The Role of Aberrant DNA Methylation in Misregulation of Gene Expression in Gonadotroph Nonfunctioning Pituitary Tumors

 ,
,
Abstract
:1. Introduction
2. Results
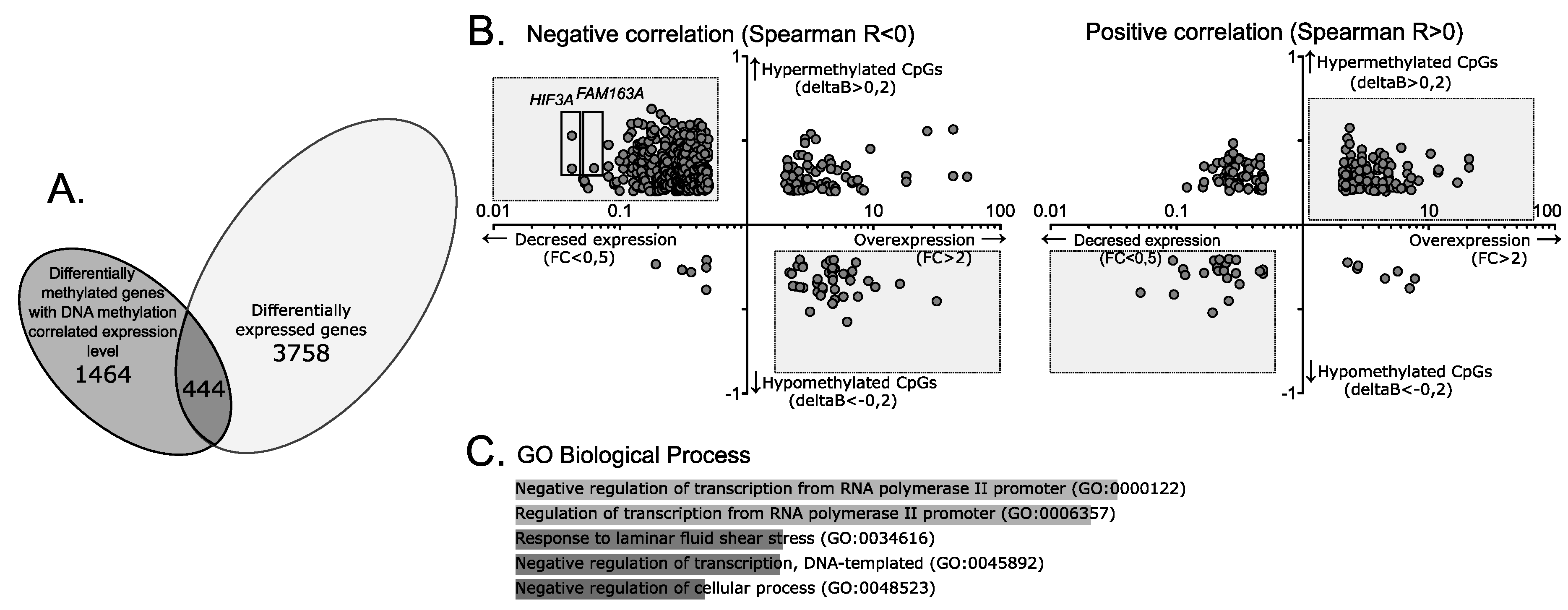
2.1. Aberrant DNA Methylation in Gene-Associated CpGs in Gonadotroph NFPAs
2.2. Gene Expression Analysis for CpGs Differentially Methylated in NFPAs and Normal Pituitary Tissue
2.3. DNA Methylation of Genes with Differential Expression Level in NFPAs and Pituitary
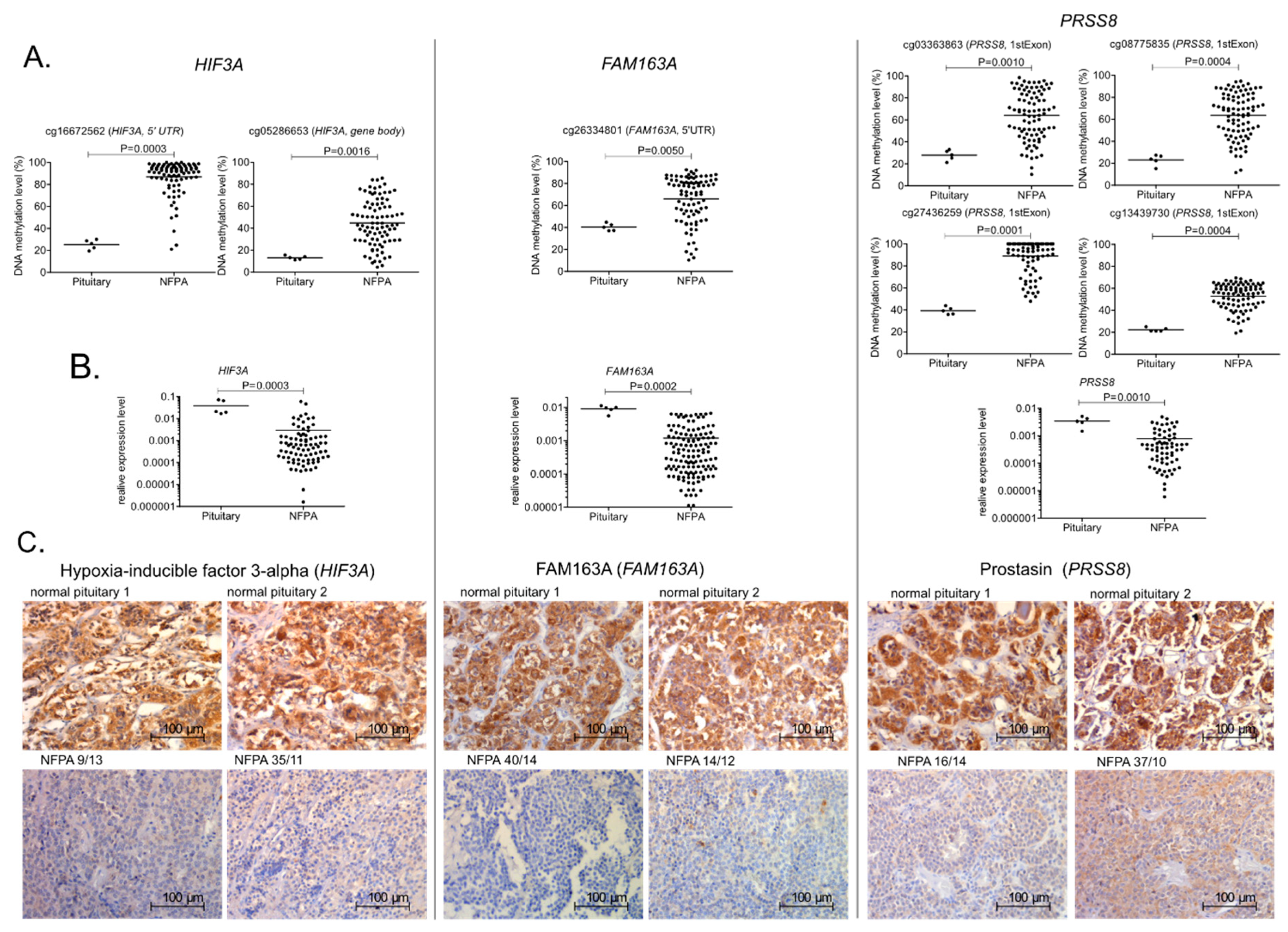
2.4. DNA Methylation and the Expression of Selected Genes
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Samples
4.2. Whole-Genome DNA Methylation Analysis
4.3. Gene Expression Analysis with Amplicon-Based Library Preparation and Next-Generation Sequencing (NGS)
4.4. Evaluation of DNA Methylation Pattern of Selected Gene Promoters
4.5. qRT-PCR Gene Expression Assessment
4.6. Immunohistochemistry
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Greenman, Y.; Naftali, S. Non-Functioning Pituitary Adenomas. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Newey, P.J.; Nesbit, M.A.; Rimmer, A.J.; Head, R.A.; Gorvin, C.M.; Attar, M.; Gregory, L.; Wass, J.A.H.; Buck, D.; Karavitaki, N.; et al. Whole-Exome Sequencing Studies of Nonfunctioning Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2013, 98, E796–E800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pease, M.; Ling, C.; Mack, W.J.; Wang, K.; Zada, G. The Role of Epigenetic Modification in Tumorigenesis and Progression of Pituitary Adenomas: A Systematic Review of the Literature. PLoS ONE 2013, 8, e82619. [Google Scholar] [CrossRef] [PubMed]
- Farrell, W.E. Pituitary Tumours: Findings from Whole Genome Analyses. Endocr. Relat. Cancer 2006, 13, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer Epigenomics: DNA Methylomes and Histone-Modification Maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Duong, C.V.; Emes, R.D.; Wessely, F.; Yacqub-Usman, K.; Clayton, R.N.; Farrell, W.E. Quantitative, Genome-Wide Analysis of the DNA Methylome in Sporadic Pituitary Adenomas. Endocr. Relat. Cancer 2012, 19, 805–816. [Google Scholar] [CrossRef]
- Ling, C.; Pease, M.; Shi, L.; Punj, V.; Shiroishi, M.S.; Commins, D.; Weisenberger, D.J.; Wang, K.; Zada, G. A Pilot Genome-Scale Profiling of DNA Methylation in Sporadic Pituitary Macroadenomas: Association with Tumor Invasion and Histopathological Subtype. PLoS ONE 2014, 9, e96178. [Google Scholar] [CrossRef]
- Salomon, M.P.; Wang, X.; Marzese, D.M.; Hsu, S.C.; Nelson, N.; Zhang, X.; Matsuba, C.; Takasumi, Y.; Ballesteros-Merino, C.; Fox, B.A.; et al. The Epigenomic Landscape of Pituitary Adenomas Reveals Specific Alterations and Differentiates among Acromegaly, Cushing’s disease and Endocrine-Inactive Subtypes. Clin. Cancer Res. 2018, 24, 4126–4136. [Google Scholar] [CrossRef]
- Gu, Y.; Zhou, X.; Hu, F.; Yu, Y.; Xie, T.; Huang, Y.; Zhao, X.; Zhang, X. Differential DNA Methylome Profiling of Nonfunctioning Pituitary Adenomas Suggesting Tumour Invasion is Correlated with Cell Adhesion. J. Neurooncol. 2016, 129, 23–31. [Google Scholar] [CrossRef]
- Kober, P.; Boresowicz, J.; Rusetska, N.; Maksymowicz, M.; Goryca, K.; Kunicki, J.; Bonicki, W.; Siedlecki, J.A.; Bujko, M. DNA Methylation Profiling in Nonfunctioning Pituitary Adenomas. Mol. Cell. Endocrinol. 2018, 473, 194–204. [Google Scholar] [CrossRef]
- Kitchen, M.O.; Bryan, R.T.; Emes, R.D.; Glossop, J.R.; Luscombe, C.; Cheng, K.K.; Zeegers, M.P.; James, N.D.; Devall, A.J.; Mein, C.A.; et al. Quantitative Genome-Wide Methylation Analysis Of High-Grade Non-Muscle Invasive Bladder Cancer. Epigenetics 2016, 11, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Klett, H.; Balavarca, Y.; Toth, R.; Gigic, B.; Habermann, N.; Scherer, D.; Schrotz-king, P.; Ulrich, A.; Schirmacher, P.; Herpel, E.; et al. Robust Prediction of Gene Regulation in Colorectal Cancer Tissues from DNA Methylation Profiles. Epigenetics 2018, 13, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.R.; Sano, T.; Yoshimoto, K.; Yamada, S.; Ishizuka, A.; Mizusawa, N.; Horiguchi, H.; Hirokawa, M.; Asa, S.L. Inactivation of RASSF1A Tumor Suppressor Gene by Aberrant Promoter Hypermethylation in Human Pituitary Adenomas. Lab. Investig. 2005, 85, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, A.; Katayama, Y.; Ogino, A.; Watanabe, T.; Yachi, K.; Ohta, T.; Komine, C.; Yokoyama, T.; Fukushima, T. Promoter Hypermethylation Profile of Cell Cycle Regulator Genes in Pituitary Adenomas. J. Neurooncol. 2007, 83, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Valiulyte, I.; Steponaitis, G.; Skiriute, D.; Tamasauskas, A.; Vaitkiene, P. Signal Transducer and Activator of Transcription 3 (STAT3) Promoter Methylation and Expression in Pituitary Adenoma. BMC Med. Genet. 2017, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Palmieri, D.; Fusco, A. HMGA2: A Pituitary Tumour Subtype-Specific Oncogene? Mol. Cell. Endocrinol. 2010, 326, 19–24. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–36. [Google Scholar] [CrossRef]
- Hellman, A.; Chess, A. Gene Body—Specific Methylation. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; DeCarvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene Body Methylation can Alter Gene Expression and is a Therapeutic Target in Cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Yang, S.L.; Wu, C.; Xiong, Z.F.; Fang, X. Progress on Hypoxia-Inducible Factor-3: Its Structure, Gene Regulation and Biological Function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Wang, Q.; Guo, Y.; Chen, Z.; Li, K.; Yang, Y.; Zhang, H.; Dong, H.; Shen, K.; Yang, W. PRSS8 Methylation and Its Significance in Esophageal Squamous Cell Carcinoma. Oncotarget 2016, 7, 28540. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, Y.; Yang, Y.; Wei, X.; Zhang, S.; Zhang, Y.; Li, K.; Yuan, M.; Guo, D.; Macias, V.; et al. PRSS8 Suppresses Colorectal Carcinogenesis and Metastasis. Oncogene 2019, 38, 497–517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jia, G.; Shi, B.; Ge, G.; Duan, H.; Yang, Y. PRSS8 is Downregulated and Suppresses Tumour Growth and Metastases in Hepatocellular Carcinoma. Cell Physiol. Biochem. 2016, 40, 757–769. [Google Scholar] [CrossRef]
- Yacqub-Usman, K.; Duong, C.V.; Clayton, R.N.; Farrell, W.E. Epigenomic Silencing of the BMP-4 Gene in Pituitary Adenomas: A Potential Target for Epidrug-Induced Re-Expression. Endocrinology 2012, 153, 3603–3612. [Google Scholar] [CrossRef]
- Palmieri, D.; D’Angelo, D.; Valentino, T.; De Martino, I.; Ferraro, A.; Wierinckx, A.; Fedele, M.; Trouillas, J.; Fusco, A. Downregulation of HMGA-Targeting microRNAs has a Critical Role in Human Pituitary Tumorigenesis. Oncogene 2012, 31, 3857–3865. [Google Scholar] [CrossRef]
- Kitchen, M.O.; Yacqub-Usman, K.; Emes, R.D.; Richardson, A.; Clayton, R.N.; Farrell, W.E. Epidrug Mediated Re-Expression of miRNA Targeting the HMGA Transcripts in Pituitary Cells. Pituitary 2015, 18, 674–684. [Google Scholar] [CrossRef]
- Li, J.; Ching, T.; Huang, S.; Garmire, L.X. Using Epigenomics Data to Predict Gene Expression in Lung Cancer. BMC Bioinformatics 2015, 16, 10. [Google Scholar] [CrossRef]
- Jiang, S.; Mortazavi, A. Integrating ChIP-seq with Other Functional Genomics Data. Brief. Funct. Genom. 2018, 17, 104–115. [Google Scholar] [CrossRef]
- García-Martínez, A.; Sottile, J.; Sá Nchez-Tejada, L.; Fajardo, C.; Cámara, R.; Lamas, C.; Barberá, V.M.; Picó, A. DNA Methylation of Tumor Suppressor Genes in Pituitary Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1272–1282. [Google Scholar] [CrossRef]
- Hauser, B.M.; Lau, A.; Gupta, S.; Bi, W.L.; Dunn, I.F. The Epigenomics of Pituitary Adenoma. Front Endocrinol. (Lausanne) 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, S.; Cheng, S.; Asa, S.L. Epigenetics of Pituitary Tumors: Pathogenetic and Therapeutic Implications. Mol. Cell Endocrinol. 2018, 469, 70–76. [Google Scholar] [CrossRef] [PubMed]
- DeLellis, R.A. Pathology and Genetics of Tumours of Endocrine Organs; IARC: Lyon, France, 2004; Volume 8, p. 230. ISBN 9283224167. [Google Scholar]
- Rhein, M.; Hagemeier, L.; Klintschar, M.; Muschler, M.; Bleich, S.; Frieling, H. DNA Methylation Results Depend on DNA Integrity- Role of Post Mortem Interval. Front Genet. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pozhitkov, A.E.; Neme, R.; Domazet-Lošo, T.; Leroux, B.G.; Soni, S.; Tautz, D.; Noble, P.A. Tracing the Dynamics of Gene Transcripts after Organismal Death. Open Biol. 2017, 7, 160267. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef]
- Nordlund, J.; Bäcklin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.-L.; Flaegstad, T.; Forestier, E.; Frost, B.-M.; Harila-Saari, A.; et al. Genome-wide Signatures of Differential DNA Methylation in Pediatric Acute Lymphoblastic Leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, 1–10. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bujko, M.; Rusetska, N.; Mikula, M. Validating Candidate Reference Genes for qRT-PCR-based Gene Expression Analysis in Nonfunctioning Pituitary Adenomas. Pituitary 2016, 19, 110–112. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIF3A | FAM163A | PRSS8 | |
|---|---|---|---|
| NFPA | |||
| Staining intensity | Number of samples (percentage) | Number of samples (percentage) | Number of samples (percentage) |
| strong (+++) | 0/42 (0%) | 0/42 (0%) | 4/42 (9.5%) |
| moderate (++) | 13/42 (30.9%) | 11/42 (26.2%) | 14/42 (33.3%) |
| weak (+) | 25/42 (59.5) | 25/42 (59.5%) | 24/42 (57.1%) |
| 0 | 4/42 (9.5%) | 6/42 (14.3%) | 0/42 (0%) |
| Normal pituitary | |||
| Staining intensity | |||
| strong (+++) | 5/5 (100%) | 5/5 (100%) | 5/5 (100%) |
| HM450K CpG Site | CpG Location | Gene | Investigation Group Spearman R; p-Value | Validation Group Spearman R; p-Value |
|---|---|---|---|---|
| cg26334801 | 5′UTR | FAM163A | −0.802; p < 0.0001 | −0.688; p < 0.0001 |
| cg16672562 | 5′UTR | HIF3A | −0.407; p = 0.0291 | −0.380; p = 0.0005 |
| cg05286653 | Gene body | HIF3A | −0.490; p = 0.0076 | −0.451; p < 0.0001 |
| cg08775835 | 1stExon | PRSS8 | −0.786; p < 0.0001 | −0.487; p < 0.0001 |
| cg13439730 | 1stExon | PRSS8 | −0.559; p = 0,0016 | −0.484; p < 0.0001 |
| cg27436259 | 1stExon | PRSS8 | −0.744; p < 0.0001 | −0.568; p < 0.0001 |
| cg03363863 | 1stExon | PRSS8 | −0.765; p < 0.0001 | −0.539; p < 0.0001 |
| Genome-Wide DNA Methylation/Whole Transcriptome Profiling | DNA Pyrosequencing/qRT-PCR | |
|---|---|---|
| NFPA patients (number of patients) | 32 | 83 |
| Age (years) | ||
| Range | 36–85 | 34–82 |
| Median | 61 | 63 |
| Gender (number of patients) | ||
| Male | 21 | 47 |
| Female | 11 | 36 |
| Histopathology (number of patients) | ||
| Gonadotroph PA | 31 | 76 |
| Null-cell/ Gonadotroph PA* | 1 | 7 |
| Clinical classification (number of patients) | ||
| Invasive NFPA | 17 | 50 |
| Non-invasive NFPA | 15 | 26 |
| Unknown | - | 7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kober, P.; Boresowicz, J.; Rusetska, N.; Maksymowicz, M.; Paziewska, A.; Dąbrowska, M.; Kunicki, J.; Bonicki, W.; Ostrowski, J.; Siedlecki, J.A.; et al. The Role of Aberrant DNA Methylation in Misregulation of Gene Expression in Gonadotroph Nonfunctioning Pituitary Tumors. Cancers 2019, 11, 1650. https://doi.org/10.3390/cancers11111650
Kober P, Boresowicz J, Rusetska N, Maksymowicz M, Paziewska A, Dąbrowska M, Kunicki J, Bonicki W, Ostrowski J, Siedlecki JA, et al. The Role of Aberrant DNA Methylation in Misregulation of Gene Expression in Gonadotroph Nonfunctioning Pituitary Tumors. Cancers. 2019; 11(11):1650. https://doi.org/10.3390/cancers11111650
Chicago/Turabian StyleKober, Paulina, Joanna Boresowicz, Natalia Rusetska, Maria Maksymowicz, Agnieszka Paziewska, Michalina Dąbrowska, Jacek Kunicki, Wiesław Bonicki, Jerzy Ostrowski, Janusz A. Siedlecki, and et al. 2019. "The Role of Aberrant DNA Methylation in Misregulation of Gene Expression in Gonadotroph Nonfunctioning Pituitary Tumors" Cancers 11, no. 11: 1650. https://doi.org/10.3390/cancers11111650