanders, J.H.A.M. Interaction of EGFR with the tumour microenvironment: Implications for radiation treatment. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 108, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogineni, V.R.; Nalla, A.K.; Gupta, R.; Dinh, D.H.; Klopfenstein, J.D.; Rao, J.S. Chk2-mediated G2/M cell cycle arrest maintains radiation resistance in malignant meningioma cells. Cancer Lett. 2011, 313, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsubhi, N.; Middleton, F.; Abdel-Fatah, T.M.A.; Stephens, P.; Doherty, R.; Arora, A.; Moseley, P.M.; Chan, S.Y.T.; Aleskandarany, M.A.; Green, A.R.; et al. Chk1 phosphorylated at serine345 is a predictor of early local recurrence and radio-resistance in breast cancer. Mol. Oncol. 2016, 10, 213–223. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, M.M.L.; Bjørås, K.Ø.; Hanssen-Bauer, A.; Solvang-Garten, K.; Otterlei, M. p38 MAPK signaling and phosphorylations in the BRCT1 domain regulate XRCC1 recruitment to sites of DNA damage. Sci. Rep. 2017, 7, 6322. [Google Scholar] [CrossRef]
- Jang, J.; Huh, Y.J.; Cho, H.-J.; Lee, B.; Park, J.; Hwang, D.-Y.; Kim, D.-W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem Cell Rep. 2017, 9, 629–641. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, J.; Ye, S.; He, M.; Ren, R.; Yuan, D.; Shao, C. SirT1 regulates radiosensitivity of hepatoma cells differently under normoxic and hypoxic conditions. Cancer Sci. 2012, 103, 1238–1244. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Tang, L.; He, Y.; Wu, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Liao, Q.; et al. BPIFB1 (LPLUNC1) inhibits radioresistance in nasopharyngeal carcinoma by inhibiting VTN expression. Cell Death Dis. 2018, 9, 432. [Google Scholar] [CrossRef] [Green Version]
- Scanlon, C.S.; Van Tubergen, E.A.; Inglehart, R.C.; D’Silva, N.J. Biomarkers of epithelial-mesenchymal transition in squamous cell carcinoma. J. Dent. Res. 2013, 92, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-K.; Jiang, X.-Y.; Zhou, X.-X.; Wang, D.-M.; Song, X.-L.; Jiang, H.-B. Upregulation of vimentin and aberrant expression of E-cadherin/beta-catenin complex in oral squamous cell carcinomas: Correlation with the clinicopathological features and patient outcome. Mod. Pathol. 2010, 23, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Jerhammar, F.; Ceder, R.; Garvin, S.; Grénman, R.; Grafström, R.C.; Roberg, K. Fibronectin 1 is a potential biomarker for radioresistance in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2010, 10, 1244–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 2007, 13, 3056–3062. [Google Scholar] [CrossRef] [PubMed]
- Brieger, J.; Kattwinkel, J.; Berres, M.; Gosepath, J.; Mann, W.J. Impact of vascular endothelial growth factor release on radiation resistance. Oncol. Rep. 2007, 18, 1597–1601. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Dok, R.; Nuyts, S. HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies. Cancers 2016, 8, 41. [Google Scholar] [CrossRef]
- Jou, A.; Hess, J. Epidemiology and Molecular Biology of Head and Neck Cancer. Oncol. Res. Treat. 2017, 40, 328–332. [Google Scholar] [CrossRef]
- Solomon, B.; Young, R.J.; Rischin, D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin. Cancer Biol. 2018, 52, 228–240. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.-X.; Zhang, J.; Wang, J.; et al. Exome Sequencing of Head and Neck Squamous Cell Carcinoma Reveals Inactivating Mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and Comparative Genomic Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, I.; Matsumura, H.; Fujii, W.; Naito, K.; Kusakabe, K.; Kiso, Y.; Kano, K. Discoidin domain receptor 2 (DDR2) regulates body size and fat metabolism in mice. Transgenic Res. 2014, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [Green Version]
- Puzio-Kuter, A.M. The Role of p53 in Metabolic Regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Hashibe, M.; Boffetta, P.; Zaridze, D.; Shangina, O.; Szeszenia-Dabrowska, N.; Mates, D.; Janout, V.; Fabiánová, E.; Bencko, V.; Moullan, N.; et al. Evidence for an important role of alcohol- and aldehyde-metabolizing genes in cancers of the upper aerodigestive tract. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2006, 15, 696–703. [Google Scholar] [CrossRef]
- D’Souza, G.; Dempsey, A. The role of HPV in head and neck cancer and review of the HPV vaccine. Prev. Med. 2011, 53 (Suppl. 1), S5–S11. [Google Scholar] [CrossRef] [Green Version]
- Mallen-St Clair, J.; Alani, M.; Wang, M.B.; Srivatsan, E.S. Human papillomavirus in oropharyngeal cancer: The changing face of a disease. Biochim. Biophys. Acta 2016, 1866, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, T.; Tribius, S.; Grob, T.J.; Meyer, F.; Busch, C.-J.; Petersen, C.; Dikomey, E.; Kriegs, M. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 107, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Gonzaléz-García, M.C.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Valverde, M.; Rojas, E.; Rodríguez-Sastre, M.A.; García-Cuellar, C.M.; Lizano, M. Human Papillomavirus Types 16 and 18 Early-expressed Proteins Differentially Modulate the Cellular Redox State and DNA Damage. Int. J. Biol. Sci. 2018, 14, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, V.M.; Filippova, M.; Filippov, V.; Payne, K.J.; Duerksen-Hughes, P. Human papillomavirus type 16 E6* induces oxidative stress and DNA damage. J. Virol. 2014, 88, 6751–6761. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Zhang, H.; Chen, G.Z.; Shin, D.M.; Doetsch, P.W. HPV16 E6 and E7 proteins induce a chronic oxidative stress response via NOX2 that causes genomic instability and increased susceptibility to DNA damage in head and neck cancer cells. Carcinogenesis 2015, 36, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Chen, X.; Mims, J.; Huang, X.; Singh, N.; Motea, E.; Planchon, S.M.; Beg, M.; Tsang, A.W.; Porosnicu, M.; Kemp, M.L.; et al. Modulators of Redox Metabolism in Head and Neck Cancer. Antioxid. Redox Signal. 2018, 29, 1660–1690. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Long, D.J.; Jaiswal, A.K. Antioxidant regulation of genes encoding enzymes that detoxify xenobiotics and carcinogens. Curr. Top. Cell. Regul. 2000, 36, 201–216. [Google Scholar] [PubMed]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef]
- Berberat, P.O.; Dambrauskas, Z.; Gulbinas, A.; Giese, T.; Giese, N.; Künzli, B.; Autschbach, F.; Meuer, S.; Büchler, M.W.; Friess, H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 3790–3798. [Google Scholar] [CrossRef]
- Ewing, P.; Wilke, A.; Eissner, G.; Holler, E.; Andreesen, R.; Gerbitz, A. Expression of heme oxygenase-1 protects endothelial cells from irradiation-induced apoptosis. Endothel. J. Endothel. Cell Res. 2005, 12, 113–119. [Google Scholar] [CrossRef]
- Yokoyama, S.; Mita, S.; Okabe, A.; Abe, M.; Ogawa, M. Prediction of radiosensitivity in human esophageal squamous cell carcinomas with heme oxygenase-1: A clinicopathological and immunohistochemical study. Oncol. Rep. 2001, 8, 355–358. [Google Scholar] [CrossRef]
- Lee, S.; Lim, M.-J.; Kim, M.-H.; Yu, C.-H.; Yun, Y.-S.; Ahn, J.; Song, J.-Y. An effective strategy for increasing the radiosensitivity of Human lung Cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radic. Biol. Med. 2012, 53, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-F.; Keng, P.C.; Shau, H.; Wu, C.-T.; Hu, Y.-C.; Liao, S.-K.; Chen, W.-C. Inhibition of lung tumor growth and augmentation of radiosensitivity by decreasing peroxiredoxin I expression. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Huang, X.; Zhang, J.; Luo, Y.; Peng, Z.; Li, S. Downregulation of peroxiredoxin I by a novel fully human phage display recombinant antibody induces apoptosis and enhances radiation sensitization in A549 lung carcinoma cells. Cancer Biother. Radiopharm. 2012, 27, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Namani, A.; Matiur Rahaman, M.; Chen, M.; Tang, X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hu, P.; Li, B.; Zhang, J.-P.; Cheng, Y.-F.; Liang, Y.-M. Role of Nrf2 signaling pathway in the radiation tolerance of patients with head and neck squamous cell carcinoma: An in vivo and in vitro study. OncoTargets Ther. 2017, 10, 1809–1819. [Google Scholar] [CrossRef]
- Barger, J.F.; Plas, D.R. Balancing biosynthesis and bioenergetics: Metabolic programs in oncogenesis. Endocr. Relat. Cancer 2010, 17, R287–R304. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef]
- Van Veelen, C.W.; Rijksen, G.; Van Ketel, B.A.; Staal, G.E. The pyruvate kinase isoenzyme shift in human gliomas: A potential marker in the treatment of gliomas. Br. J. Neurosurg. 1988, 2, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.E.; Graham, J.F.; Cummins, C.J.; Loreck, D.J.; Galarraga, J.; Van der Feen, J.; DeLaPaz, R.; Smith, B.H. Enzymes of glucose metabolism in cultured human gliomas: Neoplasia is accompanied by altered hexokinase, phosphofructokinase, and glucose-6-phosphate dehydrogenase levels. Metab. Brain Dis. 1987, 2, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Flier, J.S.; Mueckler, M.M.; Usher, P.; Lodish, H.F. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987, 235, 1492–1495. [Google Scholar] [CrossRef]
- Kunkel, M.; Moergel, M.; Stockinger, M.; Jeong, J.-H.; Fritz, G.; Lehr, H.-A.; Whiteside, T.L. Overexpression of GLUT-1 is associated with resistance to radiotherapy and adverse prognosis in squamous cell carcinoma of the oral cavity. Oral Oncol. 2007, 43, 796–803. [Google Scholar] [CrossRef]
- Yan, S.-X.; Luo, X.-M.; Zhou, S.-H.; Bao, Y.-Y.; Fan, J.; Lu, Z.-J.; Liao, X.-B.; Huang, Y.-P.; Wu, T.-T.; Wang, Q.-Y. Effect of antisense oligodeoxynucleotides glucose transporter-1 on enhancement of radiosensitivity of laryngeal carcinoma. Int. J. Med. Sci. 2013, 10, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Mims, J.; Bansal, N.; Bharadwaj, M.S.; Chen, X.; Molina, A.J.; Tsang, A.W.; Furdui, C.M. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat. Res. 2015, 183, 291–304. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Najy, A.J.; Huang, W.; Sethi, S.; Snyder, M.; Sakr, W.; Dyson, G.; Hüttemann, M.; Lee, I.; Ali-Fehmi, R.; et al. HPV-associated differential regulation of tumor metabolism in oropharyngeal head and neck cancer. Oncotarget 2017, 8, 51530–51541. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Kam, W.W.-Y.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Li, Y.-L.; Chang, J.T.; Lee, L.-Y.; Fan, K.-H.; Lu, Y.-C.; Li, Y.-C.; Chiang, C.-H.; You, G.-R.; Chen, H.-Y.; Cheng, A.-J. GDF15 contributes to radioresistance and cancer stemness of head and neck cancer by regulating cellular reactive oxygen species via a SMAD-associated signaling pathway. Oncotarget 2017, 8, 1508–1528. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Miao, L.; St Clair, D.K.; St Clair, W.H. Redox-modulated phenomena and radiation therapy: The central role of superoxide dismutases. Antioxid. Redox Signal. 2014, 20, 1567–1589. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, H.; Zhao, S.; Hong, J.; Tang, C. The effect on radioresistance of manganese superoxide dismutase in nasopharyngeal carcinoma. Oncol. Rep. 2010, 23, 1005–1011. [Google Scholar] [PubMed]
- Kroemer, G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 1997, 3, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, D.M.; Kollar, A.; Beer, K.T.; Laissue, J.; Greiner, R.H.; Djonov, V. Involvement of the hepatocyte growth factor/scatter factor receptor c-met and of Bcl-xL in the resistance of oropharyngeal cancer to ionizing radiation. Int. J. Cancer 2001, 96, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Nix, P.; Cawkwell, L.; Patmore, H.; Greenman, J.; Stafford, N. Bcl-2 expression predicts radiotherapy failure in laryngeal cancer. Br. J. Cancer 2005, 92, 2185–2189. [Google Scholar] [CrossRef] [Green Version]
- Condon, L.T.; Ashman, J.N.E.; Ell, S.R.; Stafford, N.D.; Greenman, J.; Cawkwell, L. Overexpression of Bcl-2 in squamous cell carcinoma of the larynx: A marker of radioresistance. Int. J. Cancer 2002, 100, 472–475. [Google Scholar] [CrossRef] [Green Version]
- Hüttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. Biophys. Acta 2007, 1773, 1701–1720. [Google Scholar] [CrossRef] [Green Version]
- Weljie, A.M.; Jirik, F.R. Hypoxia-induced metabolic shifts in cancer cells: Moving beyond the Warburg effect. Int. J. Biochem. Cell Biol. 2011, 43, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef]
- Hammer, S.; To, K.K.-W.; Yoo, Y.-G.; Koshiji, M.; Huang, L.E. Hypoxic suppression of the cell cycle gene CDC25A in tumor cells. Cell Cycle Georget. Tex 2007, 6, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Mechanisms of action of (n-3) fatty acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Kumlin, M.; Ingelman-Sundberg, M.; Wolk, A. Dietary long-chain n-3 fatty acids for the prevention of cancer: A review of potential mechanisms. Am. J. Clin. Nutr. 2004, 79, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [Green Version]
- Bozza, P.T.; Viola, J.P.B. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 243–250. [Google Scholar] [CrossRef]
- Corre, I.; Niaudet, C.; Paris, F. Plasma membrane signaling induced by ionizing radiation. Mutat. Res. 2010, 704, 61–67. [Google Scholar] [CrossRef]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwahara, Y.; Oikawa, T.; Ochiai, Y.; Roudkenar, M.H.; Fukumoto, M.; Shimura, T.; Ohtake, Y.; Ohkubo, Y.; Mori, S.; Uchiyama, Y.; et al. Enhancement of autophagy is a potential modality for tumors refractory to radiotherapy. Cell Death Dis. 2011, 2, e177. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guerrero-Preston, R.; Ratovitski, E.A. Phospho-ΔNp63α-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle Georget. Tex 2012, 11, 1247–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moergel, M.; Abt, E.; Stockinger, M.; Kunkel, M. Overexpression of p63 is associated with radiation resistance and prognosis in oral squamous cell carcinoma. Oral Oncol. 2010, 46, 667–671. [Google Scholar] [CrossRef]
- Zhou, X.; Münger, K. Expression of the human papillomavirus type 16 E7 oncoprotein induces an autophagy-related process and sensitizes normal human keratinocytes to cell death in response to growth factor deprivation. Virology 2009, 385, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Spangle, J.M.; Münger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragin, C.C.R.; Modugno, F.; Gollin, S.M. The epidemiology and risk factors of head and neck cancer: A focus on human papillomavirus. J. Dent. Res. 2007, 86, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Pacelli, R.; Della Vittoria Scarpati, G.; Cella, L.; Giuliano, M.; Caponigro, F.; Pepe, S. Radioresistance in head and neck squamous cell carcinoma: Biological bases and therapeutic implications. Head Neck 2015, 37, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Dimery, I.W.; Hong, W.K. Overview of combined modality therapies for head and neck cancer. J. Natl. Cancer Inst. 1993, 85, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Genden, E.M.; Ferlito, A.; Bradley, P.J.; Rinaldo, A.; Scully, C. Neck disease and distant metastases. Oral Oncol. 2003, 39, 207–212. [Google Scholar] [CrossRef]
- Hoff, C.M. Importance of hemoglobin concentration and its modification for the outcome of head and neck cancer patients treated with radiotherapy. Acta Oncol. Stockh. Swed. 2012, 51, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fryknäs, M.; Hernlund, E.; Fayad, W.; De Milito, A.; Olofsson, M.H.; Gogvadze, V.; Dang, L.; Påhlman, S.; Schughart, L.A.K.; et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat. Commun. 2014, 5, 3295. [Google Scholar] [CrossRef] [PubMed]
- Lucido, C.T.; Callejas-Valera, J.L.; Colbert, P.L.; Vermeer, D.W.; Miskimins, W.K.; Spanos, W.C.; Vermeer, P.D. β2-Adrenergic receptor modulates mitochondrial metabolism and disease progression in recurrent/metastatic HPV(+) HNSCC. Oncogenesis 2018, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Lucido, C.T.; Miskimins, W.K.; Vermeer, P.D. Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC. Cancers 2018, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Jendoubi, M.; Ballot, C.; Dammak, A.; Jonneaux, A.; Idziorek, T.; Joha, S.; Dauphin, V.; Malet-Martino, M.; Balayssac, S.; et al. Exploiting mitochondrial dysfunction for effective elimination of imatinib-resistant leukemic cells. PLoS ONE 2011, 6, e21924. [Google Scholar] [CrossRef]
- Zhao, F.; Mancuso, A.; Bui, T.V.; Tong, X.; Gruber, J.J.; Swider, C.R.; Sanchez, P.V.; Lum, J.J.; Sayed, N.; Melo, J.V.; et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene 2010, 29, 2962–2972. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, S. Metformin inhibits human breast cancer cell growth by promoting apoptosis via a ROS-independent pathway involving mitochondrial dysfunction: Pivotal role of superoxide dismutase (SOD). Cell. Oncol. Dordr. 2018, 41, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Zhi, P.; You, J.; Gao, J.-Q. Metformin synergistically suppress tumor growth with doxorubicin and reverse drug resistance by inhibiting the expression and function of P-glycoprotein in MCF7/ADR cells and xenograft models. Oncotarget 2018, 9, 2158–2174. [Google Scholar] [CrossRef]
- Zannella, V.E.; Pra, A.D.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming Metabolism with Metformin Improves Tumor Oxygenation and Radiotherapy Response. Clin. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Floridi, A.; Paggi, M.G.; Marcante, M.L.; Silvestrini, B.; Caputo, A.; De Martino, C. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J. Natl. Cancer Inst. 1981, 66, 497–499. [Google Scholar]
- Liu, Y.; Murray-Stewart, T.; Casero, R.A.; Kagiampakis, I.; Jin, L.; Zhang, J.; Wang, H.; Che, Q.; Tong, H.; Ke, J.; et al. Targeting hexokinase 2 inhibition promotes radiosensitization in HPV16 E7-induced cervical cancer and suppresses tumor growth. Int. J. Oncol. 2017, 50, 2011–2023. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta 2016, 1866, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, G.M.; van Kersen, I.; Silvestrini, B. Inhibition of the recovery from potentially lethal damage by lonidamine. Br. J. Cancer 1984, 50, 657–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magno, L.; Terraneo, F.; Bertoni, F.; Tordiglione, M.; Bardelli, D.; Rosignoli, M.T.; Ciottoli, G.B. Double-blind randomized study of lonidamine and radiotherapy in head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 45–55. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Gregorio, A.; Martínez-Ramírez, I.; Pedraza-Chaverri, J.; Lizano, M. Reprogramming of Energy Metabolism in Response to Radiotherapy in Head and Neck Squamous Cell Carcinoma. Cancers 2019, 11, 182. https://doi.org/10.3390/cancers11020182
Cruz-Gregorio A, Martínez-Ramírez I, Pedraza-Chaverri J, Lizano M. Reprogramming of Energy Metabolism in Response to Radiotherapy in Head and Neck Squamous Cell Carcinoma. Cancers. 2019; 11(2):182. https://doi.org/10.3390/cancers11020182
Chicago/Turabian StyleCruz-Gregorio, Alfredo, Imelda Martínez-Ramírez, José Pedraza-Chaverri, and Marcela Lizano. 2019. "Reprogramming of Energy Metabolism in Response to Radiotherapy in Head and Neck Squamous Cell Carcinoma" Cancers 11, no. 2: 182. https://doi.org/10.3390/cancers11020182