Abstract
Pancreatic cancer is poorly responsive to chemotherapy due to intrinsic or acquired resistance. Our previous study showed that epigenetic modifying enzymes including protein arginine methyltransferase 3 (PRMT3) are dysregulated in gemcitabine (GEM)-resistant pancreatic cancer cells. Here, we attempt to elucidate the role of PRMT3 in chemoresistance. Overexpression of PRMT3 led to increased resistance to GEM in pancreatic cancer cells, whereas reduction of PRMT3 restored GEM sensitivity in resistant cells. We identified a novel PRMT3 target, ATP-binding cassette subfamily G member 2 (ABCG2), which is known to play a critical role in drug resistance. PRMT3 overexpression upregulated ABCG2 expression by increasing its mRNA stability. Mass spectrometric analysis identified hnRNPA1 as a PRMT3 interacting protein, and methylation of hnRNPA1 at R31 by PRMT3 in vivo and in vitro. The expression of methylation-deficient hnRNPA1-R31K mutant reduced the RNA binding activity of hnRNPA1 and the expression of ABCG2 mRNA. Taken together, this provides the first evidence that PRMT3 methylates the RNA recognition motif (RRM) of hnRNPA1 and promotes the binding between hnRNPA1 and ABCG2 to enhance drug resistance. Inhibition of PRMT3 could be a novel strategy for the treatment of GEM-resistant pancreatic cancer.
1. Introduction
Pancreatic cancer is a deadly disease with a 5-year survival rate of less than 8%. Although surgical resection is a potentially curative therapy for pancreatic cancer, only about 20% of patients whose primary tumors are localized are candidates for surgery [1]. For pancreatic cancer patients with locally advanced or metastatic tumors, chemotherapy is the standard treatment. Gemcitabine (GEM) has been used as a first-line treatment for pancreatic cancer patients since the late 1990s [2]. Pancreatic cancer patients initially respond to GEM but many patients later develop resistance after prolonged treatment. Currently, several mechanisms have been proposed to mediate the acquired GEM resistance in pancreatic cancer patients, including the reduced expression of nucleoside transporter [3,4], the alterations of nucleotide metabolism enzymes [5,6], and an increased drug efflux by ABC-transporters [7,8,9].
Emerging evidence suggests that epigenetic alterations, such as DNA methylation and histone modification, play an important role in regulating drug resistance [10,11]. For instance, DNA methylation of target of methylation-induced silencing-1 (TMS1), a pro-apoptotic gene, results in reduced TMS1 expression and increased GEM resistance. Inhibition of DNA methyltransferase by 5-azacytidine restores TMS1 expression and reactivates its apoptosis-inducing activity, thereby enhancing sensitivity of cancer cells to GEM [12]. GEM is a pro-drug that requires conversion into the active form after cellular uptake [13]. Promoter methylation of deoxycytidine kinase (dCK), which is critical for GEM metabolism, has been reported to repress mRNA expression and attenuate GEM sensitivity [14]. Moreover, dCK expression has been shown to correlate with GEM efficacy [5,15]. The involvement of histone modification enzymes in GEM resistance was recently reported in which depletion of enhancer zeste homolog 2 (EZH2), a histone methyltransferase responsible for the methylation of lysine 27 of histone H3 (H3K27), induces p27Kip1 expression and sensitizes pancreatic cancer cells to GEM [16].
Post-translational modification, such as protein methylation, is another mechanism that contributes to GEM resistance. For instance, protein arginine methyltransferase 1 (PRMT1), one of the nine members of the PRMT family which transfers methyl moieties from S-adenosylmethionine (SAM) to specific arginine residues on their substrates, methylates Gli1 to promote its oncogenic activity, resulting in reduced sensitivity of cells to GEM. Gli1 transcriptional factor, which is a key mediator in the Hedgehog signaling pathway, plays a critical role in pancreatic tumorigenesis. Depletion of PRMT1 effectively reversed GEM resistance in pancreatic cancer cells [17]. In addition, we previously demonstrated that euchromatic histone lysine methyltransferase 2 (EHMT2), a H3K9 methyltransferase, induces autocrine IL-8/CXCR1/2 stimulation in pancreatic cancer cells and paracrine activation of pancreatic stellate cells (PSCs) to increase GEM resistance. Inhibition of EHMT2 resensitizes pancreatic cancer cells to GEM in vitro, and the combination of EHMT2 inhibitor and GEM can overcome drug resistance in animal models [18]. In addition to EHMT2, we found that two other members of the PRMT family, PRMT3 and PRMT6, are highly upregulated in GEM-resistant pancreatic cancer cells [18]. The contribution of PRMT3 in chemoresistance is not well understood, and the effects of PRMT6 on the cytotoxicity of doxorubicin have only been reported in one study [19]. In the current study, we attempt to further our understanding of the molecular mechanisms by which PRMT3 and PRMT6 modulate the GEM resistance in pancreatic cancer.
2. Results
2.1. PRMT3 Is Upregulated in GEM-Resistant Pancreatic Cancer Cells and Its Overexpression Increases the Resistance to Multiple Chemotherapeutic Drugs
Results from our previous PCR array to compare the expression of epigenetic-modifying enzymes in PANC-1 and GEM-resistant PANC-1-R cells identified twenty-one enzymes that were upregulated in PANC-1-R cells [18]. Notably, gene expression of PRMT3 and PRMT6, encoding protein arginine methyltransferases, was significantly upregulated in resistant cells compared with parental cells [20]. Given that little is known about the regulation of drug resistance by these two PRMTs, we sought to investigate the effects of upregulation of PRMT genes in pancreatic cancer cells. We first verified the expression of PRMTs by quantitative RT-PCR. Among the six PRMT genes examined, we observed substantially higher levels of PRMT3 and PRMT6 mRNA (Figure 1A) and protein (Figure 1B) in PANC-1-R cells compared with parental PANC-1 cells. To further establish the importance of PRMT3 and PRMT6 in GEM resistance, we generated PRMT3- and PRMT6-overexpressing stable clones from PANC-1 cells. Overexpression of PRMT3 and PRMT6 enhances cell growth (Supplementary Figure S1). Moreover, the results from cell viability assay showed that overexpression of PRMT3 and PRMT6 was more resistant to GEM compared to GFP-overexpressing PANC-1 cells (Figure 1C, left). Overexpression of PRMT3 or PRMT6 promoted anchorage-dependent cell growth. However, only PRMT3-overexpressing cells were resistant to GEM treatment (Figure 1C, right). Considering the inconsistence of PRMT6 overexpression in cell viability and anchorage-dependent cell growth, we only focus on PRMT3 and investigate its role in GEM resistance. As shown in Figure 1D, the reduction of PRMT3 resensitized PANC-1-R cells to GEM in both anchorage-dependent cell growth and cell viability assays. In addition to GEM, PRMT3-overexpressing cancer cells also showed increased resistance to irinotecan, SN-38, and topotecan, indicating that upregulation of PRMT3 contributes to multidrug resistance in pancreatic cancer cells (Figure 1E).
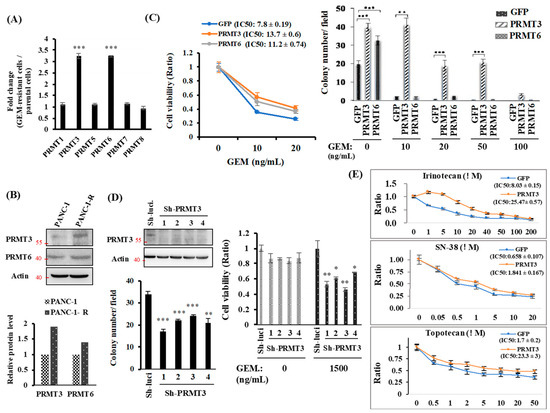
Figure 1.
The expression of PRMT3 is upregulated in GEM-resistant pancreatic cancer cells and required for GEM-resistant activity. (A) Quantitative RT-PCR analysis of PRMTs expression. Fold change of PRMTs mRNA expression in PANC-1-R normalized to parental PANC-1 cells. *** p < 0.001. (B) The protein levels of PRMT3 and PRMT6 were detected by Western blot analysis. The intensity of the bands was quantified by Image J and normalized to that of actin. (C) Left: GFP-, PRMT3- and PRMT6-ovexpressing PANC-1 cells were treated with indicated concentrations of GEM for 5 days and then subjected to MTT assay to measure cell viability. Error bars, SEM. n = 3. IC50 values of GEM. in GFP-, PRMT3- and PRMT6-ovexpressing PANC-1 cells were presented as the mean ± standard deviation. Right: GFP-, PRMT3- and PRMT6-ovexpressing PANC-1 cells were subjected to anchorage-independent growth experiment. Quantitative analysis of colony numbers is shown. Error bars, SEM. n = 3. ** p < 0.01, *** p < 0.001. (D) Left: PRMT3-reduction PANC-1-R stable clones were subjected to anchorage-independent growth assay in culture medium containing 1500 ng/mL GEM for 2–3 weeks. The number of colonies was counted under microscope. Right: PRMT3-reduction PANC-1-R cells were incubated in the presence or absence of GEM and subjected to MTT assay. Error bars, SEM. n = 3. * p < 0.05, ** p < 0.01, *** p < 0.001. (E) GFP- and PRMT3-ovexpressing PANC-1 cells were treated with indicated concentrations of irinotecan, SN-38, and topotecan, respectively. After 3 days, cells were then subjected to MTT assay to measure cell viability. Error bars, SEM. n = 3. IC50 values of each compounds in GFP- and PRMT3-overexpressing PANC1 cells were presented as the mean ± standard deviation.
2.2. PRMT3 Upregulates ABCG2 Expression
We next investigated the mechanism underlying PRMT3-mediated GEM resistance. PRMT3 is a type I PRMT, which is known to induce asymmetric dimethylarginine (ADMA) methylation. Modification of histone H4R3 by PRMT3 is associated with gene transcription regulation. Moreover, PRMT3 might directly interact with the nuclear receptor liver X receptor α (LXRα) and acts as a transcription cofactor in methyltransferase-independent manner [21]. Given that PRMT3 might function as a transcriptional regulator, we studied the gene expression profiles in GFP- and PRMT3-overexpressing cells to identify potential genes that might be regulated by PRMT3 in GEM resistance. Of these top 10 genes that demonstrated increased expression (Figure 2A), the ATP binding cassette subfamily G member 2 (ABCG2), a member of the ABC transporter superfamily, which functions as a drug efflux pump, was significantly upregulated in PRMT3-overexpressing cells and has been reported to play a role in GEM resistance in pancreatic cancer [22,23,24]. Because the dysregulation of several genes, including hENT1, dCK, cytidine deaminase (CDA), and subunits of ribonucleotide reductase, has been proposed to modulate GEM resistance [25,26,27], we first assessed the expression of those genes in GEM-resistant cells. Our data showed that the expression of hENT1, CDA, dCK, and RRM2 was not altered in various GEM-resistant cell lines (Supplementary Figure S2). Further, we did not find the altered expression of those genes in our gene microarray data. Also, we did not find the increase of the members of ABCC subfamily. Only ABCG2 was significantly upregulated. Therefore, we focused on ABCG2 and validated its gene expression. As shown in Figure 2B, the expression of ABCG2 mRNA was increased in PRMT3-overexpressing cells. In addition, ABCG2 mRNA was upregulated in two GEM-resistant pancreatic cell lines, PANC-1-R and Mia-paca-2-R, when compared to the parental cells (Figure 2B). Increase of ABCG2 proteins was also confirmed in PRMT3-overexpressing cells and PANC-1-R cells (Figure 2B). Treatment of PRMT3-overexpressng cells or PANC-1-R cells with the PRMT3 inhibitor, SGC707, significantly reduced the expression of ABCG2 (Figure 2C). To corroborate the importance of ABCG2 in GEM resistance, we first silenced ABCG2 by small interfering RNAs (siRNAs) in PRMT3-overexpressing cells and PANC-1-R cells and assayed for cell viability under GEM treatment. Loss of ABCG2 resulted in the increased sensitivity of cells to GEM (Figure 2D). Moreover, we treated PANC-1-R cells with the ABCG2 inhibitor KO143, which significantly enhanced the sensitivity of PANC-1-R cells to GEM (Figure 2E). The results suggested that the expression of ABCG2 is critical for mediating GEM resistance. The expression of PRMT3 and ABCG2 and their association were investigated in PDAC tumor tissues (N = 81) by immunohistochemical staining. Representative images of tumors with weak and strong staining of these two proteins are shown in Figure 2F. The results demonstrated a positive association between the expression of PRMT3 and ABCG2 in the tumors (p < 0.0001; Figure 2F, right). Together, these data suggested that upregulation of ABCG2 expression by PRMT3 contributes to GEM resistance in pancreatic cancer.
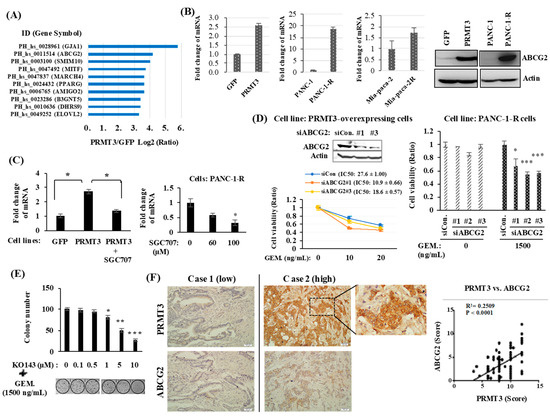
Figure 2.
ABCG2 is a target gene of PRMT3. (A) GFP- and GFP-PRMT3-overexpressing stable cells were used for gene expression microarray analysis to identify the potential target genes of PRMT3. Top ten increased genes are shown. (B) Total RNA and proteins were extracted from the indicated cell lines, and the levels of ABCG2 mRNA and proteins were determined by quantitative RT-PCR and Western blot analysis, respectively. (C) PRMT3-overexpressng cells (left) and PANC-1-R cells (right) were treated with SGC707 and the level of ABCG2 mRNA was determined by Quantitative RT-PCR analysis. * p < 0.05. (D) ABCG2 was depleted by siRNAs in PRMT3-overexpressing (left) and PANC-1-R (right) cells, respectively. These cells were cultured in the presence or absence of GEM for 5 days and then the cell viability was measured by MTT assay. Error bars, SEM. n = 3. * p < 0.05, *** p < 0.001. IC50 values of GEM. in PRMT3-ovexpressing PANC-1 cells with or without ABCG2 siRNA treatment were presented as the mean ± standard deviation. (E) PANC-1-R cells were treated with the combination of KO143 and GEM for 2 weeks. The number of colonies was counted and photographed. Error bars, SEM. n = 3. * p < 0.05, ** p < 0.01, *** p < 0.001. (F) Representative IHC staining of PRMT3 and ABCG2 in human PDAC tissues. A quantitative score for the protein expression of PRMT3 and ABCG2 was calculated from percentage of stained cells (X) multiplied by the immunostaining intensity (Y). The correlation between PRMT3 and ABCG2 in the 81 human tissue samples was determined by Pearson’s correlation coefficient.
2.3. PRMT3 Upregulates the Expression of ABCG2 via Increased mRNA Stability
Next, we determined whether PRMT3 upregulates the expression of ABCG2 via transcriptional activation. Unexpectedly, PRMT3 overexpression did not increase the promoter activity of the ABCG2 gene in PANC-1 cells (Figure 3A, left). Similarly, the ABCG2 promoter activity was not significantly altered in PANC-1-R cells (Figure 3A, right). These data suggested that PRMT3-mediated upregulation of ABCG2 expression is independent of transcriptional activation. Those results led us to speculate that PRMT3 might enhance ABCG2 expression by mediating post-transcriptional regulation. To this end, we treated GFP- and PRMT3-overexpressing cells with transcription inhibitor actinomycin D and evaluated ABCG2 mRNA levels. Compared with GFP-overexpressing cells in which ABCG2 mRNA levels decreased substantially after actinomycin D treatment for 12 h, overexpression of PRMT3 significantly prolonged ABCG2 mRNA stability, which was reversed by the PRMT3 inhibitor (Figure 3B). Consistent with the above results, we observed a marked increase in the ABCG2 mRNA stability in PANC-1-R cells, and that reduction of PRMT3 in PANC-1-R cells facilitated the degradation of ABCG2 mRNA (Figure 3C). These results supported the notion that PRMT3 upregulates ABCG2 expression by increasing its mRNA stability.
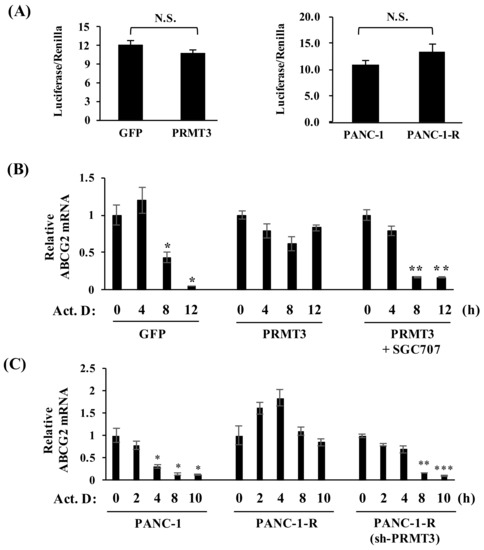
Figure 3.
Overexpression of PRMT3 enhances the stability of ABCG2 mRNA. (A) Luciferase activity of ABCG2 promoter from the indicated cells was measured after transfection for 48 h. All values represent relative firefly luciferase activity normalized to the internal control. Error bars, SEM. n = 3. N.S.: not significant. (B) The indicated cells were treated with or without SGC707 in the presence of actinomycin D for the indicated time points and the levels of ABCG2 mRNA were determined. All values represent relative mRNA level normalized to the control (0 h). Error bars, SEM. n = 3. * p < 0.05, ** p < 0.01. (C) The indicated cells were treated with actinomycin D for the indicated time points and the expression of ABCG2 mRNA was measured. All values represent relative mRNA level normalized to control (0 h). Error bars, SEM. n = 3. * p < 0.05, ** p < 0.01, *** p < 0.001.
2.4. PRMT3 Interacts with hnRNP A1
To elucidate how PRMT3 increases the stability of ABCG2 mRNA, we first searched the interacting proteins of PRMT3 to identify potential mediators involved in the regulation of mRNA stability. For this purpose, PRMT3-associated proteins were pulled down and identified by mass spectrometric analysis. A total of 293 potential PRMT3-associated proteins, including a well-known PRMT3 interaction partner, rpS2, were identified. We noticed that a number of heterogeneous nuclear ribonucleoproteins (hnRNPs), including hnRNP D and hnRNP A1, were potential PRMT3 interacting proteins (Supplementary Table S1). The hnRNP family modulates mRNA metabolism, splicing, and transcriptional and translational regulations [28,29]. To determine whether hnRNPs affect the expression of ABCG2 mRNA, we depleted hnRNP D and hnRNP A1 by short hairpin RNAs (shRNAs). However, depletion of hnRNP D had no effects on ABCG2 mRNA level (Supplementary Figure S3). We next studied hnRNP A1, another interacting partner of PRMT3 identified in our LC/MS/MS study (Figure 4A). Depletion of hnRNP A1 significantly reduced the expression of ABCG2 mRNA in both PRMT3-overexpressing cells (Figure 4B, left panel) and PANC-1-R cells (Figure 4B, right panel). Thus, we focused on hnRNPA1 in all subsequent experiments. The interaction between PRMT3 and hnRNP A1 was further validated by co-immunoprecipitation assay (Figure 4C). To assess whether hnRNP A1 is involved in GEM resistance, hnRNP A1 was depleted by shRNAs in PRMT3-overexpressing cells. These cells were subjected to cell viability assay (Figure 4D, left) and anchorage-independent growth (Figure 4D, right) in the presence or absence of GEM. The results demonstrated that reduction of hnRNP A1 increased the sensitivity to GEM in these cells. Together, these data suggest that hnRNP A1 is a PRMT3-associated protein and is involved in the regulation of ABCG2 expression and GEM resistance.
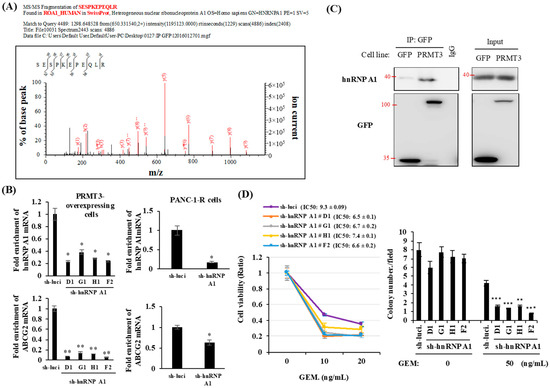
Figure 4.
HnRNP A1 is a PRMT3 interacting protein. (A) GFP- and PRMT3-ovexpressing PANC-1 cells were subjected to immunoprecipitation using the GFP antibody followed by mass spectrometric analysis. (B) PRMT3-overexpressing cells (left panel) and PANC-1-R (right panel) were transfected with hnRNP A1-targeting shRNAs. mRNA levels of hnRNP A1 and ABCG2 were measured. Error bars, SEM. n = 3. * p < 0.05, ** p < 0.01. (C) Cell lysates were collected from GFP- and PRMT3-overexpressing cells and subjected to immunoprecipitation followed by Western blotting. (D) Left: PRMT3-overexpressing cells with or without hnRNP A1 reduction were cultured in the presence or absence of GEM for 5 days and then the cell viability was measured by MTT assay. Error bars, SEM. n = 3. IC50 values of GEM in PRMT3-ovexpressing PANC-1 cells with or without hnRNP A1 shRNA treatment were presented as the mean ± standard deviation. Right: PRMT3-overexpressing cells with or without hnRNP A1 reduction were subjected to soft agar assay. Cells were treated with GEM for 3 weeks and the number of colonies was counted under microscope. Error bars, SEM. n = 3. ** p < 0.01, *** p < 0.001.
2.5. PRMT3 Methylates hnRNP A1 at Arginine 31 (R31) In Vitro and In Vivo
To determine whether hnRNP A1 is a substrate of PRMT3, we purified hnRNP A1 protein from PRMT3-overexpressing cells. Mass spectrometric analysis of the purified hnRNP A1 indicated that the arginine residue at position 31 was methylated (Figure 5A). We also carried out in vitro methylation assay by using recombinant hnRNP A1 protein and identified a methylation signal at R31 from mass spectrometric analysis (Figure 5B). To further validate the methylation status of endogenous hnRNP A1, we immunoprecipitated hnRNP A1 and detected the asymmetric dimethylarginine (ADMA) status by Western blotting. We observed a marked increase in methylation of endogenous hnRNP A1 in PRMT3-overexpressing cells (Figure 5C). hnRNP A1 shuttles between the nucleus and cytoplasm [30], and PRMT3 is predominantly located in the cytoplasm [31]. Therefore, we speculated that PRMT3 interacts and methylates hnRNP A1 in the cytoplasm. To test this possibility, we performed subcellular fractionation and immunoprecipitated hnRNP A1 from both nuclear and cytoplasmic extracts. Our result confirmed that PRMT3 interacted with hnRNP A1 in the cytoplasm (Figure 5D, left panel). In addition, the major ADMA signal was detected in the cytoplasm (Figure 5D, right panel). These results are also consistent with the IHC data showing that PRMT3 mainly appeared in the cytoplasm (Figure 2F). Mutation of hnRNP A1 R31 abolished the increase of methylation of hnRNP A1 in PRMT3-overexpressing cells, suggesting that R31 of hnRNP A1 is the major methylation site for PRMT3 in vivo (Figure 5E). Together, these results indicated that hnRNP A1 is a bona fide substrate of PRMT3, which has not been previously reported.
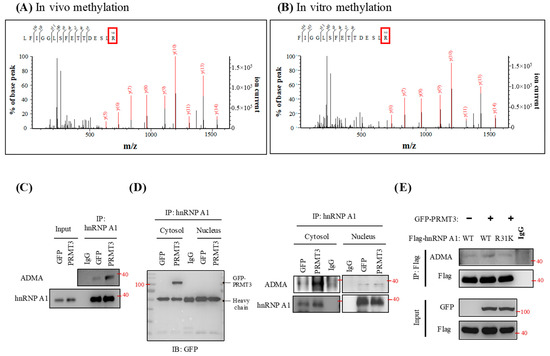
Figure 5.
PRMT3 methylates hnRNP A1 at R31. (A) hnRNP A1 protein was purified from PRMT3-overexpressing cells and the immunoprecipitated complexes were separated by SDS-PAGE. The protein bands corresponding to hnRNP A1 were excised and subjected to mass spectrometric analysis. (B) Active human full-length PRMT3 and hnRNP A1 recombinant proteins were incubated together. The samples were separated by SDS-PAGE and the protein band corresponding to hnRNP A1 was excised and subjected to mass spectrometric analysis. (C) hnRNP A1 was immunoprecipitated from GFP- and GFP-PRMT3-overexpressing cells and subjected to Western blot. The level of ADMA and hnRNP A1 was detected. (D) hnRNP A1 was immunoprecipitated from both nuclear and cytoplasmic extracts of GFP- and GFP-PRMT3-overexpressing cells, followed by Western blotting with the indicated antibodies. (E) PANC-1 cells were co-transfected with GFP, GFP-PRMT3, Flag-tagged hnRNP A1-WT, or R31K mutant. Cell lysates were subjected to immunoprecipitation followed by Western blot analysis with the indicated antibodies.
2.6. PRMT3-Mediated Methylation of hnRNP A1 Regulates the Binding of hnRNP A1 and ABCG2 mRNA
To assess whether PRMT3-methylated R31 of hnRNP A1 affects the stability of ABCG2 mRNA, we generated PANC-1 cells expressing hnRNP A1-WT, hnRNP A1-R31K, and hnRNP A1-R31F (methylation-mimicking mutant) and analyzed the ABCG2 mRNA levels. As shown in Figure 6A, the hnRNP A1- R31F mutant demonstrated increased levels of ABCG2 mRNA compared to hnRNP A1-WT- and hnRNP A1-R31K-expressing cells. Moreover, co-expression of PRMT3 and hnRNP A1-WT upregulated ABCG2 mRNA level while co-expression of PRMT3 and hnRNP A1-R31K did not show any increase. These results suggested that PRMT3 upregulated the expression of ABCG2 mRNA through R31 methylation of hnRNP A1.
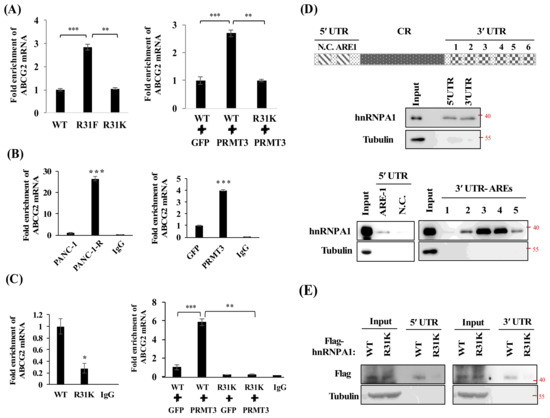
Figure 6.
PRMT3-mediated R31 methylation of hnRNP A1 regulates the binding activity of hnRNP A1 toward ABCG2 mRNA. (A) Left: total RNA was extracted from hnRNP A1-WT-, R31F- and R31K-expressing cells and the levels of ABCG2 mRNA were analyzed. Right: PANC-1 cells were cotransfected with indicated vectors. The levels of ABCG2 mRNA were determined. Error bars, SEM. n = 3. ** p < 0.01, *** p < 0.001. (B) Total cellular lysates extracted from the indicated cells were incubated with anti-hnRNP A1 or mouse IgG antibodies, respectively. The hnRNP A1-associated RNAs were eluted and analyzed by real-time PCR for ABCG2 mRNA levels. *** p < 0.001. (C) Left: total cellular lysates extracted from Flag-tagged hnRNP A1-WT, or R31K-expressing cells were incubated with anti-Flag or mouse IgG antibodies, respectively. Right: similarly, total cellular lysates extracted from PANC-1 cells which cotransfected with indicated vectors were incubated with anti-Flag or mouse IgG antibodies, respectively. The Flag-tagged hnRNP A1-associated RNAs were eluted and the levels of ABCG2 mRNA were analyzed. Error bars, SEM. n = 3. * p < 0.05, ** p < 0.01, *** p < 0.001. (D) A schematic illustrating the AREs in ABCG2 mRNA. One potential ARE in 5´UTR and six potential AREs in 3´UTR are shown. Individual ARE fragments were amplified and subjected to biotin RNA pull-down assay. The RNA-protein complexes were immunoprecipitated and analyzed by Western blot analysis using the hnRNP A1 and tubulin antibodies. (E) Flag-hnRNP A1-WT- and Flag-hnRNP A1-R31K-expressing PANC-1 cells were subjected to biotin RNA pull-down assay. The RNA-protein complexes were immunoprecipitated and analyzed by Western blot analysis using the Flag and tubulin antibodies.
HnRNP A1 protein contains two RNA-recognition motifs (RRMs) in the N-terminal region, which play important roles in the regulation of RNA-binding specificity and affinity [32,33]. Because the R31 residue is located within the first RRM of hnRNP A1, we hypothesized that PRMT3-mediated R31 methylation might affect the RNA-binding affinity of hnRNP A1 to ABCG2 mRNA. To test this possibility, we carried out RNA immunoprecipitation assay. As shown in Figure 6B, ABCG2 mRNA was significantly enriched in PANC-1-R and PRMT3-overexpressing cells. Co-expression of PRMT3 and hnRNP A1-WT significantly increased the binding between hnRNP A1-WT and ABCG2 mRNA, while co-expression of PRMT3 and hnRNP A1-R31K did not enhance the interaction (Figure 6C). To identify the binding regions of ABCG2 mRNA for hnRNP A1, we amplified the 5′-UTR and 3′-UTR of ABCG2 mRNA and labeled the RNA fragments for biotin RNA pull-down assay. Our results indicated that hnRNP A1 bound to both the 5′-UTR and 3′-UTR of ABCG2 mRNA (Figure 6D, upper). Bioinformatics analysis revealed one potential AU-rich element (ARE), the AUUUA pentamer motif which is the potential site for hnRNP A1 binding [34,35], in the 5′-UTR and six potential AREs in 3′-UTR of ABCG2 mRNA. hnRNP A1 bound to the 5’-UTR-ARE-1 but not to the negative control fragment (N.C.) that does not contain the ARE sequences (Figure 6D, bottom). In addition, hnRNP A1 bound strongly to the 3′-UTR-ARE-3 and 3′-UTR-ARE-4 sites, suggesting that these two AREs are the major binding sites for hnRNP A1 (Figure 6D, bottom). To further investigate whether PRMT3-mediated R31 methylation of hnRNP A1 regulates its binding to ABCG2 mRNA, hnRNP A1-WT- and hnRNP A1-R31K-expressing PANC-1 cells were subjected to biotin RNA pull-down assay and results showed that hnRNP A1-R31K mutant reduced the binding activity to both the 5′-UTR and 3′-UTR of ABCG2 mRNA (Figure 6E). These data suggest that PRMT3-mediated methylation of hnRNP A1 R31 enhances its binding to ABCG2 mRNA.
3. Discussion
PRMT3 belongs to type I PRMT family which attaches ADMA on its protein substrates. However, the biological functions of PRMT3 are still largely uncharacterized. The first physiological substrate of PRMT3 is the 40S ribosomal protein S2 [36], implying that PRMT3 might regulate ribosome biosynthesis and protein production. A potential role of PRMT3 in neuronal cells has been suggested by the findings that PRMT3 is highly expressed in different areas of the brain and is essential for the formation of dendritic spine in hippocampal neurons [37,38]. Recently, PRMT3 was reported to interact with LXRα to modulate hepatic lipogenesis and may participate in the onset of fatty liver [21]. The role of PRMT3 in tumorigenesis is not well characterized. Previously, studies have demonstrated that PRMT3 interacts with the tumor suppressor DAL-1/4.1B, which inhibits its methyltransferase activity in vitro and in vivo [39]. In this study, we provided the first evidence to demonstrate that PRMT3 plays an important role in GEM resistance (Figure 7, proposed model). Our results suggest that inhibition of PRMT3 might be a therapeutic strategy to improve the GEM sensitivity in pancreatic cancer treatment. Several selective and cell-permeable allosteric inhibitors of PRMT3 have been developed in the past several years [40,41]. It would be worthwhile to further evaluate the combination of PRMT3 inhibitors and GEM as a treatment option for pancreatic cancer in vivo.
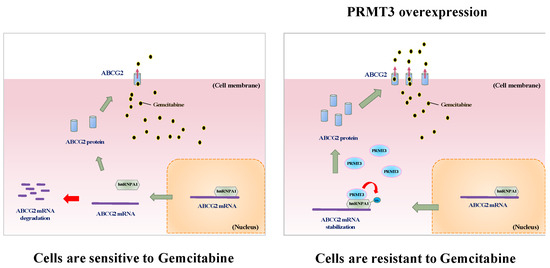
Figure 7.
Proposed model of PRMT3-mediated GEM resistance in pancreatic cancer cells. PRMT3 is upregulated in GEM-resistant pancreatic cells and its overexpression stabilizes ABCG2 mRNA via methylating arginine 31 of hnRNPA1. PRMT3-mediated ABCG2 upregulation contributes to gemcitabine resistance.
By using an S-adenosylmethionine analogue as a probe, Guo et al. identified more than 80 potential PRMT3 substrates [42]. Whether these proteins are substrates of PRMT3 in vivo has not yet been validated; however, we found an interesting candidate, hnRNP A3, from the list. Comparing the amino acid sequences of human hnRNP A1, A2, and A3 (Supplementary Figure S4), the RRM1 motif is highly conserved in this hnRNP family. In addition, the R31 residue in hnRNP A1 is also highly conserved among the three members, suggesting that this residue may be an important site for methylation and functional regulation. To date, several arginine residues of hnRNP A1, including R190, R194, R233, R218, and R225, have been reported to be methylated by PRMT1 or PRMT5 in vitro and in vivo [43,44,45,46]. All of these arginine residues are located within the RGG domain in the C-terminal region. PRMT1 is a type I methyltransferase that catalyzes ADMA on arginine residues, whereas PRMT5 is a type II methyltransferase that is responsible for catalyzing symmetric dimethylarginine (SDMA). Thus, both ADMA and SDMA occur within the RGG motif of hnRNP A1. Two recent studies demonstrated that methylation of the RGG domain might regulate mRNA expression via modulation of the internal ribosome entry site (IRES)-dependent translation [43,44]. We identified a distinct region, the RRM domain, of hnRNP A1, which can be modified via ADMA by PRMT3, and this methylation enhances the binding of hnRNP A1 to target mRNAs. Unlike the RGG domain, the functional role of arginine methylation within the N-terminal RRM domains of RNA-binding proteins has not been explored. A previous study revealed that PRMT4 methylates the RNA-binding protein HuR at one arginine residue located within the hinge region between two RRM motifs, which results in the induction of TNF-α expression via the stabilization of mRNA [47]. Our results supported the hypothesis that arginine methylation within the RRM domains of hnRNP A1 promotes its RNA-binding activity and increases the stability of its target mRNA. The RRM motifs of hnRNP A1 have also been reported to bind to telomeric DNA and be involved in telomere length maintenance [48]. It is possible that PRMT3-mediated methylation in the RRM domain of hnRNP A1 might play a role in regulating the binding of hnRNP A1 to telomeric DNA and the maintenance of telomere length.
It is well known that hnRNP A1 binds to the pre-mRNAs during the splicing process in the nucleus and might assist in the delivery of mature mRNAs to the cytoplasm. After its release from the mRNA for protein translation in the cytoplasm, hnRNP A1 interacts with nuclear import protein to be translocated back into the nucleus [30,49,50]. Because PRMT3 is highly abundant in the cytoplasm, methylation of hnRNP A1 by PRMT3 might occur primarily in the cytoplasm. So far, because there is no evidence to indicate the involvement of RRM domain of hnRNP A1 in nuclear-cytoplasmic shuttling, whether PRMT3 affects the subcellular localization of hnRNP A1 through arginine methylation of RRM domain remains unclear. In addition, it is possible that methylation of hnRNP A1 by PRMT3 increases its binding to the target mRNAs, which induces the accumulation of methylated hnRNP A1 in the cytoplasm. However, we could not exclude the possibility that PRMT3 methylates hnRNP A1 in the nucleus and the methylated hnRNP A1 forms a stable complex with target mRNAs, which is continuously exported to the cytoplasm. Therefore, the methylated hnRNP A1 is preferentially detected in the cytoplasm.
The role of ABCG2 in gemcitabine resistance is still controversial. Several studies do not support the involvement of ABCG2 in gemcitabine resistance [27,51]. However, some studies indeed demonstrated that ABCG2 contributes to gemcitabine resistance [22,23,52,53,54]. In our microarray data, we did not find the change of hENT1, hCNT1, dCK, and subunits of ribonucleotide reductase in the PRMT3-overexpressing PANC-1 cells. Only ABCG2 was significantly upregulated. Therefore, our results suggested that ABCG2 is one of the key mediators of gemcitabine resistance.
4. Material and Methods
4.1. Cell Culture and Reagents
Pancreatic cancer cell lines PANC-1 and Mia-Paca-2 were purchased from ATCC and had been re-authenticated by short tandem repeat profiling in 2014. Cells were cultured in DMEM medium with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The GEM-resistant cell line, PANC-1-R, was established from parental human PANC-1 pancreatic cancer cells as previously described [20]. PANC-1 cells with stable expressions of GFP, GFP-PRMT3, and GFP-PRMT6, were grown in the DMEM medium supplemented with 800 μg/mL G418. PANC-1-R cells with PRMT3 reduction were maintained in DMEM medium containing 1500 ng/mL of GEM and 1 μg/mL puromycin. GFP-PRMT3 stable cells with hnRNP A1 reduction were maintained in DMEM medium containing 800 μg/mL of G418 and 1 μg/mL puromycin. All cell lines were authenticated with short tandem repeat profiling. The following antibodies were used in this study: PRMT3 (Cat No. GTX23765, GeneTex, Irvine, CA, USA); PRMT6 (Cat No. A300-929A, Bethyl Laboratories, Montgomery, TX, USA); ABCG2/BCRP (Cat No. MAB4146, Millipore, Billerica, MA, USA); hnRNP A1 (Cat No. 05-1521, Millipore); GFP (Cat No. ab290, Abcam, Cambridge, UK); asymmetrical dimethyl arginine (ADMA) (Cat No. 13522, Cell Signaling Technology, Danvers, MA, USA); and Flag M2 (Cat No. F1804, Sigma, St. Louis, MO, USA). GEM (Gemzar, Lilly, Indianapolis, IN, USA) stocks were stored in aliquots at 4 °C. Irinotecan (Cat No. I1406) was obtained from Sigma. SN-38 (CAS No. 15632), Topotecan (CAS No. 14129), and PRMT3 inhibitor SGC707 (CAS No. 1687736-54-4) were purchased from Cayman Chemical (Ann Arbor, MI, USA). ABCG2 inhibitor KO143 (Cat No. SC-204030) was ordered from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
4.2. Plasmids
The pEGFP-PRMT3 and -PRMT6 plasmids were kindly provided by Dr. Mien-Chie Hung [55]. Human hnRNP A1 cDNA ORF Clone (Cat No. NM-031157) was purchased from OriGene (Rockville, MD, USA). ABCG2 promoter (pGL4-BCRP) was kindly provided by Dr. Wei-Chien Huang [56]. PRMT3 shRNA and hnRNP A1 shRNA plasmids were purchased from National RNAi Core, Taiwan. The sequences of shRNA plasmids are shown in Supplementary Table S2.
4.3. Soft Agar Colony Formation Assay
To prepare the bottom layer of agar, 0.6% agarose containing DMEM medium was added into the six-well plate and set aside for 10 min to allow agarose to solidify. Cells suspended in DMEM medium containing 0.3% agarose were plated onto the bottom layer of agar at a density of 2000 cells per well. After incubation for 2–3 weeks, colonies larger than 50 µm in diameter were counted under microscopy. The results were expressed as the means ± SE of three independent experiments.
4.4. Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted by using RNeasy mini kit and then reverse-transcribed into cDNA using the RT2 first strand kit (Qiagen, Hilden, Germany). The PCR experiments were done by 7500 Fast Real-time PCR system (Applied Biosystems, Foster City, CA, USA). The sequences of primers used for real-time PCR analysis are shown in Supplementary Table S2.
4.5. Site-Directed Mutagenesis
Flag-tagged hnRNP A1-R31K and -R31F mutants were generated using QuickChange Site-Directed Mutagenesis Kit according to the manufacturer’s protocol (Stratagene, La Jolla, CA, USA). The primers used for mutagenesis are shown in Supplementary Table S2.
4.6. Mass Spectrometry
GFP-PRMT3 and hnRNP A1 were purified from GFP-PRMT3-overexpressing cells by immunoprecipitation with GFP and hnRNP A1 antibodies, respectively. The GFP-purified complexes were subjected to in-solution digestion with trypsin and the PRMT3-interacting proteins were identified by mass spectrometry (Mithra Biotechnology Inc., New Taipei City, Taiwan). To identify the arginine residue on hnRNP A1 methylated by PRMT3, the hnRNP A1-purified complexes were separated by SDS-PAGE. The protein bands corresponding to hnRNP A1 were excised and subjected to in-gel digestion with trypsin, and the methylated arginine residue was analyzed by liquid chromatography/tandem mass spectrometry (Mithra Biotechnology Inc., New Taipei City, Taiwan).
4.7. In Vitro Methylation Assay
Active recombinant full-length human PRMT3 protein (Cat No. ab167953, Abcam) was purchased from Abcam. Human hnRNP A1 recombinant protein (Cat No. pro-1038) was purchased from PROSPEC. PRMT3 (1 μg) was incubated with hnRNP A1 (2 μg) in the presence of 40 μM S-adenosyl-L-methionine (Sigma) for 1 h at 30 °C in a final volume of 25 μL of phosphate-buffered saline. After incubation, sample was separated by SDS-PAGE, and the protein band corresponding to hnRNP A1 was excised for mass spectrometry.
4.8. RNA Immunoprecipitation (RIP) Assay
RIP assay was performed according to the manufacturer’s protocol (Cat No. 03-204, Millipore). In brief, cells were lysed with RIP lysis buffer, and cell extracts were incubated with 5 μg of normal mouse IgG or hnRNP A1 antibodies at 4 °C overnight. After incubation, 50 μL of magnetic beads were added to the immunoprecipitated complexes. The beads were washed six times with RIP wash buffer, followed by protein digestion in proteinase K buffer at 55 °C for 30 min. RNA was purified by phenol-chloroform extraction and subjected to real-time RT-PCR.
4.9. Biotinylated RNA Pull-Down Assay
Complementary DNA prepared from the mRNA of PANC-1 cells was used as a template for PCR amplification of different regions of ABCG2 mRNA. All forward primers contained the T7 RNA polymerase promoter sequence 5′-CCAAGCTTCTAATACGACTCACTATAGGGAGA-3′ (T7). All of the primer pairs were used to prepare the templates, including the 5′UTR (1-493), 3′UTR (2462-4445), 5′UTR-N.C. (1-295), 5′UTR-ARE1 (271-550), 3′UTR-ARE1 (2381-2573), 3′UTR-ARE2 (2553-2821), 3′UTR-ARE3 (2801-3062), 3′UTR-ARE4 (3276-3585), and 3′UTR-ARE5 (4027-4331) of ABCG2 mRNA. The sequences of primers are shown in Supplementary Table S2. Following PCR amplification, PCR fragments containing the T7 RNA polymerase promoter sequence were purified by gel electrophoresis. To prepare biotinylated RNA probes, the purified PCR fragments were incubated with T7 RNA polymerases (Cat No. P2075, Promega, Madison, WI, USA) and biotin RNA-labeling mixture (Cat No. 11685597910, Sigma) for 2 h at 37 °C, followed by DNase I treatment for 15 min at 30 °C to remove template DNA. The biotinylated RNA was purified by phenol-chloroform extraction and rehydrated in DEPC-treated water. Purified biotinylated RNA (500 ng) was incubated with 500 μg of cell lysates overnight at 4 °C, and RNA/proteins complexes were immunoprecipitated using the streptavidin-conjugated Dynabeads and subjected to SDS-PAGE followed by Western blot analysis using hnRNP A1 and tubulin antibodies.
4.10. Immunohistochemical (IHC) Staining and Analysis
Human PDAC tissues were obtained from patients undergoing surgical resection in National Cheng Kung University Hospital (Tainan, Taiwan) under the guidelines approved by the Institutional Review Board of National Cheng Kung University Hospital (Protocal No/IRB No: /B-ER-106-230). Written informed consent was obtained from each patient [57]. Tissue sections were stained with human PRMT3 (Cat No. GTX23765, GeneTex) and human ABCG2 (Cat No. MAB4146, Millipore) antibodies overnight at 4 °C followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. The protein signal was developed using a 3,3′-diaminobenzidine solution, and sections were evaluated under microscope by a pathologist. The staining intensity and average percentage of positive cells were analyzed using ten independent high-magnification fields. The intensity of the staining was classified into four grades: 0, negative staining; 1, weak staining; 2, moderate staining; 3, strong staining. The percentage of positive cells was categorized into the following grades: 0, <10%; 1, 10–25%; 2, 25–50%; 3, 50–75%; 4, >75%. The results (0–12) were scored by multiplying the staining intensity and the percentage of positive cells.
4.11. Statistics Analyses
The two-tailed unpaired t-test was used to assess the difference between two independent experimental groups and p-value < 0.05 was considered statistically significant. The correlation between PRMT3 and ABCG2 expression in human tissue samples was determined by Pearson’s correlation coefficient.
5. Conclusions
In summary, we identify hnRNP A1 as a novel substrate of PRMT3, and PRMT3-methylated hnRNP A1 at R31 enhances its binding to the ABCG2 mRNA, leading to upregulation of ABCG2 and resistance to GEM. Therefore, targeting PRMT3 may be a potential therapeutic approach for the treatment of GEM-resistant pancreatic cancer. (Figure 7, proposed model).
Supplementary Materials
The following are available online at http://www.mdpi.com/2072-6694/11/1/8/s1, Figure S1: Overexpression of PRMT3 and PRMT6 facilitates cell proliferation, Figure S2: The expression of hENT1, RRM2, CDA and dCK was not altered in gemcitabine-resistant cell lines, Figure S3: Depletion of hnRNP D had no effects on ABCG2 mRNA level, Figure S4: Sequence alignment of hnRNP A1, A2 and A3 proteins, Table S1: PRMT3-associated heterogeneous nuclear ribonucleoproteins (hnRNPs), Table S2: Oligonucleotide sequences.
Author Contributions
M.-C.H., M.-R.P., Y.-S.S., L.-T.C., and W.-C.H. designed the study; M.-C.H., Y.-L.T., and C.-H.T. performed research; M.-C.H., P.-Y.C., and W.-C.H. analyzed data; M.-C.H. and W.-C.H. prepared the manuscript.
Funding
This study was supported in part by the following grants: 105-2321-B-400-010, 105-2314-B-037-001, 107-2320-B-400-006-MY3 from the Ministry of Science and Technology, CA-106-PP-15 from the Ministry of Health and Welfare and NHRI-107A1-CACO-02181811.
Acknowledgments
We thank Mien-Chie Hung and Wei-Chien Huang for kindly providing the experimental materials. We also thank Wen-Yu Pan for the technique assistance.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Matano, E.; Tagliaferri, P.; Libroia, A.; Damiano, V.; Fabbrocini, A.; De Lorenzo, S.; Bianco, A.R. Gemcitabine combined with continuous infusion 5-fluorouracil in advanced and symptomatic pancreatic cancer: A clinical benefit-oriented phase II study. Br. J. Cancer 2000, 82, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Giovannetti, E.; Del Tacca, M.; Mey, V.; Funel, N.; Nannizzi, S.; Ricci, S.; Orlandini, C.; Boggi, U.; Campani, D.; Del Chiaro, M.; et al. Transcription analysis of human equilibrative nucleoside transporter-1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Res. 2006, 66, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.J.; Elsaleh, H.; Garcia, M.; Lai, R.; Ammar, A.; Regine, W.F.; Abrams, R.; Benson, A.B.; Macdonald, J.; Cass, C.E.; et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 2009, 136, 187–195. [Google Scholar] [CrossRef]
- Ohhashi, S.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Egami, T.; Yu, J.; Toma, H.; Sadatomi, S.; Nagai, E.; Tanaka, M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008, 28, 2205–2212. [Google Scholar] [PubMed]
- Hunsucker, S.A.; Spychala, J.; Mitchell, B.S. Human cytosolic 5′-nucleotidase I: Characterization and role in nucleoside analog resistance. J. Biol. Chem. 2001, 276, 10498–10504. [Google Scholar] [CrossRef]
- Xu, M.; Li, L.; Liu, Z.; Jiao, Z.; Xu, P.; Kong, X.; Huang, H.; Zhang, Y. ABCB2 (TAP1) as the downstream target of SHH signaling enhances pancreatic ductal adenocarcinoma drug resistance. Cancer Lett. 2013, 333, 152–158. [Google Scholar] [CrossRef]
- Chen, M.; Xue, X.; Wang, F.; An, Y.; Tang, D.; Xu, Y.; Wang, H.; Yuan, Z.; Gao, W.; Wei, J.; et al. Expression and promoter methylation analysis of ATP-binding cassette genes in pancreatic cancer. Oncol. Rep. 2012, 27, 265–269. [Google Scholar] [CrossRef]
- Hopper-Borge, E.; Xu, X.; Shen, T.; Shi, Z.; Chen, Z.S.; Kruh, G.D. Human multidrug resistance protein 7 (ABCC10) is a resistance factor for nucleoside analogues and epothilone B. Cancer Res. 2009, 69, 178–184. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Glasspool, R.M.; Teodoridis, J.M.; Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 2006, 94, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, K.; Miller, H.; Gordian, E.; Rocha-Lima, C.; Singal, R. Methylation-mediated silencing of TMS1 in pancreatic cancer and its potential contribution to chemosensitivity. Anticancer Res. 2010, 30, 3919–3925. [Google Scholar]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. 5). [Google Scholar] [CrossRef] [PubMed]
- Hodzic, J.; Giovannetti, E.; Diosdado, B.; Adema, A.D.; Peters, G.J. Regulation of deoxycytidine kinase expression and sensitivity to gemcitabine by micro-RNA 330 and promoter methylation in cancer cells. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, V.; Ricci, F.; Rubio-Viqueira, B.; Kulesza, P.; Yeo, C.J.; Hidalgo, M.; Klein, A.; Laheru, D.; Iacobuzio-Donahue, C.A. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: Relationship to molecular mechanisms of gemcitabine resistance and survival. Clin. Cancer Res. 2006, 12, 2492–2497. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef]
- Wang, Y.; Hsu, J.M.; Kang, Y.; Wei, Y.; Lee, P.C.; Chang, S.J.; Hsu, Y.H.; Hsu, J.L.; Wang, H.L.; Chang, W.C.; et al. Oncogenic Functions of Gli1 in Pancreatic Adenocarcinoma Are Supported by Its PRMT1-Mediated Methylation. Cancer Res. 2016, 76, 7049–7058. [Google Scholar] [CrossRef]
- Pan, M.R.; Hsu, M.C.; Luo, C.W.; Chen, L.T.; Shan, Y.S.; Hung, W.C. The histone methyltransferase G9a as a therapeutic target to override gemcitabine resistance in pancreatic cancer. Oncotarget 2016, 7, 61136–61151. [Google Scholar] [CrossRef]
- Nakakido, M.; Deng, Z.; Suzuki, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. PRMT6 increases cytoplasmic localization of p21CDKN1A in cancer cells through arginine methylation and makes more resistant to cytotoxic agents. Oncotarget 2015, 6, 30957–30967. [Google Scholar] [CrossRef]
- Pan, M.R.; Hsu, M.C.; Chen, L.T.; Hung, W.C. G9a orchestrates PCL3 and KDM7A to promote histone H3K27 methylation. Sci. Rep. 2015, 5, 18709. [Google Scholar] [CrossRef]
- Kim, D.I.; Park, M.J.; Lim, S.K.; Park, J.I.; Yoon, K.C.; Han, H.J.; Gustafsson, J.A.; Lim, J.H.; Park, S.H. PRMT3 regulates hepatic lipogenesis through direct interaction with LXRalpha. Diabetes 2015, 64, 60–71. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.; Wei, W.; Shi, M.; Xin, B.; Zhang, T.; Shen, X. Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1alpha and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 2016, 17, 188–198. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, C.; Jansen, R.; Yamaguchi, H.; de Haas, M.; van de Wetering, K.; Wijnholds, J.; Beijnen, J.; Borst, P. Contribution of the drug transporter ABCG2 (breast cancer resistance protein) to resistance against anticancer nucleosides. Mol. Cancer Ther. 2008, 7, 3092–3102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, C.Y.; Liu, T.; Wu, B.; Zhou, F.; Xiong, J.X.; Wu, H.S.; Tao, J.; Zhao, G.; Yang, M.; et al. Persistence of side population cells with high drug efflux capacity in pancreatic cancer. World J. Gastroenterol. 2008, 14, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Tanno, S.; Koizumi, K.; Nishikawa, T.; Nakamura, K.; Minoguchi, M.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br. J. Cancer 2007, 96, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Aho, U.; Nilsson, B.I.; Peters, G.J.; Pastor-Anglada, M.; Rasch, W.; Sandvold, M.L. Gemcitabine chemoresistance in pancreatic cancer: Molecular mechanisms and potential solutions. Scand. J. Gastroenterol. 2009, 44, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Vasseur, R.; Vincent, A.; Duchene, B.; Van Seuningen, I.; Jonckheere, N. The oncogenic receptor ErbB2 modulates gemcitabine and irinotecan/SN-38 chemoresistance of human pancreatic cancer cells via hCNT1 transporter and multidrug-resistance associated protein MRP-2. Oncotarget 2015, 6, 10853–10867. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef]
- Weighardt, F.; Biamonti, G.; Riva, S. The roles of heterogeneous nuclear ribonucleoproteins (hnRNP) in RNA metabolism. Bioessays 1996, 18, 747–756. [Google Scholar] [CrossRef]
- Pinol-Roma, S.; Dreyfuss, G. Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature 1992, 355, 730–732. [Google Scholar] [CrossRef]
- Tang, J.; Gary, J.D.; Clarke, S.; Herschman, H.R. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem. 1998, 273, 16935–16945. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G.; Dreyfuss, G. RNA binding specificity of hnRNP A1: Significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 1994, 13, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.M.; Jokhan, L.; Cheng, X.; Mayeda, A.; Krainer, A.R. Crystal structure of human UP1, the domain of hnRNP A1 that contains two RNA-recognition motifs. Structure 1997, 5, 559–570. [Google Scholar] [CrossRef]
- Hamilton, B.J.; Nagy, E.; Malter, J.S.; Arrick, B.A.; Rigby, W.F. Association of heterogeneous nuclear ribonucleoprotein A1 and C proteins with reiterated AUUUA sequences. J. Biol. Chem. 1993, 268, 8881–8887. [Google Scholar] [PubMed]
- Hamilton, B.J.; Burns, C.M.; Nichols, R.C.; Rigby, W.F. Modulation of AUUUA response element binding by heterogeneous nuclear ribonucleoprotein A1 in human T lymphocytes. The roles of cytoplasmic location, transcription, and phosphorylation. J. Biol. Chem. 1997, 272, 28732–28741. [Google Scholar] [CrossRef] [PubMed]
- Swiercz, R.; Cheng, D.; Kim, D.; Bedford, M.T. Ribosomal protein rpS2 is hypomethylated in PRMT3-deficient mice. J. Biol. Chem. 2007, 282, 16917–16923. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, K.; Miyata, S.; Mori, Y.; Koyama, Y.; Taneda, T.; Okuda, H.; Kousaka, A.; Tohyama, M. Immunohistochemical and western analyses of protein arginine N-methyltransferase 3 in the mouse brain. Neuroscience 2006, 141, 1971–1982. [Google Scholar] [CrossRef]
- Miyata, S.; Mori, Y.; Tohyama, M. PRMT3 is essential for dendritic spine maturation in rat hippocampal neurons. Brain Res. 2010, 1352, 11–20. [Google Scholar] [CrossRef]
- Singh, V.; Miranda, T.B.; Jiang, W.; Frankel, A.; Roemer, M.E.; Robb, V.A.; Gutmann, D.H.; Herschman, H.R.; Clarke, S.; Newsham, I.F. DAL-1/4.1B tumor suppressor interacts with protein arginine N-methyltransferase 3 (PRMT3) and inhibits its ability to methylate substrates in vitro and in vivo. Oncogene 2004, 23, 7761–7771. [Google Scholar] [CrossRef]
- Kaniskan, H.U.; Szewczyk, M.M.; Yu, Z.; Eram, M.S.; Yang, X.; Schmidt, K.; Luo, X.; Dai, M.; He, F.; Zang, I.; et al. A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angew. Chem. Int. Ed. Engl. 2015, 54, 5166–5170. [Google Scholar] [CrossRef]
- Liu, F.; Li, F.; Ma, A.; Dobrovetsky, E.; Dong, A.; Gao, C.; Korboukh, I.; Liu, J.; Smil, D.; Brown, P.J.; et al. Exploiting an allosteric binding site of PRMT3 yields potent and selective inhibitors. J. Med. Chem. 2013, 56, 2110–2124. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, R.; Zheng, W.; Chen, Y.; Blum, G.; Deng, H.; Luo, M. Profiling substrates of protein arginine N-methyltransferase 3 with S-adenosyl-L-methionine analogues. ACS Chem. Biol. 2014, 9, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Dhar, S.; Bedford, M.T. PRMT5 regulates IRES-dependent translation via methylation of hnRNP A1. Nucleic Acids Res. 2017, 45, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.L.; Lewis, S.M. Methylarginines within the RGG-Motif Region of hnRNP A1 Affect Its IRES Trans-Acting Factor Activity and Are Required for hnRNP A1 Stress Granule Localization and Formation. J. Mol. Biol. 2017, 429, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Merrill, B.M.; Rajpurohit, R.; Kumar, A.; Stone, K.L.; Papov, V.V.; Schneiders, J.M.; Szer, W.; Wilson, S.H.; Paik, W.K.; et al. Identification of N(G)-methylarginine residues in human heterogeneous RNP protein A1: Phe/Gly-Gly-Gly-Arg-Gly-Gly-Gly/Phe is a preferred recognition motif. Biochemistry 1997, 36, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Rajpurohit, R.; Lee, S.O.; Park, J.O.; Paik, W.K.; Kim, S. Enzymatic methylation of recombinant heterogeneous nuclear RNP protein A1. Dual substrate specificity for S-adenosylmethionine:histone-arginine N-methyltransferase. J. Biol. Chem. 1994, 269, 1075–1082. [Google Scholar] [PubMed]
- Li, H.; Park, S.; Kilburn, B.; Jelinek, M.A.; Henschen-Edman, A.; Aswad, D.W.; Stallcup, M.R.; Laird-Offringa, I.A. Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J. Biol. Chem. 2002, 277, 44623–44630. [Google Scholar] [CrossRef]
- LaBranche, H.; Dupuis, S.; Ben-David, Y.; Bani, M.R.; Wellinger, R.J.; Chabot, B. Telomere elongation by hnRNP A1 and a derivative that interacts with telomeric repeats and telomerase. Nat. Genet. 1998, 19, 199–202. [Google Scholar] [CrossRef]
- Pinol-Roma, S.; Dreyfuss, G. Transcription-dependent and transcription-independent nuclear transport of hnRNP proteins. Science 1991, 253, 312–314. [Google Scholar] [CrossRef]
- Reed, R.; Hurt, E. A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 2002, 108, 523–531. [Google Scholar] [CrossRef]
- Bhagwandin, V.J.; Bishop, J.M.; Wright, W.E.; Shay, J.W. The Metastatic Potential and Chemoresistance of Human Pancreatic Cancer Stem Cells. PLoS ONE 2016, 11, e0148807. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Gu, M.; Zhu, L.; Liu, J.; Xiong, Y.; Wei, Y.; Li, F. Gemcitabine upregulates ABCG2/BCRP and modulates the intracellular pharmacokinetic profiles of bioluminescence in pancreatic cancer cells. Anticancer Drugs 2016, 27, 183–191. [Google Scholar] [CrossRef]
- Gujral, T.S.; Kirschner, M.W. Hippo pathway mediates resistance to cytotoxic drugs. Proc. Natl. Acad. Sci. USA 2017, 114, E3729–E3738. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Ishiwata, T.; Hasegawa, F.; Michishita, M.; Kawai, H.; Matsuda, Y.; Arai, T.; Ishikawa, N.; Aida, J.; Takubo, K.; et al. Stemness and anti-cancer drug resistance in ATP-binding cassette subfamily G member 2 highly expressed pancreatic cancer is induced in 3D culture conditions. Cancer Sci. 2018, 109, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Chen, C.T.; Chou, C.K.; Kuo, H.P.; Li, L.Y.; Lin, C.Y.; Lee, H.J.; Wang, Y.N.; Liu, M.; Liao, H.W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011, 13, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Chao, Y.J.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).