Monoclonal Antibodies in Dermatooncology—State of the Art and Future Perspectives

 , , and
, , and 
Abstract
1. Monoclonal Antibodies
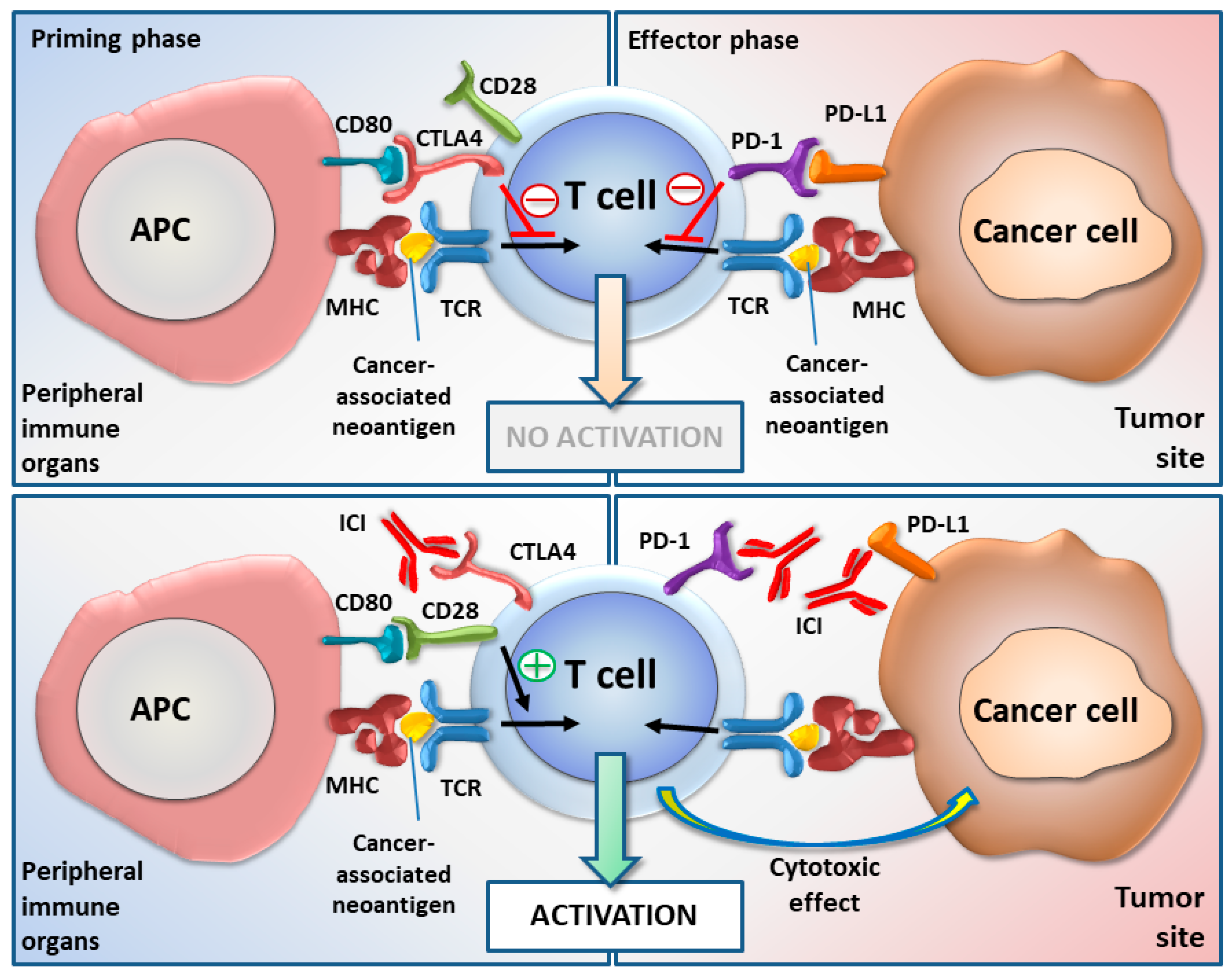
Immune Checkpoints
2. Melanoma
2.1. Checkpoint Inhibitors in Melanoma Treatment
- Cemiplimab—a small exploratory tumor biopsy-driven study to understand the relationship between biomarkers and clinical response in melanoma patients (NCT03002376)—ongoing.
- Avelumab:
- -
- Combination immunotherapy with vaccine in subjects with melanoma who have progressed on or after chemotherapy and PD-1/PD-L1 therapy (NCT03167177)—not yet recruiting.
- -
- In metastatic or locally advanced solid tumors (NCT01772004)—preliminary results show durable responses, promising survival outcomes and an acceptable safety profile in patients with previously treated metastatic melanoma [42].
- -
- In combination with other cancer immunotherapies in advanced malignancies (NCT02554812)—currently recruiting.
- Durvalumab:
- -
- Phase 1 safety and tolerability in combination with dabrafenib and trametinib or with trametinib alone (NCT02027961)—awaiting results’ publication.
- Atezolizumab—tested in numerous clinical trials.
2.2. Rituximab in Melanoma
2.3. Targeting LAG-3
2.4. Targeting TIGIT
2.5. Targeting TIM-3
2.6. Targeting B7-H3
2.7. Targeting VEGF
2.8. Targeting Chondroitin Sulfate Proteoglycan 4 (CSPG4)
3. Basal Cell Carcinoma and Cutaneous Squamous Cell Carcinoma
3.1. Checkpoint Inhibitors
3.2. EGFR-Targeting
4. Merkel-Cell Carcinoma
Checkpoint Inhibitors in MCC
5. Primary Cutaneous Lymphomas
5.1. CTCL
5.1.1. Alemtuzumab
5.1.2. Brentuximab Vedotin
5.1.3. Mogamulizumab
5.1.4. KIR3DL2 Targeting
5.1.5. Immune Checkpoint Inhibitors
5.1.6. CD47
5.2. CBCL
Rituximab
6. Problems with the Use of Checkpoint Inhibitors
6.1. Resistance to Immune Checkpoints
6.2. Response Markers for Checkpoints Inhibitors
6.3. Managing Adverse Events
7. Improving the Efficacy of ICIs
8. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Sewell, F.; Chapman, K.; Couch, J.; Dempster, M.; Heidel, S.; Loberg, L.; Maier, C.; Maclachlan, T.K.; Todd, M.; van der Laan, J.W. Challenges and opportunities for the future of monoclonal antibody development: Improving safety assessment and reducing animal use. MAbs 2017, 9, 742–755. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef]
- Stevens, N.E.; Cowin, A.J.; Kopecki, Z. Skin barrier and autoimmunity-mechanisms and novel therapeutic Approaches for autoimmune Blistering diseases of the skin. Front. Immunol. 2019, 10, 1089. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily-CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Tan, P.; Bradshaw, J.; Ledbetter, J.A.; Anasetti, C.; Damle, N.K. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 1992, 176, 1595–1604. [Google Scholar] [CrossRef]
- Lindsten, T.; Lee, K.P.; Harris, E.S.; Petryniak, B.; Craighead, N.; Reynolds, P.J.; Lombard, D.B.; Freeman, G.J.; Nadler, L.M.; Gray, G.S.; et al. Characterization of CTLA-4 structure and expression on human T cells. J. Immunol. 1993, 151, 3489–3499. [Google Scholar]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Lee, K.M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular basis of T cell inactivation by CTLA-4. Science 1998, 282, 2263–2266. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Guleria, I.; Khosroshahi, A.; Ansari, M.J.; Habicht, A.; Azuma, M.; Yagita, H.; Noelle, R.J.; Coyle, A.; Mellor, A.L.; Khoury, S.J.; et al. A critical role for the programmed death ligand 1 in fetomaternal tolerance. J. Exp. Med. 2005, 202, 231–237. [Google Scholar] [CrossRef]
- Paluch, C.; Santos, A.M.; Anzilotti, C.; Cornall, R.J.; Davis, S.J. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front Immunol. 2018, 9, 2306. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Iwai, Y.; Terawaki, S.; Honjo, T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol. 2005, 17, 133–144. [Google Scholar] [CrossRef]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef]
- Mazzarella, L.; Duso, B.A.; Trapani, D.; Belli, C.; D’Amico, P.; Ferraro, E.; Viale, G.; Curigliano, G. The evolving landscape of ‘next-generation’ immune checkpoint inhibitors: A review. Eur. J. Cancer 2019, 117, 14–31. [Google Scholar] [CrossRef]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef]
- Coit, D.G.; Thompson, J.A.; Albertini, M.R.; Barker, C.; Carson, W.E.; Contreras, C.; Daniels, G.A.; DiMaio, D.; Fields, R.C.; Fleming, M.D.; et al. Cutaneous melanoma; Version 2.2019; NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2019, 17, 367–402. [Google Scholar] [CrossRef]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef]
- Mahmoud, F.; Shields, B.; Makhoul, I.; Avaritt, N.; Wong, H.K.; Hutchins, L.F.; Shalin, S.; Tackett, A.J. Immune surveillance in melanoma: From immune attack to melanoma escape and even counterattack. Cancer Biol. 2017, 18, 451–469. [Google Scholar] [CrossRef]
- Weber, J. Overcoming immunologic tolerance to melanoma: targeting CTLA-4 with ipilimumab (MDX-010). Oncologist 2008, 13 (Suppl. 4), 16–25. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015, 16, 522–530. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Schadendorf, D.; Larkin, J.; Wolchok, J.; Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.; et al. Health-related quality of life results from the phase III CheckMate 067 study. Eur. J. Cancer 2017, 82, 80–91. [Google Scholar] [CrossRef]
- Hassel, J.C.; Heinzerling, L.; Aberle, J.; Bahr, O.; Eigentler, T.K.; Grimm, M.O.; Grunwald, V.; Leipe, J.; Reinmuth, N.; Tietze, J.K.; et al. Combined immune checkpoint blockade (anti-PD-1/anti-CTLA-4): Evaluation and management of adverse drug reactions. Cancer Treat Rev. 2017, 57, 36–49. [Google Scholar] [CrossRef]
- Ribas, A.; Camacho, L.H.; Lopez-Berestein, G.; Pavlov, D.; Bulanhagui, C.A.; Millham, R.; Comin-Anduix, B.; Reuben, J.M.; Seja, E.; Parker, C.A.; et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte–associated antigen 4 monoclonal antibody CP-675,206. J. Clin. Oncol. 2005, 23, 8968–8977. [Google Scholar] [CrossRef]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef]
- Keilholz, U.; Mehnert, J.M.; Bauer, S.; Bourgeois, H.; Patel, M.R.; Gravenor, D.; Nemunaitis, J.J.; Taylor, M.H.; Wyrwicz, L.; Lee, K.W.; et al. Avelumab in patients with previously treated metastatic melanoma: phase 1b results from the JAVELIN Solid Tumor trial. J. Immunother. Cancer 2019, 7, 12. [Google Scholar] [CrossRef]
- Zabierowski, S.E.; Herlyn, M. Melanoma stem cells: the dark seed of melanoma. J. Clin. Oncol. 2008, 26, 2890–2894. [Google Scholar] [CrossRef]
- Schlaak, M.; Schmidt, P.; Bangard, C.; Kurschat, P.; Mauch, C.; Abken, H. Regression of metastatic melanoma in a patient by antibody targeting of cancer stem cells. Oncotarget 2012, 3, 22–30. [Google Scholar] [CrossRef]
- Hillen, F.; Baeten, C.I.; van de Winkel, A.; Creytens, D.; van der Schaft, D.W.; Winnepenninckx, V.; Griffioen, A.W. Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma. Cancer Immunol. Immunother. 2008, 57, 97–106. [Google Scholar] [CrossRef]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef]
- Hussein, M.R.; Elsers, D.A.; Fadel, S.A.; Omar, A.E. Immunohistological characterisation of tumour infiltrating lymphocytes in melanocytic skin lesions. J. Clin. Pathol. 2006, 59, 316–324. [Google Scholar] [CrossRef]
- Ladanyi, A.; Kiss, J.; Mohos, A.; Somlai, B.; Liszkay, G.; Gilde, K.; Fejos, Z.; Gaudi, I.; Dobos, J.; Timar, J. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol. Immunother. 2011, 60, 1729–1738. [Google Scholar] [CrossRef]
- Garg, K.; Maurer, M.; Griss, J.; Bruggen, M.C.; Wolf, I.H.; Wagner, C.; Willi, N.; Mertz, K.D.; Wagner, S.N. Tumor-associated B cells in cutaneous primary melanoma and improved clinical outcome. Hum. Pathol. 2016, 54, 157–164. [Google Scholar] [CrossRef]
- Martinez-Rodriguez, M.; Thompson, A.K.; Monteagudo, C. A significant percentage of CD20-positive TILs correlates with poor prognosis in patients with primary cutaneous malignant melanoma. Histopathology 2014, 65, 726–728. [Google Scholar] [CrossRef]
- Meyer, S.; Fuchs, T.J.; Bosserhoff, A.K.; Hofstadter, F.; Pauer, A.; Roth, V.; Buhmann, J.M.; Moll, I.; Anagnostou, N.; Brandner, J.M.; et al. A seven-marker signature and clinical outcome in malignant melanoma: a large-scale tissue-microarray study with two independent patient cohorts. PLoS ONE 2012, 7, e38222. [Google Scholar] [CrossRef]
- Fremd, C.; Schuetz, F.; Sohn, C.; Beckhove, P.; Domschke, C. B cell-regulated immune responses in tumor models and cancer patients. Oncoimmunology 2013, 2, e25443. [Google Scholar] [CrossRef]
- Shah, S.; Divekar, A.A.; Hilchey, S.P.; Cho, H.M.; Newman, C.L.; Shin, S.U.; Nechustan, H.; Challita-Eid, P.M.; Segal, B.M.; Yi, K.H.; et al. Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Int. J. Cancer 2005, 117, 574–586. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Yanaba, K.; Tedder, T.F. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J. Immunol. 2010, 184, 4006–4016. [Google Scholar] [CrossRef]
- Perricone, M.A.; Smith, K.A.; Claussen, K.A.; Plog, M.S.; Hempel, D.M.; Roberts, B.L.; St George, J.A.; Kaplan, J.M. Enhanced efficacy of melanoma vaccines in the absence of B lymphocytes. J. Immunother. 2004, 27, 273–281. [Google Scholar] [CrossRef]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 607. [Google Scholar] [CrossRef]
- Winkler, J.K.; Schiller, M.; Bender, C.; Enk, A.H.; Hassel, J.C. Rituximab as a therapeutic option for patients with advanced melanoma. Cancer Immunol. Immunother. 2018, 67, 917–924. [Google Scholar] [CrossRef]
- Peuvrel, L.; Chiffoleau, A.; Quereux, G.; Brocard, A.; Saint-Jean, M.; Batz, A.; Jolliet, P.; Dreno, B. Melanoma and rituximab: An incidental association? Dermatology 2013, 226, 274–278. [Google Scholar] [CrossRef]
- Velter, C.; Pages, C.; Schneider, P.; Osio, A.; Brice, P.; Lebbe, C. Four cases of rituximab-associated melanoma. Melanoma Res. 2014, 24, 401–403. [Google Scholar] [CrossRef]
- Inoue, S.; Leitner, W.W.; Golding, B.; Scott, D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006, 66, 7741–7747. [Google Scholar] [CrossRef]
- van Vollenhoven, R.F.; Emery, P.; Bingham, C.O., 3rd; Keystone, E.C.; Fleischmann, R.; Furst, D.E.; Macey, K.; Sweetser, M.; Kelman, A.; Rao, R. Longterm safety of patients receiving rituximab in rheumatoid arthritis clinical trials. J. Rheumatol. 2010, 37, 558–567. [Google Scholar] [CrossRef]
- Goldberg, M.V.; Drake, C.G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R.; et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J. Immunol. 2011, 186, 5173–5183. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar] [CrossRef]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. OncoTargets 2018, 11, 7005–7009. [Google Scholar] [CrossRef]
- Wiener, Z.; Kohalmi, B.; Pocza, P.; Jeager, J.; Tolgyesi, G.; Toth, S.; Gorbe, E.; Papp, Z.; Falus, A. TIM-3 is expressed in melanoma cells and is upregulated in TGF-beta stimulated mast cells. J. Investig. Dermatol. 2007, 127, 906–914. [Google Scholar] [CrossRef]
- Baghdadi, M.; Nagao, H.; Yoshiyama, H.; Akiba, H.; Yagita, H.; Dosaka-Akita, H.; Jinushi, M. Combined blockade of TIM-3 and TIM-4 augments cancer vaccine efficacy against established melanomas. Cancer Immunol. Immunother. 2013, 62, 629–637. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Wang, J.; Chong, K.K.; Nakamura, Y.; Nguyen, L.; Huang, S.K.; Kuo, C.; Zhang, W.; Yu, H.; Morton, D.L.; Hoon, D.S. B7-H3 associated with tumor progression and epigenetic regulatory activity in cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 2050–2058. [Google Scholar] [CrossRef]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef]
- Hofmeyer, K.A.; Ray, A.; Zang, X. The contrasting role of B7-H3. Proc. Natl. Acad. Sci. USA 2008, 105, 10277–10278. [Google Scholar] [CrossRef]
- Flem-Karlsen, K.; Tekle, C.; Andersson, Y.; Flatmark, K.; Fodstad, O.; Nunes-Xavier, C.E. Immunoregulatory protein B7-H3 promotes growth and decreases sensitivity to therapy in metastatic melanoma cells. Pigment Cell Melanoma Res. 2017, 30, 467–476. [Google Scholar] [CrossRef]
- Dewing, D.; Emmett, M.; Pritchard Jones, R. The roles of angiogenesis in malignant melanoma: Trends in basic science research over the last 100 years. ISRN Oncol. 2012, 2012, 546927. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef]
- Del Vecchio, M.; Mortarini, R.; Canova, S.; Di Guardo, L.; Pimpinelli, N.; Sertoli, M.R.; Bedognetti, D.; Queirolo, P.; Morosini, P.; Perrone, T.; et al. Bevacizumab plus fotemustine as first-line treatment in metastatic melanoma patients: clinical activity and modulation of angiogenesis and lymphangiogenesis factors. Clin. Cancer Res. 2010, 16, 5862–5872. [Google Scholar] [CrossRef]
- von Moos, R.; Seifert, B.; Simcock, M.; Goldinger, S.M.; Gillessen, S.; Ochsenbein, A.; Michielin, O.; Cathomas, R.; Schlappi, M.; Moch, H.; et al. First-line temozolomide combined with bevacizumab in metastatic melanoma: a multicentre phase II trial (SAKK 50/07). Ann. Oncol. 2012, 23, 531–536. [Google Scholar] [CrossRef]
- Corrie, P.G.; Marshall, A.; Nathan, P.D.; Lorigan, P.; Gore, M.; Tahir, S.; Faust, G.; Kelly, C.G.; Marples, M.; Danson, S.J.; et al. Adjuvant bevacizumab for melanoma patients at high risk of recurrence: survival analysis of the AVAST-M trial. Ann. Oncol. 2018, 29, 1843–1852. [Google Scholar] [CrossRef]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Davis, T.; Henry-Spires, R.; MacRae, S.; Willman, A.; et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef]
- Price, M.A.; Colvin Wanshura, L.E.; Yang, J.; Carlson, J.; Xiang, B.; Li, G.; Ferrone, S.; Dudek, A.Z.; Turley, E.A.; McCarthy, J.B. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011, 24, 1148–1157. [Google Scholar] [CrossRef]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin sulfate proteoglycan 4 and its potential as an antibody immunotherapy target across different tumor types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef]
- Bander, T.S.; Nehal, K.S.; Lee, E.H. Cutaneous Squamous Cell Carcinoma: Updates in Staging and Management. Dermatol. Clin. 2019, 37, 241–251. [Google Scholar] [CrossRef]
- Gandhi, S.A.; Kampp, J. Skin cancer epidemiology, detection, and management. Med. Clin. N. Am. 2015, 99, 1323–1335. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Fisher, M.S.; Kripke, M.L. Suppressor T lymphocytes control the development of primary skin cancers in ultraviolet-irradiated mice. Science 1982, 216, 1133–1134. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to Immunotherapy in diverse cancers. Mol. Cancer 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Belai, E.B.; de Oliveira, C.E.; Gasparoto, T.H.; Ramos, R.N.; Torres, S.A.; Garlet, G.P.; Cavassani, K.A.; Silva, J.S.; Campanelli, A.P. PD-1 blockage delays murine squamous cell carcinoma development. Carcinogenesis 2014, 35, 424–431. [Google Scholar] [CrossRef]
- Lipson, E.J.; Bagnasco, S.M.; Moore, J., Jr.; Jang, S.; Patel, M.J.; Zachary, A.A.; Pardoll, D.M.; Taube, J.M.; Drake, C.G. Tumor regression and allograft rejection after administration of Anti-PD-1. N. Engl. J. Med. 2016, 374, 896–898. [Google Scholar] [CrossRef]
- Winkler, J.K.; Schneiderbauer, R.; Bender, C.; Sedlaczek, O.; Frohling, S.; Penzel, R.; Enk, A.; Hassel, J.C. Anti-programmed cell death-1 therapy in nonmelanoma skin cancer. Br. J. Dermatol. 2017, 176, 498–502. [Google Scholar] [CrossRef]
- Fischer, S.; Ali, O.H.; Jochum, W.; Kluckert, T.; Flatz, L.; Siano, M. Anti-PD-1 Therapy leads to near-complete remission in a patient with metastatic basal cell carcinoma. Oncol. Res. Treat. 2018, 41, 391–394. [Google Scholar] [CrossRef]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J. Immunother. Cancer 2017, 5, 23. [Google Scholar] [CrossRef]
- Ikeda, S.; Goodman, A.M.; Cohen, P.R.; Jensen, T.J.; Ellison, C.K.; Frampton, G.; Miller, V.; Patel, S.P.; Kurzrock, R. Metastatic basal cell carcinoma with amplification of PD-L1: exceptional response to anti-PD1 therapy. NPJ Genom. Med. 2016, 1. [Google Scholar] [CrossRef]
- Borradori, L.; Sutton, B.; Shayesteh, P.; Daniels, G.A. Rescue therapy with anti-programmed cell death protein 1 inhibitors of advanced cutaneous squamous cell carcinoma and basosquamous carcinoma: preliminary experience in five cases. Br. J. Dermatol. 2016, 175, 1382–1386. [Google Scholar] [CrossRef]
- Cannon, J.G.D.; Russell, J.S.; Kim, J.; Chang, A.L.S. A case of metastatic basal cell carcinoma treated with continuous PD-1 inhibitor exposure even after subsequent initiation of radiotherapy and surgery. JAAD Case Rep. 2018, 4, 248–250. [Google Scholar] [CrossRef][Green Version]
- Kudchadkar, R.R.; Yushak, M.L.; Lawson, D.H.; Delman, K.A.; Lowe, M.C.; Goings, M.; McBrien, S.; Mckellar, M.; Sieja, K.; Maynard, N.; et al. Phase II trial of pembrolizumab (MK-3475) in metastatic cutaneous squamous cell carcinoma (cSCC). J. Clin. Oncol. 2018, 36, 9543. [Google Scholar] [CrossRef]
- Chang, A.L.S.; Tran, D.C.; Cannon, J.G.D.; Li, S.; Jeng, M.; Patel, R.; Van der Bokke, L.; Pague, A.; Brotherton, R.; Rieger, K.E.; et al. Pembrolizumab for advanced basal cell carcinoma: An investigator-initiated, proof-of-concept study. J. Am. Acad. Dermatol. 2019, 80, 564–566. [Google Scholar] [CrossRef]
- Falchook, G.S.; Leidner, R.; Stankevich, E.; Piening, B.; Bifulco, C.; Lowy, I.; Fury, M.G. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN2810. J. Immunother. Cancer 2016, 4, 70. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Markham, A.; Duggan, S. Cemiplimab: First Global Approval. Drugs 2018, 78, 1841–1846. [Google Scholar] [CrossRef]
- Khan, M.H.; Alam, M.; Yoo, S. Epidermal growth factor receptor inhibitors in the treatment of nonmelanoma skin cancers. Dermatol. Surg. 2011, 37, 1199–1209. [Google Scholar] [CrossRef]
- Eder, J.; Simonitsch-Klupp, I.; Trautinger, F. Treatment of unresectable squamous cell carcinoma of the skin with epidermal growth factor receptor antibodies—A case series. Eur. J. Dermatol. 2013, 23, 658–662. [Google Scholar] [CrossRef]
- Bauman, J.E.; Eaton, K.D.; Martins, R.G. Treatment of recurrent squamous cell carcinoma of the skin with cetuximab. Arch. Dermatol. 2007, 143, 889–892. [Google Scholar] [CrossRef]
- Seber, S.; Gonultas, A.; Ozturk, O.; Yetisyigit, T. Recurrent squamous cell carcinoma of the skin treated successfully with single agent cetuximab therapy. OncoTargets 2016, 9, 945–948. [Google Scholar] [CrossRef]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef] [PubMed]
- Wollina, U. Update of cetuximab for non-melanoma skin cancer. Expert Opin. Biol. 2014, 14, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Kalapurakal, S.J.; Malone, J.; Robbins, K.T.; Buescher, L.; Godwin, J.; Rao, K. Cetuximab in refractory skin cancer treatment. J. Cancer 2012, 3, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Chang, Y.; Moore, P.S. MCV and Merkel cell carcinoma: a molecular success story. Curr. Opin. Virol. 2012, 2, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Hardee, M.E.; Cornelius, L.A.; Hutchins, L.F.; Becker, J.C.; Gao, L. Merkel cell carcinoma: Epidemiology, target, and therapy. Curr. Dermatol. Rep. 2014, 3, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Youlden, D.R.; Youl, P.H.; Peter Soyer, H.; Fritschi, L.; Baade, P.D. Multiple primary cancers associated with Merkel cell carcinoma in Queensland, Australia, 1982–2011. J. Investig. Dermatol. 2014, 134, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.A.; Dores, G.M.; Curtis, R.E.; Anderson, W.F.; Travis, L.B. Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1545–1549. [Google Scholar] [CrossRef]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef]
- Buell, J.F.; Trofe, J.; Hanaway, M.J.; Beebe, T.M.; Gross, T.G.; Alloway, R.R.; First, M.R.; Woodle, E.S. Immunosuppression and Merkel cell cancer. Transpl. Proc. 2002, 34, 1780–1781. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Bononi, I.; Puozzo, A.; Govoni, M.; Foschi, V.; Lanza, G.; Gafa, R.; Gaboriaud, P.; Touze, F.A.; Selvatici, R.; et al. Merkel cell carcinomas arising in autoimmune disease affected patients treated with biologic drugs, including Anti-TNF. Clin. Cancer Res. 2017, 23, 3929–3934. [Google Scholar] [CrossRef]
- Iyer, J.G.; Blom, A.; Doumani, R.; Lewis, C.; Tarabadkar, E.S.; Anderson, A.; Ma, C.; Bestick, A.; Parvathaneni, U.; Bhatia, S.; et al. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 2016, 5, 2294–2301. [Google Scholar] [CrossRef]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International workshop on merkel cell carcinoma research working group. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef]
- Paulson, K.G.; Iyer, J.G.; Blom, A.; Warton, E.M.; Sokil, M.; Yelistratova, L.; Schuman, L.; Nagase, K.; Bhatia, S.; Asgari, M.M.; et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J. Investig. Dermatol. 2013, 133, 642–646. [Google Scholar] [CrossRef]
- Afanasiev, O.K.; Yelistratova, L.; Miller, N.; Nagase, K.; Paulson, K.; Iyer, J.G.; Ibrani, D.; Koelle, D.M.; Nghiem, P. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin. Cancer Res. 2013, 19, 5351–5360. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with pembrolizumab in advanced merkel-cell carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbe, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Becker, J.C.; Lorenz, E.; Ugurel, S.; Eigentler, T.K.; Kiecker, F.; Pfohler, C.; Kellner, I.; Meier, F.; Kahler, K.; Mohr, P.; et al. Evaluation of real-world treatment outcomes in patients with distant metastatic Merkel cell carcinoma following second-line chemotherapy in Europe. Oncotarget 2017, 8, 79731–79741. [Google Scholar] [CrossRef]
- Cowey, C.L.; Mahnke, L.; Espirito, J.; Helwig, C.; Oksen, D.; Bharmal, M. Real-world treatment outcomes in patients with metastatic Merkel cell carcinoma treated with chemotherapy in the USA. Future Oncol. 2017, 13, 1699–1710. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Hunger, M.; Brohl, A.S.; Nghiem, P.; Bhatia, S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; et al. Early objective response to avelumab treatment is associated with improved overall survival in patients with metastatic Merkel cell carcinoma. Cancer Immunol. Immunother. 2019, 68, 609–618. [Google Scholar] [CrossRef]
- Topalian, S.L.; Bhatia, S.; Kudchadkar, R.R.; Amin, A.; Sharfman, W.H.; Lebbe, C.; Delord, J.-P.; Shinohara, M.M.; Baxi, S.S.; Chung, C.H.; et al. Nivolumab (Nivo) as neoadjuvant therapy in patients with resectable Merkel cell carcinoma (MCC) in CheckMate 358. J. Clin. Oncol. 2018, 36, 9505. [Google Scholar] [CrossRef]
- Becker, J.C.; Hassel, J.C.; Menzer, C.; Kähler, K.C.; Eigentler, T.K.; Meier, F.E.; Berking, C.; Gutzmer, R.; Mohr, P.; Kiecker, F.; et al. Adjuvant ipilimumab compared with observation in completely resected Merkel cell carcinoma (ADMEC): A randomized, multicenter DeCOG/ADO study. J. Clin. Oncol. 2018, 36, 9527. [Google Scholar] [CrossRef]
- Slingluff, C.L.; Dasilva, D.; Schwarzenberger, P.; Ricciardi, T.; Macri, M.J.; Ryan, A.; Venhaus, R.R.; Bhardwaj, N. Phase 1/2 study of in situ vaccination with tremelimumab + intravenous (IV) durvalumab + poly-ICLC in patients with select relapsed, advanced cancers with measurable, biopsy-accessible tumors. J. Clin. Oncol. 2017, 35, TPS3106. [Google Scholar] [CrossRef]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef]
- Wilcox, R.A. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2017, 92, 1085–1102. [Google Scholar] [CrossRef]
- Scarisbrick, J.J.; Quaglino, P.; Prince, H.M.; Papadavid, E.; Hodak, E.; Bagot, M.; Servitje, O.; Berti, E.; Ortiz-Romero, P.; Stadler, R.; et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br. J. Dermatol. 2018. [Google Scholar] [CrossRef]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.H.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A.; et al. Cutaneous lymphoma international consortium study of outcome in advanced stages of mycosis fungoides and sezary syndrome: Effect of specific prognostic markers on survival and development of a prognostic model. J. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef]
- Janiga, J.; Kentley, J.; Nabhan, C.; Abdulla, F. Current systemic therapeutic options for advanced mycosis fungoides and Sezary syndrome. Leuk. Lymphoma 2018, 59, 562–577. [Google Scholar] [CrossRef]
- Guenova, E.; Hoetzenecker, W.; Rozati, S.; Levesque, M.P.; Dummer, R.; Cozzio, A. Novel therapies for cutaneous T-cell lymphoma: what does the future hold? Expert. Opin. Investig. Drugs 2014, 23, 457–467. [Google Scholar] [CrossRef]
- Saulite, I.; Hoetzenecker, W.; Weidinger, S.; Cozzio, A.; Guenova, E.; Wehkamp, U. Sezary syndrome and atopic dermatitis: Comparison of immunological aspects and targets. Biomed. Res. Int. 2016, 2016, 9717530. [Google Scholar] [CrossRef]
- Photiou, L.; van der Weyden, C.; McCormack, C.; Miles Prince, H. Systemic treatment options for advanced-stage mycosis fungoides and sezary syndrome. Curr. Oncol. Rep. 2018, 20, 32. [Google Scholar] [CrossRef]
- Buggins, A.G.; Mufti, G.J.; Salisbury, J.; Codd, J.; Westwood, N.; Arno, M.; Fishlock, K.; Pagliuca, A.; Devereux, S. Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood 2002, 100, 1715–1720. [Google Scholar]
- Clark, R.A.; Watanabe, R.; Teague, J.E.; Schlapbach, C.; Tawa, M.C.; Adams, N.; Dorosario, A.A.; Chaney, K.S.; Cutler, C.S.; Leboeuf, N.R.; et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci. Transl. Med. 2012, 4, 117ra117. [Google Scholar] [CrossRef]
- Watanabe, R.; Teague, J.E.; Fisher, D.C.; Kupper, T.S.; Clark, R.A. Alemtuzumab therapy for leukemic cutaneous T-cell lymphoma: diffuse erythema as a positive predictor of complete remission. JAMA Dermatol. 2014, 150, 776–779. [Google Scholar] [CrossRef]
- Grover, N.S.; Savoldo, B. Challenges of driving CD30-directed CAR-T cells to the clinic. BMC Cancer 2019, 19, 203. [Google Scholar] [CrossRef]
- Ansell, S.M. Brentuximab vedotin. Blood 2014, 124, 3197–3200. [Google Scholar] [CrossRef]
- Ansell, S.M. Brentuximab vedotin: delivering an antimitotic drug to activated lymphoma cells. Expert. Opin. Investig. Drugs 2011, 20, 99–105. [Google Scholar] [CrossRef]
- Prince, H.M.; Gautam, A.; Kim, Y.H. Brentuximab vedotin: targeting CD30 as standard in CTCL. Oncotarget 2018, 9, 11887–11888. [Google Scholar] [CrossRef]
- Prince, H.M.; Kim, Y.H.; Horwitz, S.M.; Dummer, R.; Scarisbrick, J.; Quaglino, P.; Zinzani, P.L.; Wolter, P.; Sanches, J.A.; Ortiz-Romero, P.L.; et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet 2017, 390, 555–566. [Google Scholar] [CrossRef]
- Lewis, D.J.; Talpur, R.; Huen, A.O.; Tetzlaff, M.T.; Duvic, M. Brentuximab vedotin for patients with refractory lymphomatoid papulosis: An analysis of phase 2 results. JAMA Dermatol. 2017, 153, 1302–1306. [Google Scholar] [CrossRef]
- Duvic, M.; Tetzlaff, M.T.; Gangar, P.; Clos, A.L.; Sui, D.; Talpur, R. Results of a phase II trial of brentuximab vedotin for CD30+ cutaneous T-cell lymphoma and lymphomatoid papulosis. J. Clin. Oncol. 2015, 33, 3759–3765. [Google Scholar] [CrossRef]
- Poon, L.M.; Kwong, Y.L. Complete remission of refractory disseminated NK/T cell lymphoma with brentuximab vedotin and bendamustine. Ann. Hematol. 2016, 95, 847–849. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Advani, R.H.; Bartlett, N.L.; Jacobsen, E.D.; Sharman, J.P.; O’Connor, O.A.; Siddiqi, T.; Kennedy, D.A.; Oki, Y. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood 2014, 123, 3095–3100. [Google Scholar] [CrossRef]
- Ferenczi, K.; Fuhlbrigge, R.C.; Pinkus, J.; Pinkus, G.S.; Kupper, T.S. Increased CCR4 expression in cutaneous T cell lymphoma. J. Investig. Dermatol. 2002, 119, 1405–1410. [Google Scholar] [CrossRef]
- Ishida, T.; Utsunomiya, A.; Iida, S.; Inagaki, H.; Takatsuka, Y.; Kusumoto, S.; Takeuchi, G.; Shimizu, S.; Ito, M.; Komatsu, H.; et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin. Cancer Res. 2003, 9, 3625–3634. [Google Scholar]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.; Zinzani, P.L.; et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2018, 19, 1192–1204. [Google Scholar] [CrossRef]
- Suzuki, Y.; Saito, M.; Ishii, T.; Urakawa, I.; Matsumoto, A.; Masaki, A.; Ito, A.; Kusumoto, S.; Suzuki, S.; Hiura, M.; et al. Mogamulizumab treatment elicits autoantibodies attacking the skin in patients with adult T cell leukemia-lymphoma. Clin Cancer Res. 2019. [Google Scholar] [CrossRef]
- Honda, T.; Hishizawa, M.; Kataoka, T.R.; Ohmori, K.; Takaori-Kondo, A.; Miyachi, Y.; Kabashima, K. Stevens-Johnson syndrome associated with mogamulizumab-induced deficiency of regulatory T cells in an adult T-cell leukaemia Patient. Acta Dermatol. Venereol. 2015, 95, 606–607. [Google Scholar] [CrossRef]
- Ishida, T.; Ito, A.; Sato, F.; Kusumoto, S.; Iida, S.; Inagaki, H.; Morita, A.; Akinaga, S.; Ueda, R. Stevens-Johnson Syndrome associated with mogamulizumab treatment of adult T-cell leukemia/lymphoma. Cancer Sci. 2013, 104, 647–650. [Google Scholar] [CrossRef]
- Bonnet, P.; Battistella, M.; Roelens, M.; Ram-Wolff, C.; Herms, F.; Frumholtz, L.; Bouaziz, J.D.; Brice, P.; Moins-Teisserenc, H.; Bagot, M.; et al. Association of autoimmunity and long-term complete remission in patients with Sezary syndrome treated with mogamulizumab. Br. J. Dermatol. 2019, 180, 419–420. [Google Scholar] [CrossRef]
- Hosoi, H.; Mushino, T.; Nishikawa, A.; Hashimoto, H.; Murata, S.; Hatanaka, K.; Tamura, S.; Hanaoka, N.; Shimizu, N.; Sonoki, T. Severe graft-versus-host disease after allogeneic hematopoietic stem cell transplantation with residual mogamulizumab concentration. Int. J. Hematol. 2018, 107, 717–719. [Google Scholar] [CrossRef]
- Fuji, S.; Inoue, Y.; Utsunomiya, A.; Moriuchi, Y.; Uchimaru, K.; Choi, I.; Otsuka, E.; Henzan, H.; Kato, K.; Tomoyose, T.; et al. Pretransplantation anti-CCR4 antibody mogamulizumab against adult T-cell leukemia/lymphoma is associated with significantly increased risks of severe and corticosteroid-refractory graft-versus-host disease, nonrelapse mortality, and overall mortality. J. Clin. Oncol. 2016, 34, 3426–3433. [Google Scholar] [CrossRef]
- Ishida, T.; Jo, T.; Takemoto, S.; Suzushima, H.; Suehiro, Y.; Choi, I.; Yoshimitsu, M.; Saburi, Y.; Nosaka, K.; Utsunomiya, A.; et al. Follow-up of a randomised phase II study of chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: impact on allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2019, 184, 479–483. [Google Scholar] [CrossRef]
- Dai, J.; Almazan, T.H.; Hong, E.K.; Khodadoust, M.S.; Arai, S.; Weng, W.K.; Kim, Y.H. Potential association of anti-CCR4 antibody mogamulizumab and graft-vs-host disease in patients with mycosis fungoides and sezary syndrome. JAMA Dermatol. 2018, 154, 728–730. [Google Scholar] [CrossRef]
- Fuji, S.; Shindo, T. Friend or foe? Mogamulizumab in allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia/lymphoma. Stem Cell Investig. 2016, 3, 70. [Google Scholar] [CrossRef]
- Schmitt, C.; Marie-Cardine, A.; Bensussan, A. Therapeutic Antibodies to KIR3DL2 and Other Target Antigens on Cutaneous T-Cell Lymphomas. Front. Immunol. 2017, 8, 1010. [Google Scholar] [CrossRef]
- Battistella, M.; Leboeuf, C.; Ram-Wolff, C.; Hurabielle, C.; Bonnafous, C.; Sicard, H.; Bensussan, A.; Bagot, M.; Janin, A. KIR3DL2 expression in cutaneous T-cell lymphomas: expanding the spectrum for KIR3DL2 targeting. Blood 2017, 130, 2900–2902. [Google Scholar] [CrossRef]
- Hurabielle, C.; Thonnart, N.; Ram-Wolff, C.; Sicard, H.; Bensussan, A.; Bagot, M.; Marie-Cardine, A. Usefulness of KIR3DL2 to diagnose, follow-up, and manage the treatment of patients with Sezary syndrome. Clin. Cancer Res. 2017, 23, 3619–3627. [Google Scholar] [CrossRef]
- Sicard, H.; Bonnafous, C.; Morel, A.; Bagot, M.; Bensussan, A.; Marie-Cardine, A. A novel targeted immunotherapy for CTCL is on its way: Anti-KIR3DL2 mAb IPH4102 is potent and safe in non-clinical studies. Oncoimmunology 2015, 4, e1022306. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marie-Cardine, A.; Viaud, N.; Thonnart, N.; Joly, R.; Chanteux, S.; Gauthier, L.; Bonnafous, C.; Rossi, B.; Blery, M.; Paturel, C.; et al. IPH4102, a humanized KIR3DL2 antibody with potent activity against cutaneous T-cell lymphoma. Cancer Res. 2014, 74, 6060–6070. [Google Scholar] [CrossRef] [PubMed]
- Bagot, M.; Porcu, P.; Marie-Cardine, A.; Battistella, M.; William, B.M.; Vermeer, M.; Whittaker, S.; Rotolo, F.; Ram-Wolff, C.; Khodadoust, M.S.; et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol. 2019. [Google Scholar] [CrossRef]
- Samimi, S.; Benoit, B.; Evans, K.; Wherry, E.J.; Showe, L.; Wysocka, M.; Rook, A.H. Increased programmed death-1 expression on CD4+ T cells in cutaneous T-cell lymphoma: implications for immune suppression. Arch. Dermatol. 2010, 146, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Cetinozman, F.; Jansen, P.M.; Vermeer, M.H.; Willemze, R. Differential expression of programmed death-1 (PD-1) in Sezary syndrome and mycosis fungoides. Arch. Dermatol. 2012, 148, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Wada, D.A.; Wilcox, R.A.; Harrington, S.M.; Kwon, E.D.; Ansell, S.M.; Comfere, N.I. Programmed death 1 is expressed in cutaneous infiltrates of mycosis fungoides and Sezary syndrome. Am. J. Hematol. 2011, 86, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Guenova, E.; Watanabe, R.; Teague, J.E.; Desimone, J.A.; Jiang, Y.; Dowlatshahi, M.; Schlapbach, C.; Schaekel, K.; Rook, A.H.; Tawa, M.; et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin. Cancer Res. 2013, 19, 3755–3763. [Google Scholar] [CrossRef]
- Rubio Gonzalez, B.; Zain, J.; Rosen, S.T.; Querfeld, C. Tumor microenvironment in mycosis fungoides and Sezary syndrome. Curr. Opin. Oncol. 2016, 28, 88–96. [Google Scholar] [CrossRef]
- Torrealba, M.P.; Manfrere, K.C.; Miyashiro, D.R.; Lima, J.F.; de, M.O.L.; Pereira, N.Z.; Cury-Martins, J.; Pereira, J.; Duarte, A.J.S.; Sato, M.N.; et al. Chronic activation profile of circulating CD8+ T cells in Sezary syndrome. Oncotarget 2018, 9, 3497–3506. [Google Scholar] [CrossRef]
- Querfeld, C.; Leung, S.; Myskowski, P.L.; Curran, S.A.; Goldman, D.A.; Heller, G.; Wu, X.; Kil, S.H.; Sharma, S.; Finn, K.J.; et al. Primary T cells from cutaneous T-cell lymphoma skin explants display an exhausted immune checkpoint profile. Cancer Immunol. Res. 2018, 6, 900–909. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef]
- Dulos, J.; Carven, G.J.; van Boxtel, S.J.; Evers, S.; Driessen-Engels, L.J.; Hobo, W.; Gorecka, M.A.; de Haan, A.F.; Mulders, P.; Punt, C.J.; et al. PD-1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J. Immunother. 2012, 35, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.; Rook, A.H.; Porcu, P.; Foss, F.M.; Moskowitz, A.J.; Shustov, A.R.; Shanbhag, S.; Sokol, L.; Shine, R.; Fling, S.P.; et al. Pembrolizumab for treatment of relapsed/refractory mycosis fungoides and Sezary syndrome: Clinical efficacy in a Citn multicenter phase 2 study. Blood 2016, 128, 181. [Google Scholar]
- Khodadoust, M.S.; Rook, A.; Porcu, P.; Foss, F.M.; Moskowitz, A.J.; Shustov, A.R.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Li, S.; et al. Durable responses with pembrolizumab in relapsed/refractory mycosis fungoides and Sézary syndrome: Final results from a phase 2 multicenter study. Blood 2018, 132, 2896. [Google Scholar] [CrossRef]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Ollinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017, 552, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.A.; Feldman, A.L.; Wada, D.A.; Yang, Z.Z.; Comfere, N.I.; Dong, H.; Kwon, E.D.; Novak, A.J.; Markovic, S.N.; Pittelkow, M.R.; et al. B7-H1 (PD-L1, CD274) suppresses host immunity in T-cell lymphoproliferative disorders. Blood 2009, 114, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.K.; Wilson, A.J.; Gibson, H.M.; Hafner, M.S.; Hedgcock, C.J.; Berger, C.L.; Edelson, R.L.; Lim, H.W. Increased expression of CTLA-4 in malignant T-cells from patients with mycosis fungoides -- cutaneous T cell lymphoma. J. Investig. Dermatol. 2006, 126, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Anzengruber, F.; Ignatova, D.; Schlaepfer, T.; Chang, Y.T.; French, L.E.; Pascolo, S.; Contassot, E.; Bobrowicz, M.; Hoetzenecker, W.; Guenova, E. Divergent LAG-3 versus BTLA, TIGIT, and FCRL3 expression in Sezary syndrome. Leuk. Lymphoma 2019, 1–9. [Google Scholar] [CrossRef]
- Ansell, S.; Gutierrez, M.E.; Shipp, M.A.; Gladstone, D.; Moskowitz, A.; Borello, I.; Popa-Mckiver, M.; Farsaci, B.; Zhu, L.; Lesokhin, A.M.; et al. A phase 1 study of nivolumab in combination with ipilimumab for relapsed or refractory hematologic malignancies (CheckMate 039). Blood 2016, 128, 183. [Google Scholar]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, Y.; Gao, P.; Yao, Z. Targeting CD47: the achievements and concerns of current studies on cancer immunotherapy. J. Thorac. Dis. 2017, 9, E168–E174. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.D.S.; Banerjee, S.; Kruglov, O.; Viller, N.N.; Horwitz, S.M.; Lesokhin, A.; Zain, J.; Querfeld, C.; Chen, R.; Okada, C.; et al. Targeting CD47 in Sezary syndrome with SIRPalphaFc. Blood Adv. 2019, 3, 1145–1153. [Google Scholar] [CrossRef]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPalpha axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Thompson, J.; Taylor, M.; Pillai, R.; Johnson, L.D.; Catalano, T.; Petrova, P.S.; Uger, R.A.; Irwin, M.; Sievers, E.L.; et al. A single direct intratumoral injection of TTI-621 (SIRPαFc) induces antitumor activity in patients with relapsed/refractory mycosis fungoides and Sézary syndrome: Preliminary findings employing an immune checkpoint inhibitor blocking the CD47 “Do Not Eat” signal. Blood 2017, 130, 4076. [Google Scholar]
- Folkes, A.S.; Feng, M.; Zain, J.M.; Abdulla, F.; Rosen, S.T.; Querfeld, C. Targeting CD47 as a cancer therapeutic strategy: the cutaneous T-cell lymphoma experience. Curr. Opin. Oncol. 2018, 30, 332–337. [Google Scholar] [CrossRef]
- Varricchi, G.; Marone, G.; Mercurio, V.; Galdiero, M.R.; Bonaduce, D.; Tocchetti, C.G. Immune checkpoint inhibitors and cardiac toxicity: An emerging issue. Curr. Med. Chem. 2018, 25, 1327–1339. [Google Scholar] [CrossRef]
- Wilcox, R.A. Cutaneous B-cell lymphomas: 2019 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2018, 93, 1427–1430. [Google Scholar] [CrossRef]
- Senff, N.J.; Noordijk, E.M.; Kim, Y.H.; Bagot, M.; Berti, E.; Cerroni, L.; Dummer, R.; Duvic, M.; Hoppe, R.T.; Pimpinelli, N.; et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood 2008, 112, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Trautinger, F.; Eder, J.; Assaf, C.; Bagot, M.; Cozzio, A.; Dummer, R.; Gniadecki, R.; Klemke, C.D.; Ortiz-Romero, P.L.; Papadavid, E.; et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sezary syndrome—Update 2017. Eur. J. Cancer 2017, 77, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Hodak, E.; Zinzani, P.L.; Specht, L.; Ladetto, M.; Committee, E.G. Primary cutaneous lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv30–iv40. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Guarino, M.; Ortiz-Romero, P.L.; Fernandez-Misa, R.; Montalban, C. Rituximab in the treatment of primary cutaneous B-cell lymphoma: A review. Actas Dermosifiliogr. 2014, 105, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Penate, Y.; Hernandez-Machin, B.; Perez-Mendez, L.I.; Santiago, F.; Rosales, B.; Servitje, O.; Estrach, T.; Fernandez-Guarino, M.; Calzado, L.; Acebo, E.; et al. Intralesional rituximab in the treatment of indolent primary cutaneous B-cell lymphomas: an epidemiological observational multicentre study. The Spanish Working Group on Cutaneous Lymphoma. Br. J. Dermatol. 2012, 167, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Vakeva, L.; Ranki, A.; Malkonen, T. Intralesional rituximab treatment for primary cutaneous B-cell lymphoma: Nine finnish cases. Acta Dermatol. Venereol. 2016, 96, 396–397. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seto, T.; Sam, D.; Pan, M. Mechanisms of primary and secondary resistance to immune checkpoint inhibitors in cancer. Med. Sci. 2019, 7, 14. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transcriptomic features of response to anti-PD-1 Therapy in metastatic melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [PubMed]
- Wargo, J.A.; Gopalakrishnan, V.; Spencer, C.; Karpinets, T.; Reuben, A.; Andrews, M.C.; Tetzlaff, M.T.; Lazar, A.; Hwu, P.; Hwu, W.-J.; et al. Association of the diversity and composition of the gut microbiome with responses and survival (PFS) in metastatic melanoma (MM) patients (pts) on anti-PD-1 therapy. J. Clin. Oncol. 2017, 35, 3008. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, R.; Liu, F.; Lee, S.A.; Zhang, L. Modulation of gut microbiota: A novel paradigm of enhancing the efficacy of programmed death-1 and programmed death ligand-1 blockade therapy. Front. Immunol. 2018, 9, 374. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Zimmer, L.; Apuri, S.; Eroglu, Z.; Kottschade, L.A.; Forschner, A.; Gutzmer, R.; Schlaak, M.; Heinzerling, L.; Krackhardt, A.M.; Loquai, C.; et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur. J. Cancer 2017, 75, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Kitano, S.; Nakayama, T.; Yamashita, M. Biomarkers for immune checkpoint inhibitors in melanoma. Front. Oncol. 2018, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.A.; Levesque, M.P.; Cheng, P.F. Melanoma immunotherapy: Next-generation biomarkers. Front. Oncol. 2018, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Young, G.D.; McMiller, T.L.; Chen, S.; Salas, J.T.; Pritchard, T.S.; Xu, H.; Meeker, A.K.; Fan, J.; Cheadle, C.; et al. Differential expression of immune-regulatory genes associated with PD-L1 display in melanoma: Implications for PD-1 pathway blockade. Clin. Cancer Res. 2015, 21, 3969–3976. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Kollmann, D.; Ignatova, D.; Jedamzik, J.; Chang, Y.T.; Jomrich, G.; Baierl, A.; Kazakov, D.; Michal, M.; French, L.E.; Hoetzenecker, W.; et al. PD-L1 expression is an independent predictor of favorable outcome in patients with localized esophageal adenocarcinoma. Oncoimmunology 2018, 7, e1435226. [Google Scholar] [CrossRef] [PubMed]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017, 171, 934–949 e916. [Google Scholar] [CrossRef] [PubMed]
- Grigg, C.; Rizvi, N.A. PD-L1 biomarker testing for non-small cell lung cancer: truth or fiction? J. Immunother. Cancer 2016, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Khunger, M.; Jain, P.; Rakshit, S.; Pasupuleti, V.; Hernandez, A.V.; Stevenson, J.; Pennell, N.A.; Velcheti, V. Safety and efficacy of PD-1/PD-L1 inhibitors in treatment-naive and chemotherapy-refractory patients with non-small-cell lung cancer: A systematic review and meta-analysis. Clin. Lung Cancer 2018, 19, e335–e348. [Google Scholar] [CrossRef]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a biomarker in malignant melanoma treated with checkpoint blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Kaskel, P.; Berking, C.; Sander, S.; Volkenandt, M.; Peter, R.U.; Krahn, G. S-100 protein in peripheral blood: a marker for melanoma metastases: a prospective 2-center study of 570 patients with melanoma. J. Am. Acad. Dermatol. 1999, 41, 962–969. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Martin-Liberal, J.; Lee, A.; Chauhan, D.; Gore, M.; Larkin, J. Prognostic score for patients with advanced melanoma treated with ipilimumab. Eur. J. Cancer 2015, 51, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.B.; McGlynn, K.A.; Devesa, S.S.; Freedman, N.D.; Anderson, W.F. Sex disparities in cancer mortality and survival. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Kugel, C.H., 3rd; Douglass, S.M.; Webster, M.R.; Kaur, A.; Liu, Q.; Yin, X.; Weiss, S.A.; Darvishian, F.; Al-Rohil, R.N.; Ndoye, A.; et al. Age correlates with response to Anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin. Cancer Res. 2018, 24, 5347–5356. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body fatness and cancer—Viewpoint of the IARC working group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2019, 176, 404. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Tanemura, A.; Tada, Y.; Katayama, I.; Kumanogoh, A.; Nishikawa, H. Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int. Immunol. 2018, 30, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, Y.; Otsuka, A.; Nakashima, C.; Seidel, J.A.; Kitoh, A.; Dainichi, T.; Nakajima, S.; Sawada, Y.; Matsushita, S.; Aoki, M.; et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. Oncoimmunology 2016, 5, e1248327. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Fassler, M.; Diem, S.; Mangana, J.; Hasan Ali, O.; Berner, F.; Bomze, D.; Ring, S.; Niederer, R.; Del Carmen Gil Cruz, C.; Perez Shibayama, C.I.; et al. Antibodies as biomarker candidates for response and survival to checkpoint inhibitors in melanoma patients. J. Immunother. Cancer 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Sullivan, R.J.; Ott, P.A.; Carlino, M.S.; Khushalani, N.I.; Ye, F.; Guminski, A.; Puzanov, I.; Lawrence, D.P.; Buchbinder, E.I.; et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol. 2016, 2, 234–240. [Google Scholar] [CrossRef]
- Tocut, M.; Brenner, R.; Zandman-Goddard, G. Autoimmune phenomena and disease in cancer patients treated with immune checkpoint inhibitors. Autoimmun. Rev. 2018, 17, 610–616. [Google Scholar] [CrossRef]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol 2019. [Google Scholar] [CrossRef]
- Wang, D.Y.; Johnson, D.B.; Davis, E.J. Toxicities Associated With PD-1/PD-L1 Blockade. Cancer J. 2018, 24, 36–40. [Google Scholar] [CrossRef]
- Haanen, J.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K.; Committee, E.G. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv264–iv266. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, C.O., 3rd; Brogdon, C.; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high Tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Boada, A.; Carrera, C.; Segura, S.; Collgros, H.; Pasquali, P.; Bodet, D.; Puig, S.; Malvehy, J. Cutaneous toxicities of new treatments for melanoma. Clin. Transl. Oncol. 2018, 20, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.B.; Macdonald, B.; Golitz, L.E.; LoRusso, P.; Sekulic, A. Cutaneous adverse effects of targeted therapies: Part II: Inhibitors of intracellular molecular signaling pathways. J. Am. Acad. Dermatol. 2015, 72, 221–236, quiz 237-228. [Google Scholar] [CrossRef] [PubMed]
- Minkis, K.; Garden, B.C.; Wu, S.; Pulitzer, M.P.; Lacouture, M.E. The risk of rash associated with ipilimumab in patients with cancer: a systematic review of the literature and meta-analysis. J. Am. Acad Dermatol. 2013, 69, e121–e128. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Hwang, S.J.; Carlos, G.; Wakade, D.; Byth, K.; Kong, B.Y.; Chou, S.; Carlino, M.S.; Kefford, R.; Fernandez-Penas, P. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: A single-institution cohort. J. Am. Acad. Dermatol. 2016, 74, 455–461 e451. [Google Scholar] [CrossRef]
- Freeman-Keller, M.; Kim, Y.; Cronin, H.; Richards, A.; Gibney, G.; Weber, J.S. Nivolumab in resected and unresectable metastatic melanoma: Characteristics of immune-related adverse events and association with outcomes. Clin. Cancer Res. 2016, 22, 886–894. [Google Scholar] [CrossRef]
- Nakamura, Y.; Teramoto, Y.; Asami, Y.; Matsuya, T.; Adachi, J.I.; Nishikawa, R.; Yamamoto, A. Nivolumab therapy for treatment-related vitiligo in a patient with relapsed metastatic melanoma. JAMA Dermatol. 2017, 153, 942–944. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Boussemart, L.; Mateus, C.; Routier, E.; Boutros, C.; Cazenave, H.; Viollet, R.; Thomas, M.; Roy, S.; Benannoune, N.; et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016, 152, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kumar, A.B.; Finnes, H.; Markovic, S.N.; Park, S.; Dronca, R.S.; Dong, H. Combining immune checkpoint inhibitors with conventional cancer therapy. Front. Immunol. 2018, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, Y.; Cheng, Y.; Wei, Y.; Wei, X. Immune checkpoint blockade and its combination therapy with small-molecule inhibitors for cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 199–224. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Brunetti, O.; Azzariti, A.; Galetta, D.; Nardulli, P.; Leonetti, F.; Silvestris, N. Strategies to improve cancer immune checkpoint inhibitors efficacy, other than abscopal effect: A systematic review. Cancers 2019, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular pathways: next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef] [PubMed]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcgammaRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell 2015, 28, 543. [Google Scholar] [CrossRef] [PubMed]
- Goletz, C.; Lischke, T.; Harnack, U.; Schiele, P.; Danielczyk, A.; Ruhmann, J.; Goletz, S. Glyco-engineered anti-human programmed death-ligand 1 antibody mediates stronger CD8 T cell activation than its normal glycosylated and non-glycosylated counterparts. Front. Immunol. 2018, 9, 1614. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Li, K.; Zhang, T. FcgammaR-binding is an important functional attribute for immune checkpoint antibodies in cancer immunotherapy. Front. Immunol. 2019, 10, 292. [Google Scholar] [CrossRef]
- Trenevska, I.; Li, D.; Banham, A.H. Therapeutic antibodies against intracellular tumor antigens. Front. Immunol. 2017, 8, 1001. [Google Scholar] [CrossRef]
- Dubrovsky, L.; Dao, T.; Gejman, R.S.; Brea, E.J.; Chang, A.Y.; Oh, C.Y.; Casey, E.; Pankov, D.; Scheinberg, D.A. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology 2016, 5, e1049803. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, I.; Hendriks, M.; van Ginkel, R.J.; Samplonius, D.F.; Bremer, E.; Helfrich, W. Bispecific antibody approach for improved melanoma-selective PD-L1 immune checkpoint blockade. J. Investig. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Name of the Agent | Molecular Target | Isotype | Source | Mechanism of Action | Trade Name | Registration Status in Dermatooncology | Most Relevant Clinical Trials |
|---|---|---|---|---|---|---|---|
| Ipilimumab | CTLA-4 | IgG1 | human | Triggers ADCC Blocks inhibitory APC—T cell interaction | Yervoy | 2011—melanoma | NCT00636168 in MM, NCT01844505 in MM |
| Tremelimumab (CP-675206) | CTLA-4 | IgG2 | human | Reduced ADCC activity Blocks inhibitory APC—T cell interaction | NA | In clinical trials in melanoma | NCT00257205 in MM, NCT01704287 in MM |
| Cemiplimab REGN2810 (SAR439684) | PD-1 | IgG4 | human | Reduced ADCC activity Blocks inhibitory APC—T cell as well as tumor cell–T cell interaction | Libtayo | 2018—CSCC | NCT03002376 in MM, NCT02383212 in BCC and CSCC, NCT02760498 in CSCC |
| Nivolumab | PD-1 | IgG4 | human | Reduced ADCC activity Blocks inhibitory APC—T cell as well as tumor cell–T cell interaction | Opdivo | 2014—melanoma | NCT01844505 in MM, NCT01927419 in MM |
| Pembrolizumab (initially known as lambrolizumab) | PD-1 | IgG4 | humanized | Reduced ADCC activity Blocks inhibitory APC—T cell as well as tumor cell–T cell interaction | Keytruda | 2014—melanoma | NCT02362594 in MM, NCT02243579 in CTLC |
| Atezolizumab | PD-L1 | IgG1 | humanized | Reduced ADCC activity Blocks inhibitory APC—T cell as well as tumor cell–T cell interaction | Tecentriq | In clinical trials in CTCL and melanoma | NCT03357224 in CTCL, NCT04020809 in MM, NCT01656642 in MM |
| Avelumab | PD-L1 | IgG1 | human | Triggers ADCC Blocks inhibitory APC—T cell as well as tumor cell–T cell interaction | Bavencio | 2017—MCC | NCT01772004 in MM, NCT02155647 in MCC |
| Durvalumab (MEDI4736) | PD-L1 | IgG1 | human | Reduced ADCC activity Blocks inhibitory APC—T cell as well as tumor cell–T cell interaction | Imfinzi | In clinical trials in CTCL, melanoma and MCC | NCT02027961 in MM, NCT02643303 in CTCL |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobrowicz, M.; Zagozdzon, R.; Domagala, J.; Vasconcelos-Berg, R.; Guenova, E.; Winiarska, M. Monoclonal Antibodies in Dermatooncology—State of the Art and Future Perspectives. Cancers 2019, 11, 1420. https://doi.org/10.3390/cancers11101420
Bobrowicz M, Zagozdzon R, Domagala J, Vasconcelos-Berg R, Guenova E, Winiarska M. Monoclonal Antibodies in Dermatooncology—State of the Art and Future Perspectives. Cancers. 2019; 11(10):1420. https://doi.org/10.3390/cancers11101420
Chicago/Turabian StyleBobrowicz, Malgorzata, Radoslaw Zagozdzon, Joanna Domagala, Roberta Vasconcelos-Berg, Emmanuella Guenova, and Magdalena Winiarska. 2019. "Monoclonal Antibodies in Dermatooncology—State of the Art and Future Perspectives" Cancers 11, no. 10: 1420. https://doi.org/10.3390/cancers11101420
APA StyleBobrowicz, M., Zagozdzon, R., Domagala, J., Vasconcelos-Berg, R., Guenova, E., & Winiarska, M. (2019). Monoclonal Antibodies in Dermatooncology—State of the Art and Future Perspectives. Cancers, 11(10), 1420. https://doi.org/10.3390/cancers11101420