The Role of Merlin/NF2 Loss in Meningioma Biology
by
 , and
, and
Sungho Lee
1, 
Patrick J. Karas
1,
Caroline C. Hadley
1,
James C. Bayley V
1,
A. Basit Khan
1,
Ali Jalali
1,
Alex D. Sweeney
2,
Tiemo J. Klisch
3,4 and
Akash J. Patel
1,2,3,*
1
Department of Neurosurgery, Baylor College of Medicine, Houston, TX 77030, USA
2
Department of Otolaryngology-Head and Neck Surgery, Baylor College of Medicine, Houston, TX 77030, USA
3
Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA
4
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA
*
Author to whom correspondence should be addressed.
Cancers 2019, 11(11), 1633; https://doi.org/10.3390/cancers11111633
Submission received: 13 September 2019
/
Revised: 17 October 2019
/
Accepted: 19 October 2019
/
Published: 24 October 2019
(This article belongs to the Special Issue New Insights into Neurofibromatosis)
Abstract
:Mutations in the neurofibromin 2 (NF2) gene were among the first genetic alterations implicated in meningioma tumorigenesis, based on analysis of neurofibromatosis type 2 (NF2) patients who not only develop vestibular schwannomas but later have a high incidence of meningiomas. The NF2 gene product, merlin, is a tumor suppressor that is thought to link the actin cytoskeleton with plasma membrane proteins and mediate contact-dependent inhibition of proliferation. However, the early recognition of the crucial role of NF2 mutations in the pathogenesis of the majority of meningiomas has not yet translated into useful clinical insights, due to the complexity of merlin’s many interacting partners and signaling pathways. Next-generation sequencing studies and increasingly sophisticated NF2-deletion-based in vitro and in vivo models have helped elucidate the consequences of merlin loss in meningioma pathogenesis. In this review, we seek to summarize recent findings and provide future directions toward potential therapeutics for this tumor.
1. Introduction
Meningiomas are the most common type of primary intracranial tumors, currently classified by the World Health Organization (WHO) as benign (grade I), atypical (grade II), or malignant (grade III). Over the years, our understanding of the molecular underpinnings of these tumors has been greatly accelerated by advancements in next-generation sequencing (NGS) technology. It has been thought for some time that the protein encoded by the neurofibromin 2 (NF2) gene is a key to understand these brain tumors. The NF2 gene resides on the long arm of chromosome 22 (chr22q) and encodes a 69 kDa protein called merlin (moesin-ezrin-radixin-like protein), which is a part of the Band 4.1 FERM gene family [1]. Merlin is a cytoskeleton scaffolding protein that links actin filaments, transmembrane receptors, and intracellular signaling molecules to regulate several essential pathways controlling proliferation and survival. These include the hippo pathway, mammalian target of rapamycin (mTOR)/PI3K/AKT pathway, and receptor tyrosine kinases (RTKs) [2,3]. In this review, we summarize the role of NF2 loss in meningioma pathogenesis and its impact on meningioma biology based on the known functions of merlin.
2. Evidence Linking NF2 and Meningiomas—Inherited Disorders
2.1. Neurofibromatosis Type 2 (NF2)
The first indication that meningiomas may have a genetic contribution came from neurofibromatosis type 2 (NF2) [4,5]. NF2 is a rare autosomal dominant tumor syndrome with an estimated birth incidence of 1 in 33,000 [6], resulting from biallelic inactivation of NF2. Development of bilateral vestibular schwannomas is a pathognomonic feature present in approximately 60% of cases [7]. However, considerable heterogeneity in clinical presentation has led to the development of additional diagnostic criteria [8] (Table 1). Other lesions encountered in NF2 include non-vestibular schwannomas, meningiomas, ependymomas, and congenital cataracts.
The NF2 mutational spectrum in NF2 is vast; most variations do not recur. Nonsense (39%) and frameshift (27%) mutations in the NF2 gene are most frequent in NF2 patients, with splice site (25%) and non-truncating mutations (7%) making up a smaller fraction of mutations [9]. Nonsense and frameshift mutations that truncate the protein are associated with a more severe disease phenotype, including increased frequency of multiple and recurring meningiomas [10,11,12,13,14]. Somatic mosaicism occurs in up to 33% of NF2 patients, which results in a milder phenotype and lower risk of transmission to offspring [15,16,17]. Mutations may only be detectable in the tumor tissue, emphasizing the importance of analyzing surgical specimens.
Intracranial meningiomas affect about half of NF2 patients [8,10,18,19], and spinal meningiomas are seen in approximately 20% of the patients [20]. Over half of these patients have multiple meningiomas that exhibit heterogeneous behavior and an asynchronous growth pattern [21]. While the majority of syndromic meningiomas remain stable in size or grow minimally over time, few tumors, including tumors that appear de novo, grow more rapidly and therefore are more frequently resected [22]. Furthermore, several distinct histological subtypes were seen in patients who underwent resection of multiple meningiomas, suggesting that NF2 inactivation is an early tumorigenic event that occurs prior to commitment to a specific histopathologic subtype. Subsequent studies in sporadic meningiomas demonstrated that up to 60% of these cases exhibit inactivation of NF2 by somatic mutation, epigenetic inactivation, or allelic loss of chr22q [23,24,25,26]. These findings suggest that NF2 loss is a critical event in the development of a subpopulation of meningiomas.
2.2. Schwannomatosis
Schwannomatosis is characterized by the development of multiple schwannomas in the absence of other NF2-defining lesions such as bilateral vestibular schwannomas or ependymomas [27]. It is a rarer disorder than NF2, with an estimated incidence of 1/40,000 to 1/70,000 [28]. Germline mutations in SWItch/Sucrose Non-Fermentable (SWI/SNF)-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1) or leucine-zipper-like transcriptional regulator 1 (LZTR1) predispose to the disorder [29,30]. Both genes are located on chr22q in proximity to NF2. In the presence of SMARCB1 or LZTR1 mutations, there is also biallelic NF2 loss of function, via acquired somatic mutations and loss of heterozygosity (LOH) [31]. Multiple meningiomas occur in 5% of schwannomatosis cases [32], but only in association with SMARCB1 mutations [33,34,35], hinting at a potential interaction between NF2 and SMARCB1 in meningioma pathogenesis, later validated in NGS studies.
3. Insights from NGS Studies
Over the past decade, several groups have leveraged the wide availability and reduced cost of NGS to characterize meningiomas, providing additional insights into the mutation landscape of meningiomas from early genetic studies (Table 2).
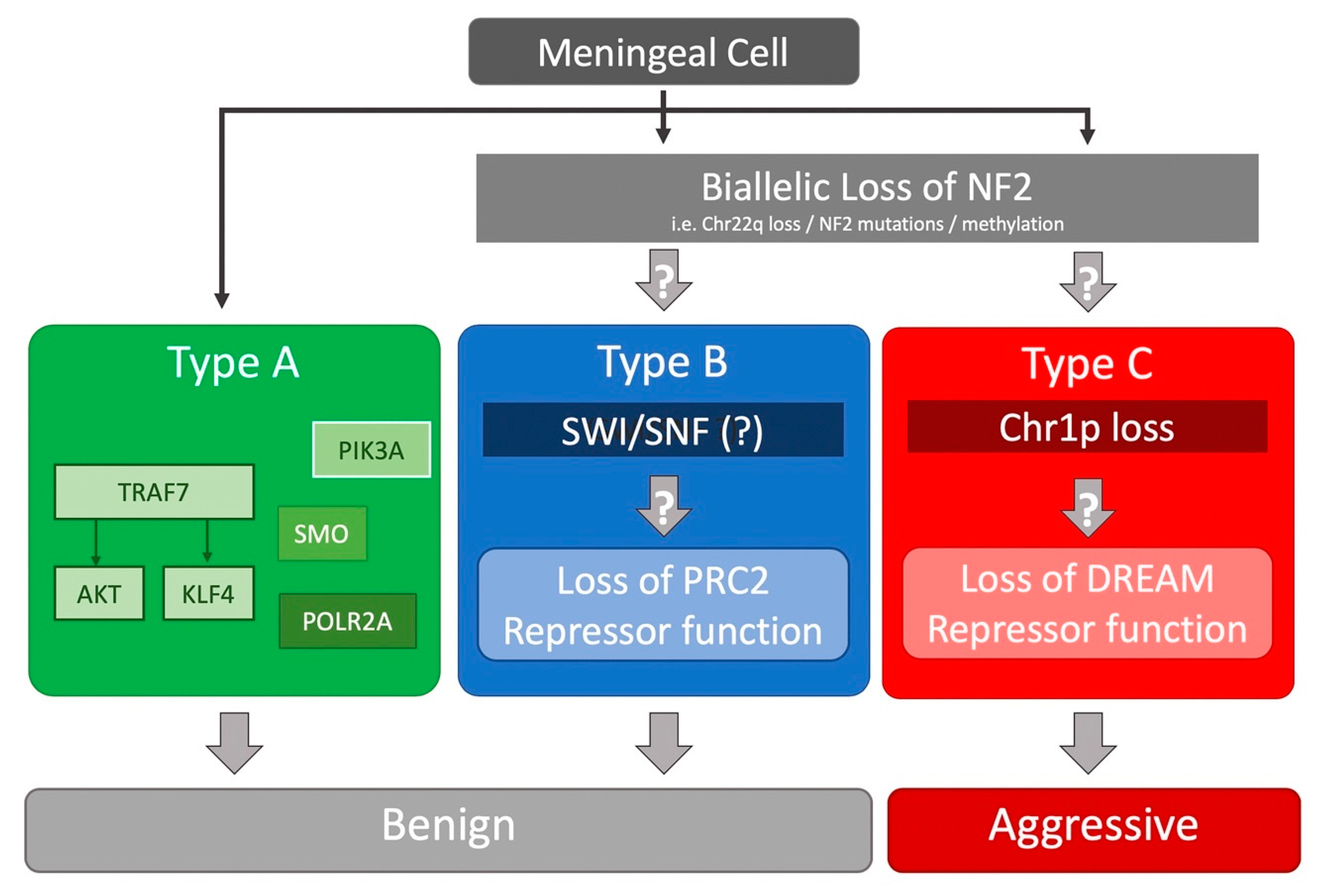
Several whole-exome sequencing (WES) studies identified recurrent somatic mutations in NF2, TNF receptor-associated factor 7 (TRAF7), Krupple-like factor 4 (KLF4), v-akt murine thymoma viral oncogene homolog 1 (AKT1), smoothened, frizzled family receptor (SMO), and RNA polymerase II subunit A (POLR2A) in benign (WHO grade I) tumors [36,37,39]. Interestingly, a significant proportion (6.4%) of NF2-mutated tumors harbored recurrent mutations in SMARCB1 [39]. While KLF4 or AKT1 mutations almost always co-occurred with TRAF7 mutations, they did not occur together; SMO-mutated meningiomas all harbored the activating L412F mutation [36]. All secretory meningiomas carried TRAF7/KLF4 mutations [36,38]. One study of 18 radiation-induced meningiomas found that nearly all had chr22q LOH, with a majority also having NF2 loss via fusion events [40]. All of these studies have found that chr22q/NF2 loss does not co-occur with TRAF7, AKT1, KLF4, SMO, or POLR2A mutations, which were all found in benign meningiomas (Figure 1) [36,37,38,39,40].
On the other hand, a different mutational landscape is found in high-grade (WHO grade II and III) meningiomas, which demonstrate higher recurrence and shorter survival compared to benign meningiomas [26,44,45,46]. Two recent NGS studies of high-grade meningiomas found more chromosomal abnormalities compared to benign meningiomas, similar to other aggressive cancers [41,42]. Most high-grade meningiomas were characterized by NF2 loss, without any other significantly recurring somatic mutations, in contrast to benign meningiomas [41,42].
Bi et al. examined 36 paired samples of meningiomas undergoing malignant progression and found NF2 loss in 73% of the cases in both low- and high-grade samples from the same patient, emphasizing that NF2 loss is an early event in meningioma progression. Losses of chromosome arms 1p, 6q, 14q, 3p, 10q, 18q, and 19q were additionally seen in these tumors. Interestingly, recurrent tumors from the same patient demonstrated 75% overlap of arm-level somatic copy number variations (CNV), suggesting that these chromosomal losses are again an early and essential feature in high-grade meningiomas.
Harmanci et al. found that the majority of atypical meningiomas had NF2 loss and fell into two categories: those with significant chromosomal losses (“CNV-high”) and those with SMARCB1 mutations (“CNV-low”). However, they did not find any differences in the transcriptional profiles of these two groups, finding all of these tumors significantly enriched for cell cycle processes, including upregulation of E2F and FOXM1 networks. Additionally, they found NF2 mutant tumors demonstrated increased methylation of polycomb repressive complex 2 (PRC2) target genes, driven by the upregulation of EZH2, the catalytic subunit. Vasudevan et al. sought targetable pathways in high-grade meningiomas and found that elevated FOXM1 expression is associated with poor clinical outcomes [47].
Recently, we undertook a large-scale RNA-seq based study of 160 tumors of all grades to further refine our understanding of meningioma biology [43] (Figure 1). Using only transcriptional profiling, we found three types of meningioma. Subsequent analysis of their genomic landscape revealed one type (type A) was composed of only benign meningiomas with mutations in TRAF7, AKT1, KLF4, SMO. The other two types had chr22q/NF2 loss (type B and C) but no other known mutations. Interestingly, these two types had very different molecular and clinical characteristics, arguing that merlin inactivation sets the stage for additional tumorigenic events that have dramatically different consequences. While type A and B tumors did not recur after complete resection, type C tumors (a mixture of WHO grade I, II, and III tumors) behaved “aggressively”, recurring frequently despite complete resection. An important outcome of this was that the RNA-seq based classification system could identify WHO grade I meningiomas that behaved aggressively despite their benign histopathologic features.
Type B (a mixture of grade I and II tumors) meningiomas were characterized by chr22q/NF2 loss and co-occurrence of SMARCB1 mutations [43]. The atypical meningiomas that Harmanci et al. referred to as “CNV-low” likely represent type B tumors [42]. Gene set enrichment analysis (GSEA) suggests that type B tumors are characterized by loss or dysfunction of the repressive PRC2 complex, which is responsible for H3K27 di- and trimethylation and subsequent chromatin silencing. In type B tumors, co-immunoprecipitation of EZH1, a core subunit of the PRC2 complex, did not pull down the other critical PRC2 subunits such as EED and SUZ12, suggesting a failed assembly of the PRC2 complex. In addition, a portion of type B tumors harbored SMARCB1 mutations. SMARCB1, whose gene is also encoded on chr22q, is a critical component of the SWI/SNF complex. The SWI/SNF and PRC2 chromatin remodeling complexes have a close and complex interconnectivity in regulating the chromatin state [48,49]. How these complexes are dysfunctional in type B meningiomas remains unknown, which highlights the need for further studies to understand the complex biology underlying these tumors. It will be interesting to understand how the di- and trimethylation profiles in the three types are altered.
On the other hand, type C meningiomas, also with NF2 loss, had a significant burden of chromosomal gains/losses, most commonly loss of chr22q and chr1p together, as well as significantly shorter recurrence-free survival, despite over half of these tumors being WHO grade I [43]. The “CNV-high” atypical meningiomas reported by Harmanci et al. likely correspond to these tumors [42]. GSEA showed that the target genes of the DREAM complex, including FOXM1 and MYBL2, were significantly enriched in type C tumors, when compared to the other two types. The DREAM complex is a highly conserved master regulator of the cell cycle which can alternate between a repressive form that inhibits cell cycle gene expression and an activated form that promotes the progression of the cell cycle, on the basis of the interaction of the MuvB core with RB-like proteins (e.g., E2F2) or FOXM1 with MYBL2, respectively [50]. These findings provide further clarity to the observation that increase in the cell cycle-focused E2F2 transcriptional network and elevated expression of FOXM1 are associated with high-grade meningiomas [42,47]. Interestingly, while RBBP4, one of the MuvB core complex members, is located on chr1p, its expression was not significantly changed in type C tumors. How chr1p loss might lead to the switch from the repressor form of the DREAM complex to the activator form remains to be discovered.
Taken together with previous work, these results suggest that chr22q/NF2 loss is a requisite for the development of aggressive meningiomas which also harbor chr1p losses. NF2 mutation and chromosomal losses may be two distinct processes that work in parallel, but they are early events in tumorigenesis. This also explains the findings highlighted by Dewan et al. in which two tumors within a single NF2 patient can have dramatically different clinical courses [51].
4. Merlin Signaling
4.1. Molecular Conformation
Early insights into merlin function were gleaned from its sequence homology to the FERM family of proteins. Canonical ERM proteins are comprised of a N-terminal FERM domain, followed by an alpha-helix domain and a C-terminal domain. Typically, ERM proteins are maintained in a dormant state by an intramolecular association between N-terminal FERM and C-terminal domains. Upon phosphorylation of a conserved threonine residue at the C-terminal domain by Rho kinase, ERM proteins undergo a conformation change, unmasking sites at the C-terminal domain for the binding of F-actin and other membrane proteins, thereby rendering it active [52,53].
While bearing some similarity to other ERM proteins, the process by which merlin undergoes conformational change and the relative importance of its open and closed state to its scaffolding and tumor suppressor function remain a source of controversy. Traditionally, phosphorylation of merlin at Serine 518 by p21 activated kinase 1 (PAK1) or protein kinase A (PKA) versus dephosphorylation mediated by MYPT1–PP1 was believed to mediate the transition between its open and closed conformations [54,55,56]. Initial studies demonstrated that the tumor suppressor activity of merlin was dependent upon the non-phosphorylated, closed conformation of merlin. However, detailed structural analyses of merlin using fluorescence energy transfer analysis showed that the hyperphosphorylation of Serine 518 or the expression of phosphomimetic and non-phosphorylatable S518D and S518A mutations had only subtle effects on the conformation of merlin [57]. Moreover, studies based on small-angle neutron scattering and immunoprecipitation showed that the phosphorylation of Serine 518 stabilized the closed form rather than promoting an open conformation as initially thought, and that the phosphorylated form of merlin was nonetheless able to interact with its target proteins [58]. Two recent studies reported the interaction between merlin and phosphatidylinositol 4,5-bisphosphate (PIP2), which promotes its open conformation and anti-proliferative activity, whose effects may be distinct from those mediated by phosphorylation at Serine 518 [57,59]. Clearly, the mechanism of the conformational change of merlin is different from that of typical FERM proteins. Merlin’s activity might depend on other factors and, hence, remains an interesting and active area of investigation.
4.2. Contact Inhibition
Merlin functions as a tumor suppressor in a wide range of cancers. However, relatively few studies have investigated its molecular mechanisms specific to meningioma pathogenesis. Consequently, the understanding of merlin signaling in meningiomas needs to be supplemented by insights from studies in other cancers and cell types.
Early studies suggested that merlin’s tumor suppressor activity is related to its contact inhibition of proliferation. In various cell types, merlin is upregulated and hypophosphorylated with an increasing degree of cell confluency [54]. Dephosphorylated, active merlin preferentially interacts with and inhibits CD44, a cell-to-cell adhesion molecule and receptor for hyaluronan, an abundant extracellular matrix (ECM) component [60]. Treatment of sub-confluent schwannoma cells with hyaluronan rapidly induced dephosphorylation of merlin and inhibited cell growth, which was abolished by mutation in the hyaluronan binding domain of CD44. CD44 also constitutively associates with various RTKs that mediate growth factor signaling. Consistent with this notion, siRNA-mediated knockdown of merlin in schwannoma cells increased the levels of ErbB2/ErbB3 RTK, suggesting that merlin normally functions to reduce the availability of RTKs at the plasma membrane [61].
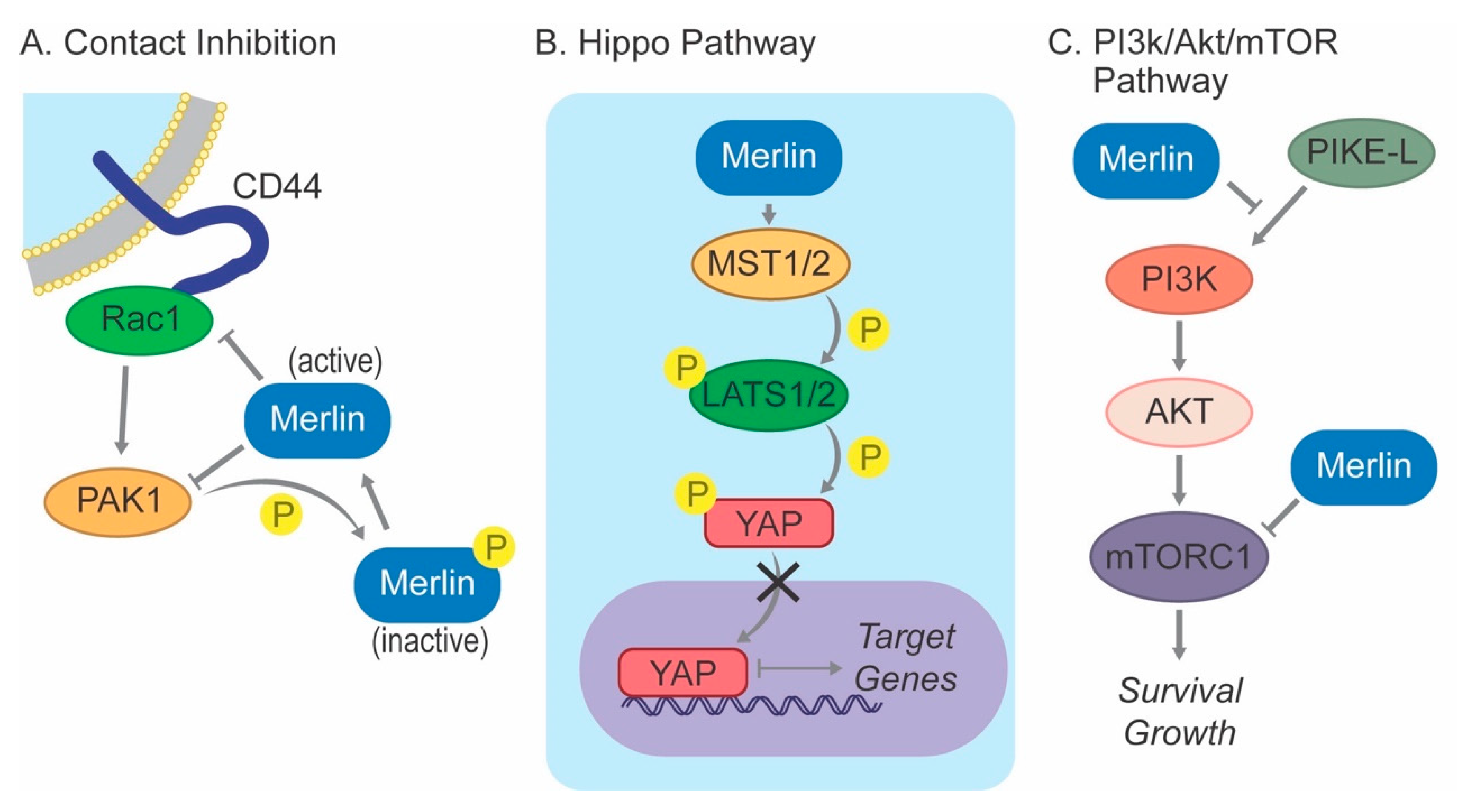
In addition, merlin-mediated contact inhibition is critically regulated by a reciprocal interaction with Ras-related C3 botulinum toxin substrate 1 (RAC1) and its downstream kinase PAK1 (Figure 2). In confluent cells, activation of PAK1 by RAC1 is sufficient to release the cells from contact inhibition [55]. However, active (dephosphorylated) merlin suppresses the recruitment of RAC1 to the plasma membrane, preventing the activation of RAC1 and PAK1. On the other hand, PAK1 phosphorylates merlin at Serine 518, and its subsequent inactivation prevents the translocation to the plasma membrane, mitigating the inhibitory effect of merlin on RAC1. Merlin-deficient meningioma cell lines demonstrate increased expression of PAK1 compared to normal arachnoid cap cells, and knocking down PAK1 expression using doxycycline-inducible shRNA or treatment with PAK1 inhibitors inhibited cell proliferation in vitro and tumor growth in xenograft models [62].
4.3. Hippo Pathway
The regulation of the Hippo pathway by merlin is better characterized in meningiomas. This evolutionarily conserved signaling pathway, first identified in Drosophila melanogaster, inhibits cell proliferation and promotes apoptosis to limit organ size during normal development and suppress tumorigenesis [63]. It relies upon a kinase cascade including macrophage-stimulating 1/2 (MST1/2), salvador family WW domain-containing protein 1 (SAV1, also called WW45), and large tumor suppressor 1/2 (LATS1/2) to phosphorylate yes-associated protein (YAP), leading to the sequestration of this key transcriptional coactivator from the nucleus, thereby inhibiting the transcription of target genes associated with proliferation and survival (Figure 2).
The suppression of merlin using NF2 siRNA in established meningioma cell lines inhibited contact-dependent inhibition of growth and promoted cell cycle progression in association with increased levels and elevated nuclear co-localization of YAP [64]. In addition, nuclear YAP immunoreactivity was revealed in 92% of merlin-negative tumors, further suggesting that merlin is a negative regulator of the Hippo pathway in meningiomas. A separate study also confirmed a complementary pattern of merlin expression and nuclear YAP expression, although, unexpectedly, nuclear YAP expression was found even in merlin-positive tumors [65]. Furthermore, nuclear YAP expression was increased when meningioma cell lines were plated at sparse cell density and in less rigid extracellular matrix, suggesting that inhibition of YAP-mediated Hippo signaling pathway by merlin is dependent on cell-to-cell contact and upstream adhesion molecules.
4.4. PI3K/AKT/mTOR Pathway
Phosphoinositide 3-kinase (PI3K)/AKT signaling is involved in the regulation of cell growth and proliferation [66]. Growth factor stimulation triggers the production of phosphatidylinositol (3,4,5) triphosphate (PIP3) by PI3K, leading to the phosphorylation and activation of downstream AKT at the plasma membrane and subsequent activation of mammalian target of rapamycin complex (mTORC), resulting in the translation of target proteins. It has been shown that merlin inhibits the activation of PI3K by binding phosphatidylinositol 3-kinase enhancer-L (PIKE-L) [67]. The finding that a subset of meningiomas have activating E17K mutations in AKT supports a crucial role of this pathway in meningioma biology [36,37,39,43]. High-grade meningiomas showed higher levels of phosphorylated AKT compared to benign tumors, further supporting a role for this pathway in merlin-driven meningioma pathogenesis [68]. Conversely, inhibition of AKT phosphorylation decreased meningioma growth in several in vitro studies [69,70].
Merlin has also been identified as a negative regulator of mTORC1 (Figure 2). This association was initially suspected from the observation that cultured primary human merlin-deficient meningioma cells exhibited a strikingly enlarged morphology compared to non-neoplastic arachnoid cap cells from the same patient [71], bearing similarity to tuberous sclerosis, wherein mutations in TSC1 and TSC2 lead to aberrant activation of mTORC1 [72]. Additional studies demonstrated that merlin-deficient primary meningioma cell lines and tumors exhibit constitutive activation of mTORC1, and conversely, exogenous expression of wild-type, but not mutant, merlin inhibited mTORC1 signaling [73]. Interestingly, merlin-deficient mTORC1 activation was independent of upstream PI3K/Akt and ERK signaling, which traditionally activate this signaling in response to various mitogenic stimuli. Therefore, the non-canonical mechanism by which the loss of merlin induces mTORC1 signaling is unknown.
mTORC1 inhibition is a validated therapeutic strategy in various types of cancers, and several orally bioavailable mTORC1 inhibitors are currently FDA-approved, including temisirolimus and everolimus. Despite the incomplete understanding of the interaction between merlin and mTORC1, mTORC1 inhibitors have been tested in various in vitro and in vivo meningioma models and patients. Temisirolimus and everolimus treatment significantly decreased viability and proliferation of a meningioma cell line in a concentration-dependent manner, and temisirolimus significantly reduced tumor burden in xenograft models [74]. Interestingly, shRNA-mediated downregulation of merlin rendered the meningioma cells more resistant to mTORC1 inhibition, presumably due to merlin deficiency-mediated constitutive upregulation of mTORC1 activity. In a small prospective phase 2 trial of 17 patients with progressive or refractory symptomatic meningiomas, concurrent treatment with bevacizumab and everolimus demonstrated overall median progression-free survival of 22 months [75]. An additional trial with the mTORC1/2 inhibitor AZD2014 is currently ongoing for patients with neurofibromatosis type 2-associated meningiomas (NCT 02831257) and recurrent high-grade meningiomas (NCT 03071874).
5. Animal Models
5.1. Xenograft Models
Xenograft models rely upon the implantation of human meningioma cells into immunocompromised mice. Their usefulness as a tool to investigate the role of merlin in meningioma pathogenesis is limited by several factors. Primary cells isolated from surgical samples, especially from benign tumors, do not reliably generate tumors, and the methods for their intracranial delivery have not been standardized [76]. In our experience, intracranial implantation of high-grade meningiomas can lead to xenograft formation; however, these do not serially transplant. Thus, large amounts of the original tumor are necessary to continue to develop numerous xenografts from a single tumor.
Most investigators have therefore relied upon established cell lines for xenograft experiments, such as the well-characterized BenMen1 line derived from a WHO grade I meningothelial meningioma which recapitulates key histologic and genetic features of the parent tumor, including NF2 mutation [60,61]. However, this cell line has been retrovirally transduced with the human telomerase reverse transcriptase (hTERT) gene, in order to bypass senescence, with unclear alterations in underlying tumor biology [77]. The less well characterized HBL-52 cell line was derived from a transitional grade I optic canal meningioma, but this cell line harbors the TRAF7 driver mutation [78,79,80]. Other cell lines are derived from high-grade meningiomas such as the IOMM-Lee and CH-157MN cell lines, and these cell lines demonstrate the genomic instability seen in more aggressive parent tumors [61,64,65]. However, the IOMM-Lee cell line has intact NF2, rendering it unsuitable for studies of merlin function [79]. Overall, these cell lines have no comparable controls, and their predetermined or laboratory-altered genetics are unlikely to account for the full complexity of their real-life counterparts. Finally, due to the immunocompromised nature of the host, potentially important immune interactions cannot be analyzed.
5.2. Genetically Engineered Mouse Models (GEMM)
Homozygous deletion of Nf2 in mice is embryonically lethal, and heterozygous Nf2 knockout mice develop osteosarcomas but not meningiomas [81]. Therefore, initial efforts to create a Nf2 deletion-based GEMM of meningiomas relied upon a conditional knockout approach. Given the lack of known arachnoid-specific promoters at the time, an adenovirus encoding recombinant Cre was injected into the subdural space of mice harboring two copies of floxed Nf2 allele (Nf2flox/flox), driving arachnoid-specific deletion of Nf2. Remarkably, this was sufficient to induce a range of benign meningioma encompassing the transitional, meningothelial, and fibroblastic subtypes, although only a minority of injected mice ultimately developed meningiomas [82]. Additional studies demonstrated that the arachnoid-specific deletion of cyclin-dependent kinase inhibitor (Cdkn2ab), frequently deleted in high-grade meningiomas, increased meningioma frequency and the development of grade II and III meningiomas in mice [83]. Shortly after these initial studies, prostaglandin D2 synthase (PTGDS) was identified as a specific marker for meningioma precursor cells. By crossing transgenic mice expressing Cre under the PTGDS promoter to Nf2flox/flox mice, biallelic Nf2 inactivation in meningioma precursor cells was achieved, without the need for exogenous Cre delivery [84]. This resulted in the generation of meningothelial and fibroblastic meningiomas in the majority of animals. Taken together, these results provide proof for a fundamental role of merlin in meningioma induction and provide ideal models for further investigation into merlin signaling, especially incorporating insights from recent genetic studies based on next-generation sequencing.
6. Conclusions
Merlin/NF2 loss is a key driver in the development of both syndromic and most sporadic meningiomas. Next-generation sequencing studies have provided a framework for an increasingly sophisticated categorization of meningiomas into two groups: non-NF2 mutants and NF2 mutants. It is intriguing, however, that the two types of NF2-inactivated meningiomas (type B and C) seem to have different underlying molecular mechanisms and dramatically different clinical outcomes. How the loss of merlin function leads to two very different biological dysregulations is yet to be investigated. These insights need to be further investigated through large-scale NGS studies and, more importantly, biochemical and molecular studies to reveal therapeutically relevant targets.
Author Contributions
Conceptualization, S.L. and A.J.P.; methodology, S.L. and A.J.P.; formal analysis, S.L., P.J.K., C.C.H., J.C.B.V., A.B.K., A.J., A.D.S., T.J.K., and A.J.P.; investigation, S.L., P.J.K., C.C.H., J.C.B.V., A.B.K., A.J., A.D.S., T.J.K., and A.J.P.; writing—original draft preparation, S.L., P.J.K., and A.J.P.; writing—review and editing, S.L., P.J.K., C.C.H., J.C.B.V., A.B.K., A.J., A.D.S., T.J.K., and A.J.P.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Asthagiri, A.R.; Parry, D.M.; Butman, J.A.; Kim, H.J.; Tsilou, E.T.; Zhuang, Z.; Lonser, R.R. Neurofibromatosis type 2. Lancet Lond. Engl. 2009, 373, 1974–1986. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, I.; Yu, Q. Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr. Protein Pept. Sci. 2010, 11, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Trofatter, J.A.; MacCollin, M.M.; Rutter, J.L.; Murrell, J.R.; Duyao, M.P.; Parry, D.M.; Eldridge, R.; Kley, N.; Menon, A.G.; Pulaski, K. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993, 72, 791–800. [Google Scholar] [CrossRef]
- Rouleau, G.A.; Merel, P.; Lutchman, M.; Sanson, M.; Zucman, J.; Marineau, C.; Hoang-Xuan, K.; Demczuk, S.; Desmaze, C.; Plougastel, B. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 1993, 363, 515–521. [Google Scholar] [CrossRef]
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. A 2010, 152A, 327–332. [Google Scholar] [CrossRef]
- Baser, M.E.; Friedman, J.M.; Wallace, A.J.; Ramsden, R.T.; Joe, H.; Evans, D.G.R. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology 2002, 59, 1759–1765. [Google Scholar] [CrossRef]
- Evans, D.G.; Huson, S.M.; Donnai, D.; Neary, W.; Blair, V.; Newton, V.; Harris, R. A clinical study of type 2 neurofibromatosis. Q. J. Med. 1992, 84, 603–618. [Google Scholar]
- Ahronowitz, I.; Xin, W.; Kiely, R.; Sims, K.; MacCollin, M.; Nunes, F.P. Mutational spectrum of the NF2 gene: A meta-analysis of 12 years of research and diagnostic laboratory findings. Hum. Mutat. 2007, 28, 1–12. [Google Scholar] [CrossRef]
- Parry, D.M.; Eldridge, R.; Kaiser-Kupfer, M.I.; Bouzas, E.A.; Pikus, A.; Patronas, N. Neurofibromatosis 2 (NF2): Clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am. J. Med. Genet. 1994, 52, 450–461. [Google Scholar] [CrossRef]
- Kluwe, L.; Mautner, V.F. A missense mutation in the NF2 gene results in moderate and mild clinical phenotypes of neurofibromatosis type 2. Hum. Genet. 1996, 97, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Ruttledge, M.H.; Andermann, A.A.; Phelan, C.M.; Claudio, J.O.; Han, F.Y.; Chretien, N.; Rangaratnam, S.; MacCollin, M.; Short, P.; Parry, D.; et al. Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am. J. Hum. Genet. 1996, 59, 331–342. [Google Scholar] [PubMed]
- Kluwe, L.; MacCollin, M.; Tatagiba, M.; Thomas, S.; Hazim, W.; Haase, W.; Mautner, V.F. Phenotypic variability associated with 14 splice-site mutations in the NF2 gene. Am. J. Med. Genet. 1998, 77, 228–233. [Google Scholar] [CrossRef]
- Baser, M.E.; Kuramoto, L.; Joe, H.; Friedman, J.M.; Wallace, A.J.; Gillespie, J.E.; Ramsden, R.T.; Evans, D.G.R. Genotype-phenotype correlations for nervous system tumors in neurofibromatosis 2: A population-based study. Am. J. Hum. Genet. 2004, 75, 231–239. [Google Scholar] [CrossRef]
- Evans, D.G.; Wallace, A.J.; Wu, C.L.; Trueman, L.; Ramsden, R.T.; Strachan, T. Somatic mosaicism: A common cause of classic disease in tumor-prone syndromes? Lessons from type 2 neurofibromatosis. Am. J. Hum. Genet. 1998, 63, 727–736. [Google Scholar]
- Kluwe, L.; Mautner, V.; Heinrich, B.; Dezube, R.; Jacoby, L.B.; Friedrich, R.E.; MacCollin, M. Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J. Med. Genet. 2003, 40, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Moyhuddin, A.; Baser, M.E.; Watson, C.; Purcell, S.; Ramsden, R.T.; Heiberg, A.; Wallace, A.J.; Evans, D.G.R. Somatic mosaicism in neurofibromatosis 2: Prevalence and risk of disease transmission to offspring. J. Med. Genet. 2003, 40, 459–463. [Google Scholar] [CrossRef]
- Mautner, V.F.; Lindenau, M.; Baser, M.E.; Hazim, W.; Tatagiba, M.; Haase, W.; Samii, M.; Wais, R.; Pulst, S.M. The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery 1996, 38, 880–885; discussion 885–886. [Google Scholar] [CrossRef]
- Otsuka, G.; Saito, K.; Nagatani, T.; Yoshida, J. Age at symptom onset and long-term survival in patients with neurofibromatosis Type 2. J. Neurosurg. 2003, 99, 480–483. [Google Scholar] [CrossRef]
- Mautner, V.F.; Tatagiba, M.; Lindenau, M.; Fünsterer, C.; Pulst, S.M.; Baser, M.E.; Kluwe, L.; Zanella, F.E. Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. AJR Am. J. Roentgenol. 1995, 165, 951–955. [Google Scholar] [CrossRef]
- Dirks, M.S.; Butman, J.A.; Kim, H.J.; Wu, T.; Morgan, K.; Tran, A.P.; Lonser, R.R.; Asthagiri, A.R. Long-term natural history of neurofibromatosis Type 2–associated intracranial tumors. J. Neurosurg. 2012, 117, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, S.; Bah, A.B.; Henin, D.; Parfait, B.; Grayeli, A.B.; Sterkers, O.; Kalamarides, M. Long-term follow-up of 287 meningiomas in neurofibromatosis type 2 patients: Clinical, radiological, and molecular features. Neuro-oncology 2012, 14, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Ruttledge, M.H.; Sarrazin, J.; Rangaratnam, S.; Phelan, C.M.; Twist, E.; Merel, P.; Delattre, O.; Thomas, G.; Nordenskjöld, M.; Collins, V.P. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat. Genet. 1994, 6, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Giordano, M.J.; Fishback, A.S.; Guha, A. Loss of merlin expression in sporadic meningiomas, ependymomas and schwannomas. Neurology 1997, 49, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Lomas, J.; Bello, M.J.; Arjona, D.; Alonso, M.E.; Martinez-Glez, V.; Lopez-Marin, I.; Amiñoso, C.; de Campos, J.M.; Isla, A.; Vaquero, J.; et al. Genetic and epigenetic alteration of the NF2 gene in sporadic meningiomas: NF2 in Meningiomas. Genes. Chromosomes Cancer 2005, 42, 314–319. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Perry, A.; Reifenberger, G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 2006, 5, 1045–1054. [Google Scholar] [CrossRef]
- MacCollin, M.; Chiocca, E.A.; Evans, D.G.; Friedman, J.M.; Horvitz, R.; Jaramillo, D.; Lev, M.; Mautner, V.F.; Niimura, M.; Plotkin, S.R.; et al. Diagnostic criteria for schwannomatosis. Neurology 2005, 64, 1838–1845. [Google Scholar] [CrossRef]
- Koontz, N.A.; Wiens, A.L.; Agarwal, A.; Hingtgen, C.M.; Emerson, R.E.; Mosier, K.M. Schwannomatosis: The overlooked neurofibromatosis? AJR Am. J. Roentgenol. 2013, 200, W646–W653. [Google Scholar] [CrossRef]
- Hulsebos, T.J.M.; Plomp, A.S.; Wolterman, R.A.; Robanus-Maandag, E.C.; Baas, F.; Wesseling, P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am. J. Hum. Genet. 2007, 80, 805–810. [Google Scholar] [CrossRef]
- Piotrowski, A.; Xie, J.; Liu, Y.F.; Poplawski, A.B.; Gomes, A.R.; Madanecki, P.; Fu, C.; Crowley, M.R.; Crossman, D.K.; Armstrong, L.; et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 2014, 46, 182–187. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Farschtschi, S.; Mautner, V.-F.; Cooper, D.N. The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis. Hum. Genet. 2017, 136, 129–148. [Google Scholar] [CrossRef] [PubMed]
- Merker, V.L.; Esparza, S.; Smith, M.J.; Stemmer-Rachamimov, A.; Plotkin, S.R. Clinical features of schwannomatosis: A retrospective analysis of 87 patients. Oncologist 2012, 17, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Bacci, C.; Sestini, R.; Provenzano, A.; Paganini, I.; Mancini, I.; Porfirio, B.; Vivarelli, R.; Genuardi, M.; Papi, L. Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics 2010, 11, 73–80. [Google Scholar] [CrossRef]
- Melean, G.; Velasco, A.; Hernández-Imaz, E.; Rodríguez-Álvarez, F.J.; Martín, Y.; Valero, A.; Hernández-Chico, C. RNA-based analysis of two SMARCB1 mutations associated with familial schwannomatosis with meningiomas. Neurogenetics 2012, 13, 267–274. [Google Scholar] [CrossRef] [PubMed]
- van den Munckhof, P.; Christiaans, I.; Kenter, S.B.; Baas, F.; Hulsebos, T.J.M. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics 2012, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avşar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Reuss, D.E.; Piro, R.M.; Jones, D.T.W.; Simon, M.; Ketter, R.; Kool, M.; Becker, A.; Sahm, F.; Pusch, S.; Meyer, J.; et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013, 125, 351–358. [Google Scholar] [CrossRef]
- Clark, V.E.; Harmancı, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.; Suppiah, S.; Tonge, P.D.; Jalali, S.; Danesh, A.; Bruce, J.P.; Mamatjan, Y.; Klironomos, G.; Gonen, L.; Au, K.; et al. Therapeutic radiation for childhood cancer drives structural aberrations of NF2 in meningiomas. Nat. Commun. 2017, 8, 186. [Google Scholar] [CrossRef]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic landscape of high-grade meningiomas. NPJ Genom. Med. 2017, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Harmancı, A.S.; Youngblood, M.W.; Clark, V.E.; Coşkun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat. Commun. 2017, 8, 14433. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Wan, Y.-W.; Al-Ouran, R.; Revelli, J.-P.; Cardenas, M.F.; Oneissi, M.; Xi, L.; Jalali, A.; Magnotti, J.F.; Muzny, D.M.; et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed]
- Modha, A.; Gutin, P.H. Diagnosis and treatment of atypical and anaplastic meningiomas: A review. Neurosurgery 2005, 57, 538–550; discussion 538–550. [Google Scholar] [CrossRef] [PubMed]
- Pearson, B.E.; Markert, J.M.; Fisher, W.S.; Guthrie, B.L.; Fiveash, J.B.; Palmer, C.A.; Riley, K. Hitting a moving target: Evolution of a treatment paradigm for atypical meningiomas amid changing diagnostic criteria. Neurosurg. Focus 2008, 24, E3. [Google Scholar] [CrossRef] [PubMed]
- Aghi, M.K.; Carter, B.S.; Cosgrove, G.R.; Ojemann, R.G.; Amin-Hanjani, S.; Martuza, R.L.; Curry, W.T.; Barker, F.G. Long-term recurrence rates of atypical meningiomas after gross total resection with or without postoperative adjuvant radiation. Neurosurgery 2009, 64, 56–60; discussion 60. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Braunstein, S.E.; Phillips, J.J.; Pekmezci, M.; Tomlin, B.A.; Wu, A.; Reis, G.F.; Magill, S.T.; Zhang, J.; Feng, F.Y.; et al. Comprehensive Molecular Profiling Identifies FOXM1 as a Key Transcription Factor for Meningioma Proliferation. Cell Rep. 2018, 22, 3672–3683. [Google Scholar] [CrossRef] [Green Version]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef]
- Kadoch, C.; Copeland, R.A.; Keilhack, H. PRC2 and SWI/SNF Chromatin Remodeling Complexes in Health and Disease. Biochemistry 2016, 55, 1600–1614. [Google Scholar] [CrossRef]
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef]
- Dewan, R.; Pemov, A.; Dutra, A.S.; Pak, E.D.; Edwards, N.A.; Ray-Chaudhury, A.; Hansen, N.F.; Chandrasekharappa, S.C.; Mullikin, J.C.; Asthagiri, A.R.; et al. First insight into the somatic mutation burden of neurofibromatosis type 2-associated grade I and grade II meningiomas: A case report comprehensive genomic study of two cranial meningiomas with vastly different clinical presentation. BMC Cancer 2017, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Gary, R.; Bretscher, A. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol. Biol. Cell 1995, 6, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.A.; Reczek, D.; Bretscher, A.; Karplus, P.A. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 2000, 101, 259–270. [Google Scholar] [CrossRef]
- Shaw, R.J.; Paez, J.G.; Curto, M.; Yaktine, A.; Pruitt, W.M.; Saotome, I.; O’Bryan, J.P.; Gupta, V.; Ratner, N.; Der, C.J.; et al. The Nf2 Tumor Suppressor, Merlin, Functions in Rac-Dependent Signaling. Dev. Cell 2001, 1, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Lopez-Lago, M.; Giancotti, F.G. Merlin/ NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol. 2005, 171, 361–371. [Google Scholar] [CrossRef]
- Jin, H.; Sperka, T.; Herrlich, P.; Morrison, H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature 2006, 442, 576–579. [Google Scholar] [CrossRef] [Green Version]
- Hennigan, R.F.; Foster, L.A.; Chaiken, M.F.; Mani, T.; Gomes, M.M.; Herr, A.B.; Ip, W. Fluorescence Resonance Energy Transfer Analysis of Merlin Conformational Changes. Mol. Cell. Biol. 2010, 30, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Ali Khajeh, J.; Ju, J.H.; Atchiba, M.; Allaire, M.; Stanley, C.; Heller, W.T.; Callaway, D.J.E.; Bu, Z. Molecular Conformation of the Full-Length Tumor Suppressor NF2/Merlin—A Small-Angle Neutron Scattering Study. J. Mol. Biol. 2014, 426, 2755–2768. [Google Scholar] [CrossRef]
- Chinthalapudi, K.; Mandati, V.; Zheng, J.; Sharff, A.J.; Bricogne, G.; Griffin, P.R.; Kissil, J.; Izard, T. Lipid binding promotes the open conformation and tumor-suppressive activity of neurofibromin 2. Nat. Commun. 2018, 9, 1388. [Google Scholar] [CrossRef]
- Morrison, H. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001, 15, 968–980. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, Z.; Brown, C.M.; Patel, A.K.; Ryan, A.F.; Ongkeko, R.; Doherty, J.K. Merlin knockdown in human Schwann cells: Clues to vestibular schwannoma tumorigenesis. Otol. Neurotol. Off. Publ. Am. Otol. Soc. Am. Neurotol. Soc. Eur. Acad. Otol. Neurotol. 2010, 31, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.-Y.; Dong, B.; Duron, S.G.; Campbell, D.A.; Ong, C.C.; Hoeflich, K.P.; Chang, L.-S.; Welling, D.B.; Yang, Z.; Chernoff, J. Group I Paks as therapeutic targets in NF2-deficient meningioma. Oncotarget 2015, 6, 1981. [Google Scholar] [CrossRef] [PubMed]
- Watt, K.I.; Harvey, K.F.; Gregorevic, P. Regulation of Tissue Growth by the Mammalian Hippo Signaling Pathway. Front. Physiol. 2017, 8, 942. [Google Scholar] [CrossRef] [PubMed]
- Striedinger, K.; VandenBerg, S.R.; Baia, G.S.; McDermott, M.W.; Gutmann, D.H.; Lal, A. The Neurofibromatosis 2 Tumor Suppressor Gene Product, Merlin, Regulates Human Meningioma Cell Growth by Signaling through YAP. Neoplasia 2008, 10, 1204–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanahashi, K.; Natsume, A.; Ohka, F.; Motomura, K.; Alim, A.; Tanaka, I.; Senga, T.; Harada, I.; Fukuyama, R.; Sumiyoshi, N.; et al. Activation of Yes-Associated Protein in Low-Grade Meningiomas Is Regulated by Merlin, Cell Density, and Extracellular Matrix Stiffness. J. Neuropathol. Exp. Neurol. 2015, 74, 704–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The relation between PI3K/AKT signalling pathway and cancer. Gene 2019, 698, 120–128. [Google Scholar] [CrossRef]
- Rong, R.; Tang, X.; Gutmann, D.H.; Ye, K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc. Natl. Acad. Sci. USA 2004, 101, 18200–18205. [Google Scholar] [CrossRef] [Green Version]
- Mawrin, C. Different Activation of Mitogen-Activated Protein Kinase and Akt Signaling Is Associated with Aggressive Phenotype of Human Meningiomas. Clin. Cancer Res. 2005, 11, 4074–4082. [Google Scholar] [CrossRef]
- Bush, M.L.; Oblinger, J.; Brendel, V.; Santarelli, G.; Huang, J.; Akhmametyeva, E.M.; Burns, S.S.; Wheeler, J.; Davis, J.; Yates, C.W.; et al. AR42, a novel histone deacetylase inhibitor, as a potential therapy for vestibular schwannomas and meningiomas. Neuro-oncology 2011, 13, 983–999. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Roth, P.; Sahm, F.; Burghardt, I.; Schuknecht, B.; Rushing, E.J.; Regli, L.; Lindemann, J.P.; von Deimling, A. Durable Control of Metastatic AKT1-Mutant WHO Grade 1 Meningothelial Meningioma by the AKT Inhibitor, AZD5363. J. Natl. Cancer Inst. 2017, 109, 1–4. [Google Scholar] [CrossRef]
- James, M.F.; Lelke, J.M.; Maccollin, M.; Plotkin, S.R.; Stemmer-Rachamimov, A.O.; Ramesh, V.; Gusella, J.F. Modeling NF2 with human arachnoidal and meningioma cell culture systems: NF2 silencing reflects the benign character of tumor growth. Neurobiol. Dis. 2008, 29, 278–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, D.J. Tuberous sclerosis: From tubers to mTOR. Ann. Hum. Genet. 2003, 67, 87–96. [Google Scholar] [CrossRef] [PubMed]
- James, M.F.; Han, S.; Polizzano, C.; Plotkin, S.R.; Manning, B.D.; Stemmer-Rachamimov, A.O.; Gusella, J.F.; Ramesh, V. NF2/Merlin Is a Novel Negative Regulator of mTOR Complex 1, and Activation of mTORC1 Is Associated with Meningioma and Schwannoma Growth. Mol. Cell. Biol. 2009, 29, 4250–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachow, D.; Andrae, N.; Kliese, N.; Angenstein, F.; Stork, O.; Wilisch-Neumann, A.; Kirches, E.; Mawrin, C. mTORC1 Inhibitors Suppress Meningioma Growth in Mouse Models. Clin. Cancer Res. 2013, 19, 1180–1189. [Google Scholar] [CrossRef]
- Shih, K.C.; Chowdhary, S.; Rosenblatt, P.; Weir, A.B.; Shepard, G.C.; Williams, J.T.; Shastry, M.; Burris, H.A.; Hainsworth, J.D. A phase II trial of bevacizumab and everolimus as treatment for patients with refractory, progressive intracranial meningioma. J. Neurooncol. 2016, 129, 281–288. [Google Scholar] [CrossRef]
- McCutcheon, I.E.; Friend, K.E.; Gerdes, T.M.; Zhang, B.-M.; Wildrick, D.M.; Fuller, G.N. Intracranial injection of human meningioma cells in athymic mice: An orthotopic model for meningioma growth. J. Neurosurg. 2000, 92, 306–314. [Google Scholar] [CrossRef]
- Püttmann, S.; Senner, V.; Braune, S.; Hillmann, B.; Exeler, R.; Rickert, C.H.; Paulus, W. Establishment of a benign meningioma cell line by hTERT-mediated immortalization. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 1163–1171. [Google Scholar] [CrossRef]
- Akat, K.; Mennel, H.-D.; Kremer, P.; Gassler, N.; Bleck, C.K.E.; Kartenbeck, J. Molecular characterization of desmosomes in meningiomas and arachnoidal tissue. Acta Neuropathol. (Berl.) 2003, 106, 337–347. [Google Scholar] [CrossRef]
- Mei, Y.; Bi, W.L.; Greenwald, N.F.; Agar, N.Y.; Beroukhim, R.; Dunn, G.P.; Dunn, I.F. Genomic profile of human meningioma cell lines. PLoS ONE 2017, 12, e0178322. [Google Scholar] [CrossRef]
- Akat, K.; Bleck, C.K.E.; Lee, Y.-M.A.; Haselmann-Weiss, U.; Kartenbeck, J. Characterization of a novel type of adherens junction in meningiomas and the derived cell line HBL-52. Cell Tissue Res. 2008, 331, 401–412. [Google Scholar] [CrossRef]
- McClatchey, A.I.; Saotome, I.; Mercer, K.; Crowley, D.; Gusella, J.F.; Bronson, R.T.; Jacks, T. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev. 1998, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalamarides, M. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002, 16, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Peyre, M.; Stemmer-Rachamimov, A.; Clermont-Taranchon, E.; Quentin, S.; El-Taraya, N.; Walczak, C.; Volk, A.; Niwa-Kawakita, M.; Karboul, N.; Giovannini, M.; et al. Meningioma progression in mice triggered by Nf2 and Cdkn2ab inactivation. Oncogene 2013, 32, 4264–4272. [Google Scholar] [CrossRef]
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Niwa-Kawakita, M.; Chareyre, F.; Taranchon, E.; Han, Z.-Y.; Martinelli, C.; Lusis, E.A.; Hegedus, B.; Gutmann, D.H.; et al. Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene 2011, 30, 2333–2344. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Summary of the mutational landscape of meningiomas based on several next-generation sequencing studies.
Figure 1.
Summary of the mutational landscape of meningiomas based on several next-generation sequencing studies.

Figure 2.
Merlin signaling. (A) Active, de-phosphorylated Merlin inhibits Rac1 and PAK1, key mediators of contact inhibition; (B) Merlin activates the Hippo pathway, leading to YAP phosphorylation and its sequestration from the nucleus; (C) Merlin is a negative regulator of PIKE-L and mTORC1.
Figure 2.
Merlin signaling. (A) Active, de-phosphorylated Merlin inhibits Rac1 and PAK1, key mediators of contact inhibition; (B) Merlin activates the Hippo pathway, leading to YAP phosphorylation and its sequestration from the nucleus; (C) Merlin is a negative regulator of PIKE-L and mTORC1.


{kind=link}
{kind=link}
Table 1.
Manchester clinical diagnostic criteria for neurofibromatosis type 2 (NF2).
| Diagnostic Criteria | Additional Findings Needed |
|---|---|
| Bilateral vestibular schwannomas | None. |
| Family history of NF2 | Unilateral vestibular schwannoma, OR at least two of: meningioma, schwannoma, glioma, neurofibroma, cataract. |
| Unilateral vestibular schwannoma | At least two of: meningioma, schwannoma, glioma, neurofibroma, cataract. |
| Multiple meningiomas | Unilateral vestibular schwannoma, OR at least two of: schwannoma, glioma neurofibroma, cataract. |
Table 2.
Summary of next-generation sequencing (NGS) studies.
| Study | Tumor Type (n) | Genetic Alterations | Key Findings |
|---|---|---|---|
| Clark et al. (2013) [36] | WHO I/II (243/57) | NF2/ch22q loss TRAF7/KLF4 TRAF7/AKT1 SMO | Mutually exclusive non-NF2 driver mutations. NF2 tumors are more aggressive. Non-NF2 tumors are benign and localize to medial skull base. |
| Brastianos et al. (2013) [37] | WHO I/II/III (47/15/3) | NF2/ch22q loss SMO AKT1 | As above. |
| Reuss et al. (2013) [38] | Secretory (30) | TRAF7/KLF4 | All secretory meningiomas carried the KLF4 K409Q mutation. |
| Clark et al. (2016) [39] | WHO I/II/III/? (552/214/7/2) | POLR2A SMARCB1 | Identification of POLR2A driver mutation. SMARCB1 and NF2 mutations co-occur. |
| Agnihotri et al. (2017) [40] | Radiation-induced (31) | NF2/ch22q loss | NF2 gene rearrangements common in radiation-induced tumors. Non-NF2 driver mutations not observed. |
| Bi et al. (2017) [41] | WHO I/II/III (75/113/21) | NF2/ch22q loss Genomic instability | NF2/ch22q loss and genomic disruptions occur early in progression and remain consistent over time. |
| Harmanci et al. (2017) [42] | WHO I/II/III/? (548/211/7/9) | NF2/genomic instability NF2/SMARCB1 | NF2 is the sole driver mutation in atypical meningiomas and occurs in conjunction with genomic instability or SMARCB1 mutations. |
| Patel et al. (2019) [43] | WHO I/II/III (119/33/5) | Loss of PRC2 or DREAM complex repression | Transcriptional signatures identified a sole subgroup with recurring tumors, characterized by DREAM target genes activation. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, S.; Karas, P.J.; Hadley, C.C.; Bayley V, J.C.; Khan, A.B.; Jalali, A.; Sweeney, A.D.; Klisch, T.J.; Patel, A.J. The Role of Merlin/NF2 Loss in Meningioma Biology. Cancers 2019, 11, 1633. https://doi.org/10.3390/cancers11111633
AMA Style
Lee S, Karas PJ, Hadley CC, Bayley V JC, Khan AB, Jalali A, Sweeney AD, Klisch TJ, Patel AJ. The Role of Merlin/NF2 Loss in Meningioma Biology. Cancers. 2019; 11(11):1633. https://doi.org/10.3390/cancers11111633
Chicago/Turabian StyleLee, Sungho, Patrick J. Karas, Caroline C. Hadley, James C. Bayley V, A. Basit Khan, Ali Jalali, Alex D. Sweeney, Tiemo J. Klisch, and Akash J. Patel. 2019. "The Role of Merlin/NF2 Loss in Meningioma Biology" Cancers 11, no. 11: 1633. https://doi.org/10.3390/cancers11111633
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.