Paroxetine Induces Apoptosis of Human Breast Cancer MCF-7 Cells through Ca2+-and p38 MAP Kinase-Dependent ROS Generation

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
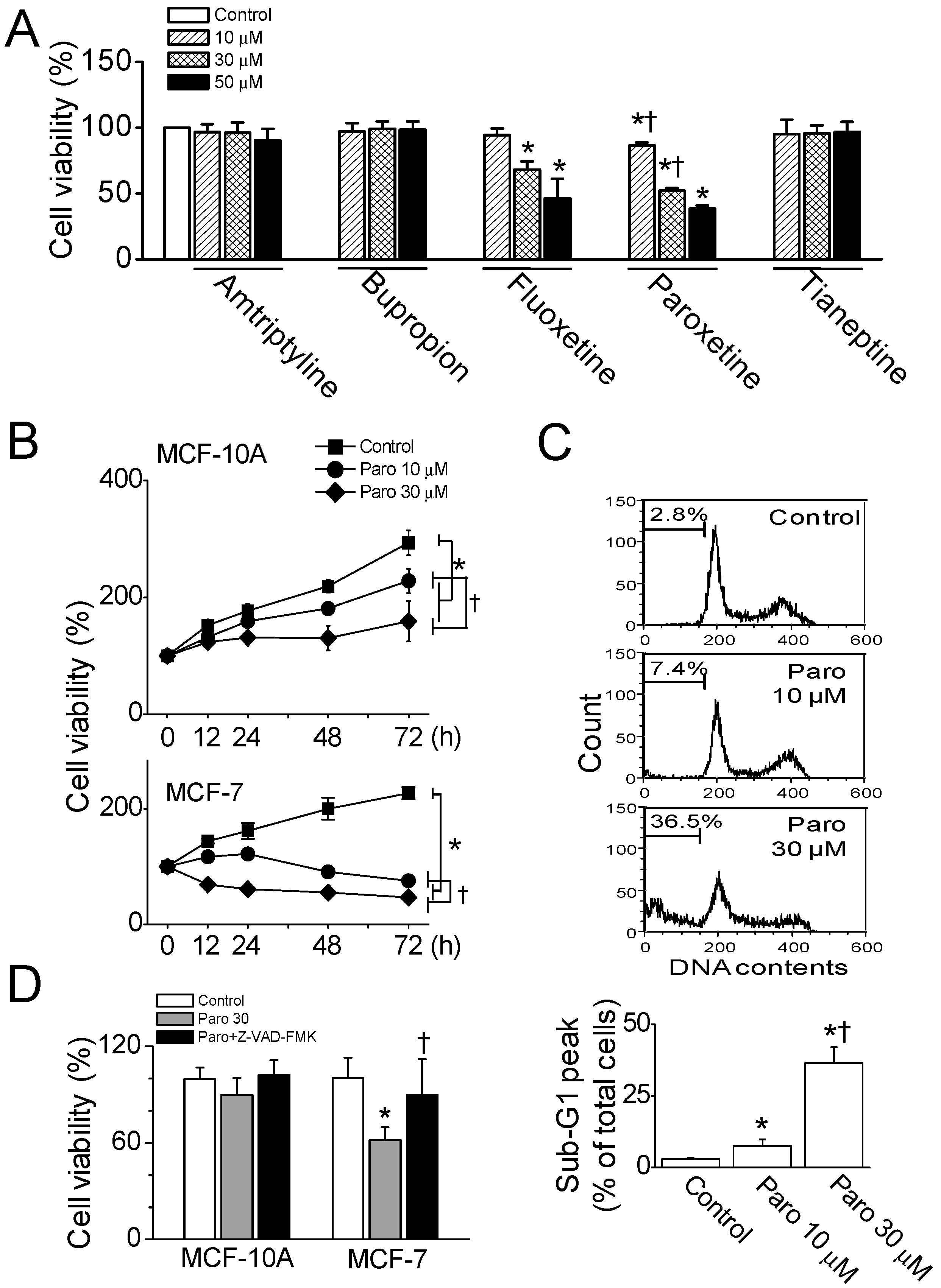
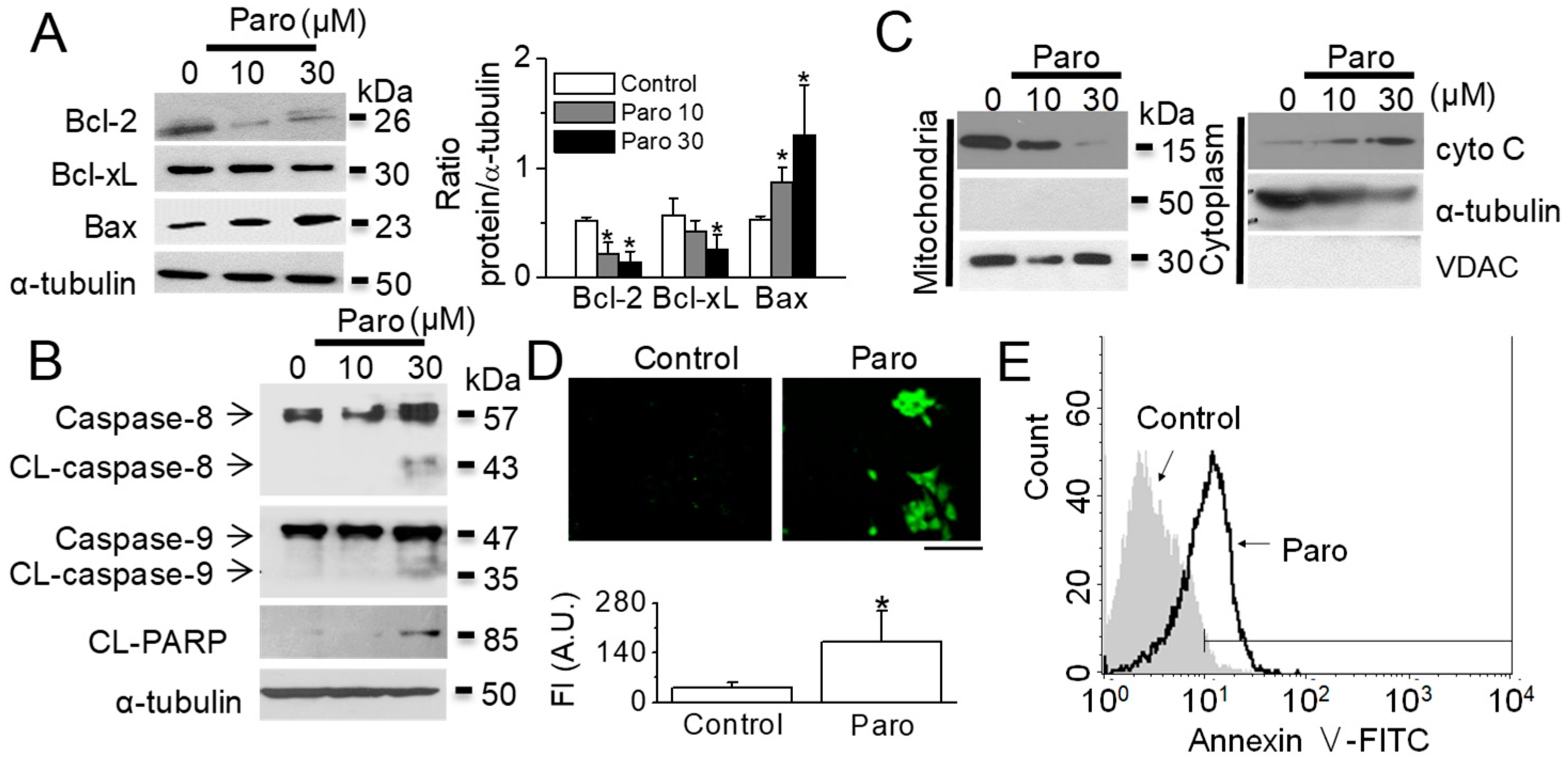
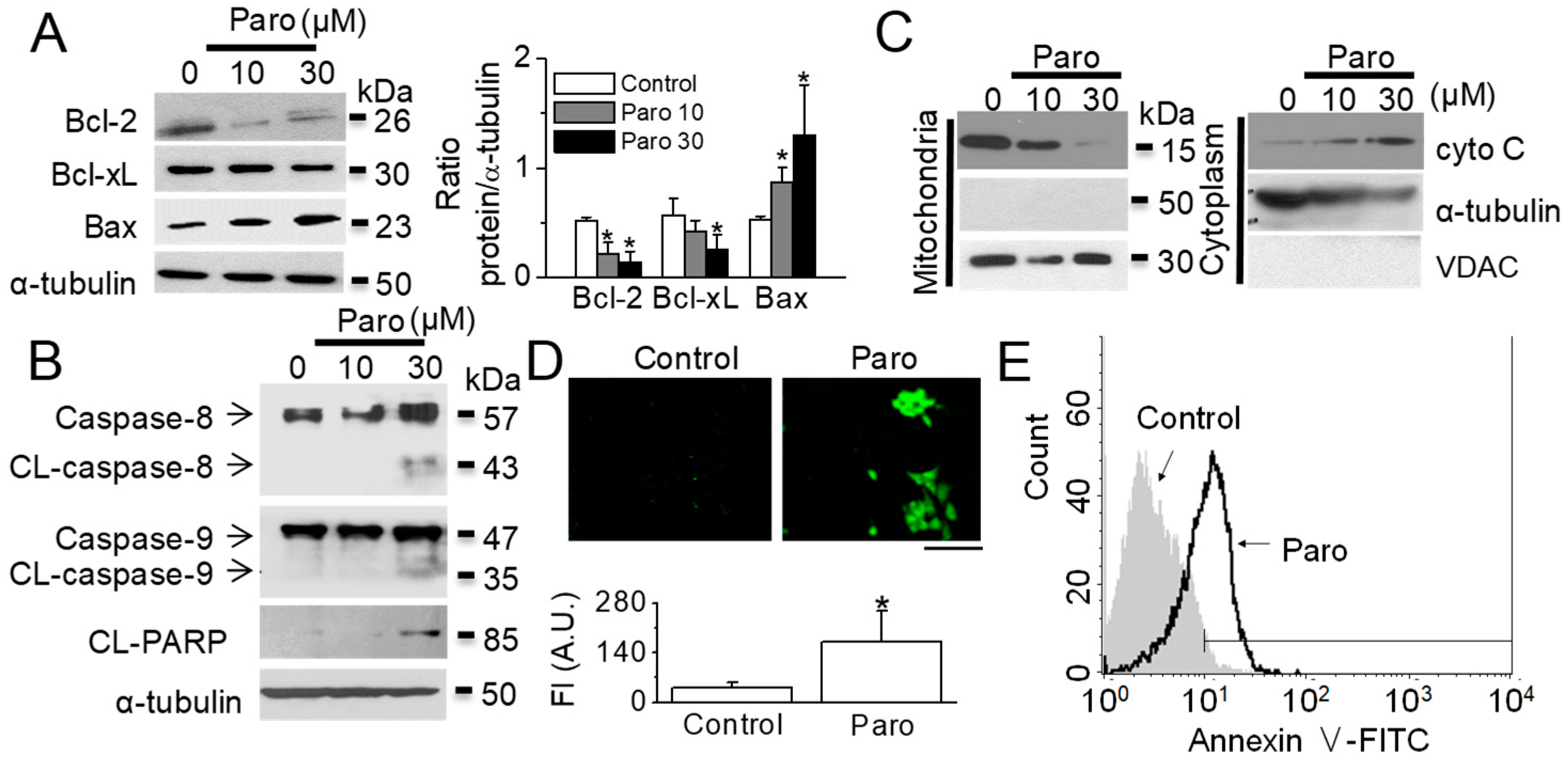
2.1. Apoptotic Death of Breast Cancer MCF-7 Cells Induced by Paroxetine Treatment
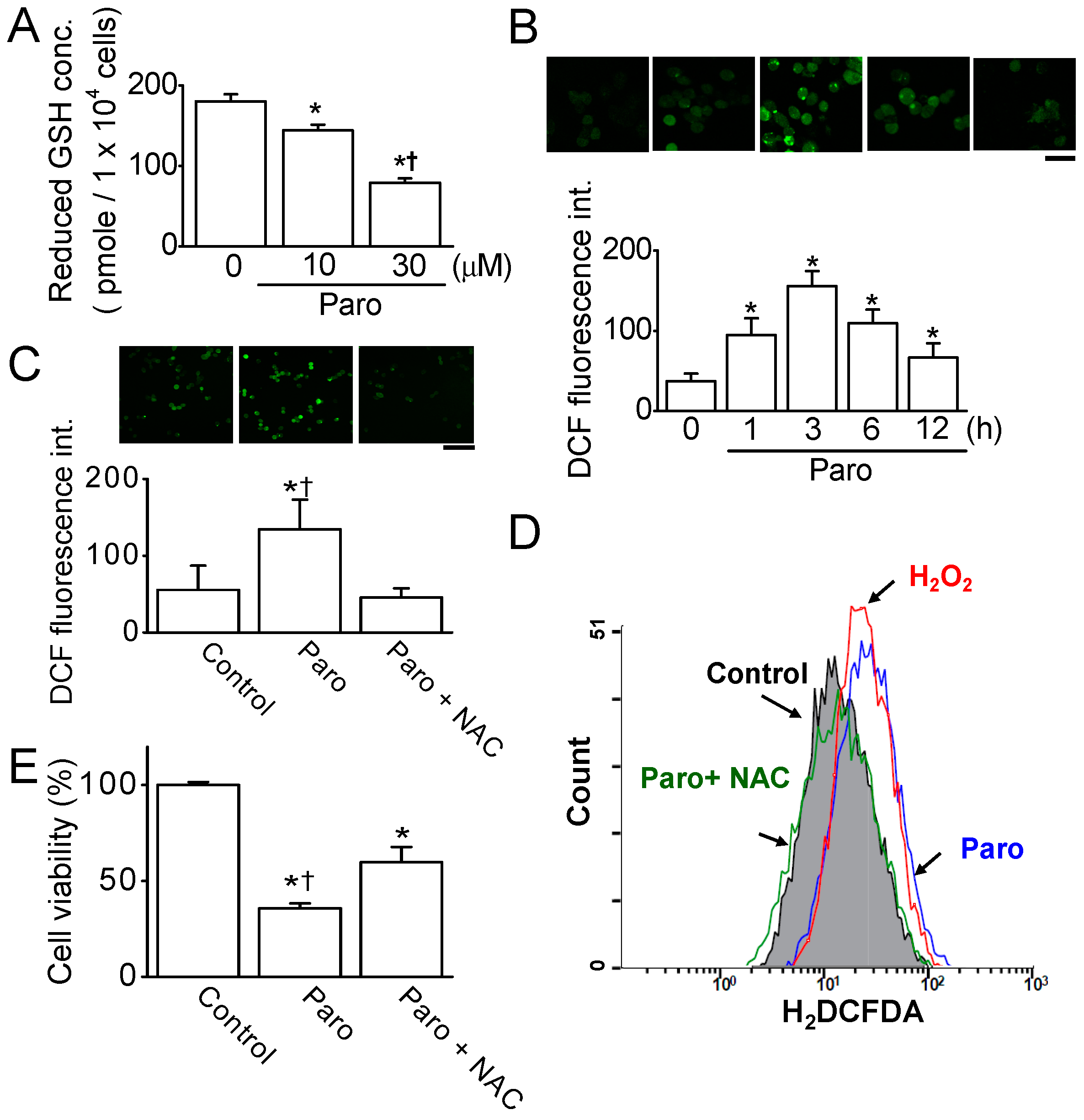
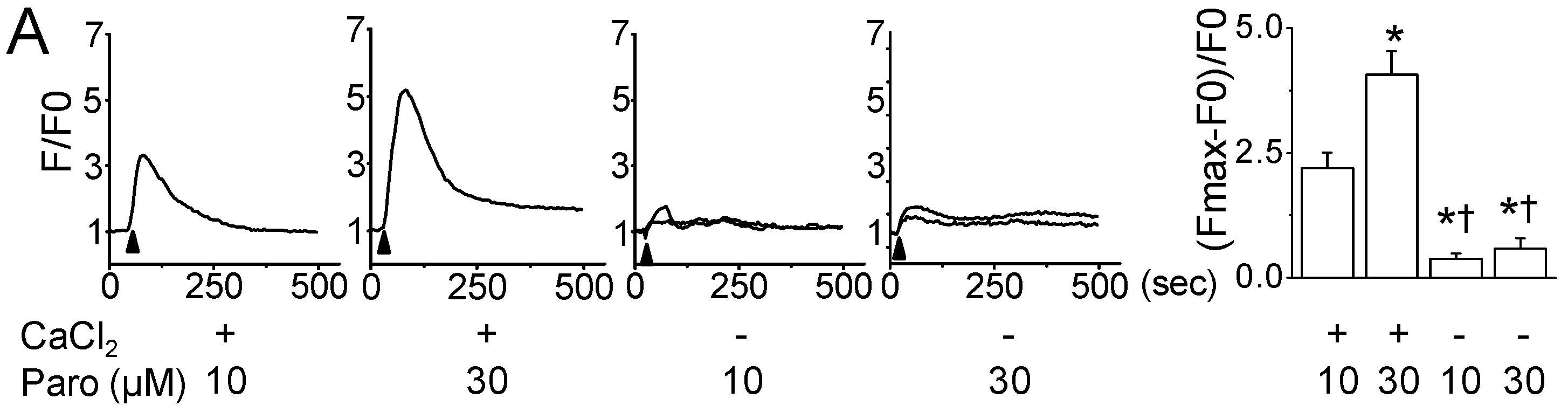
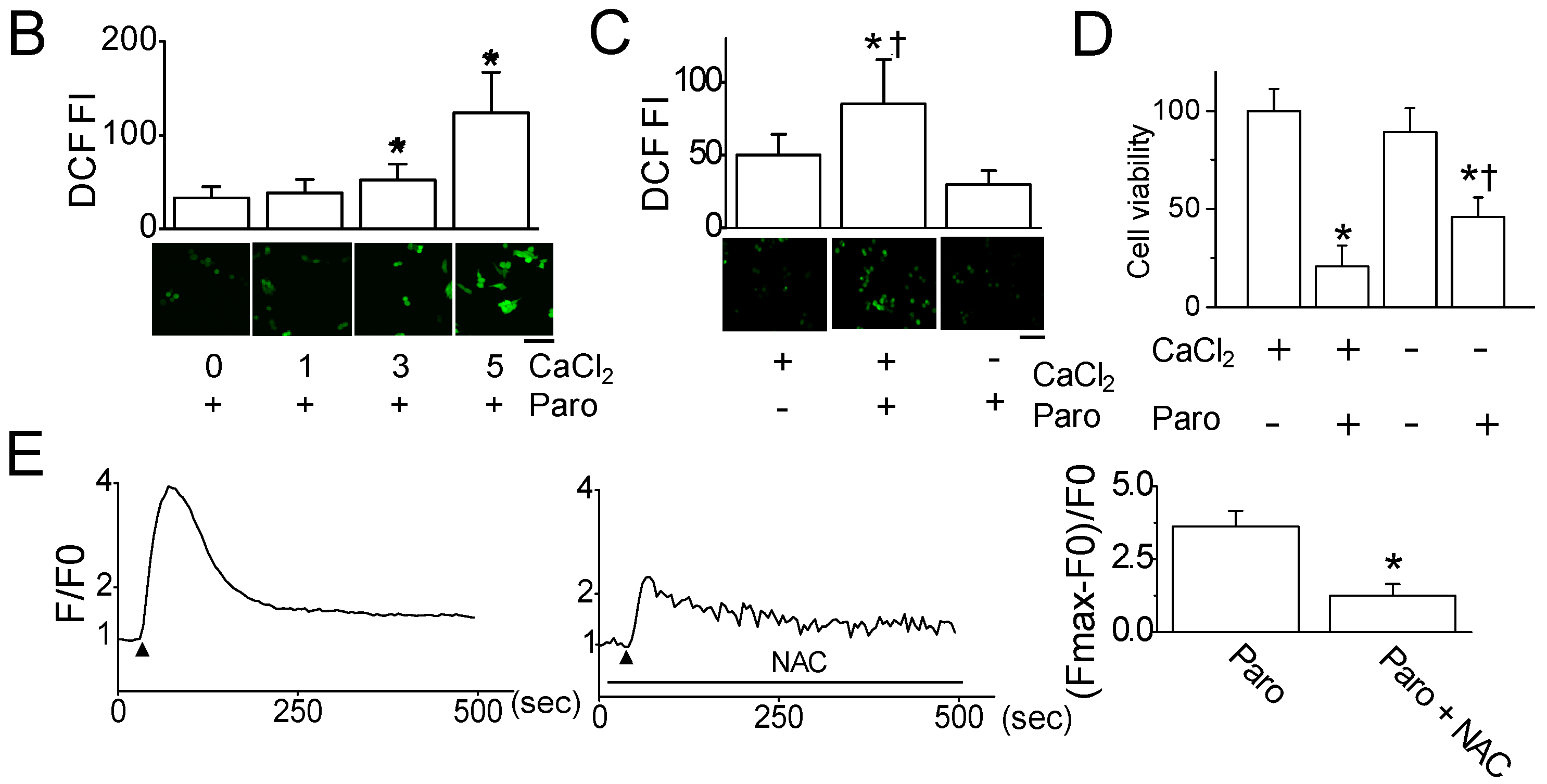
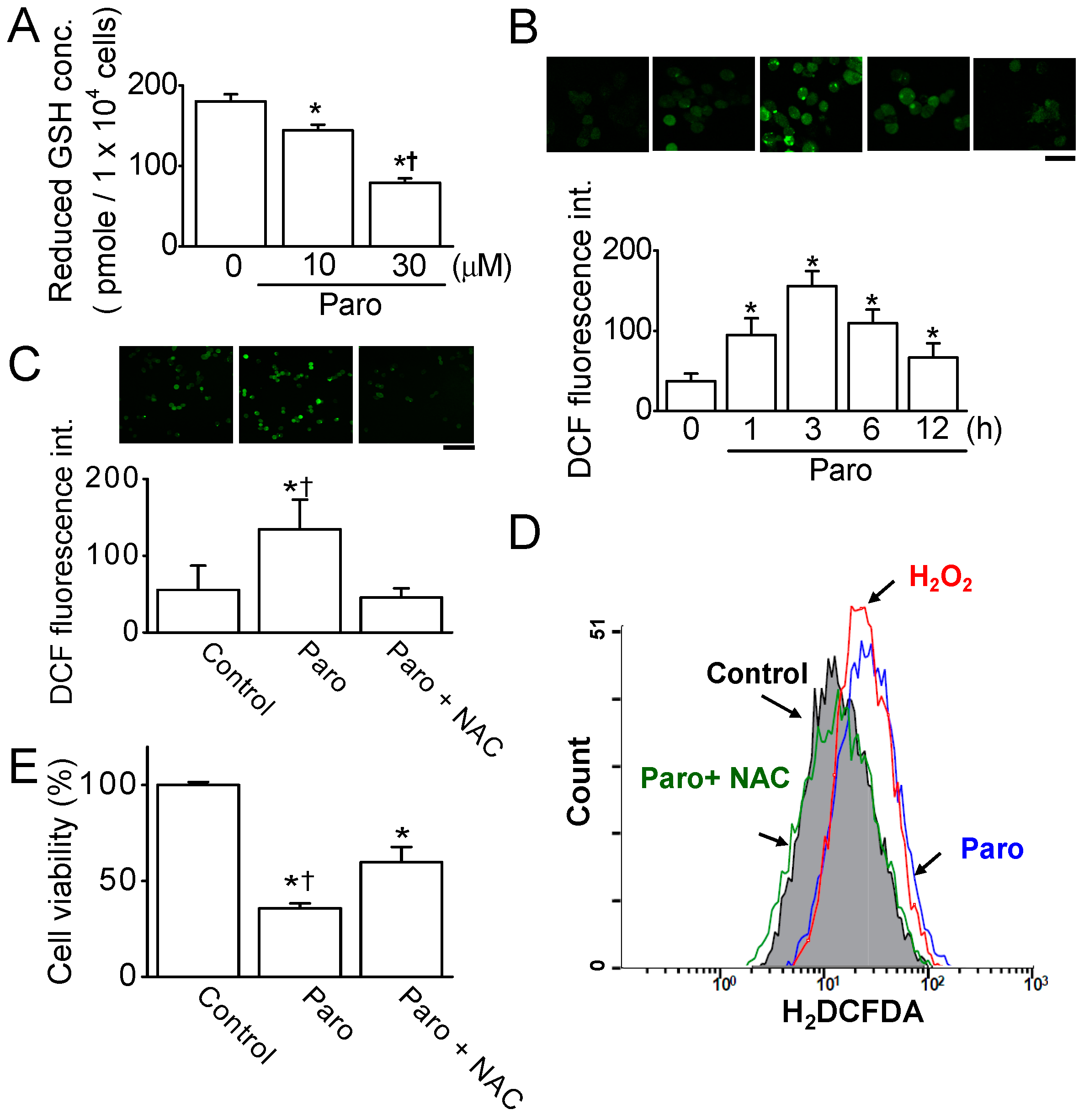
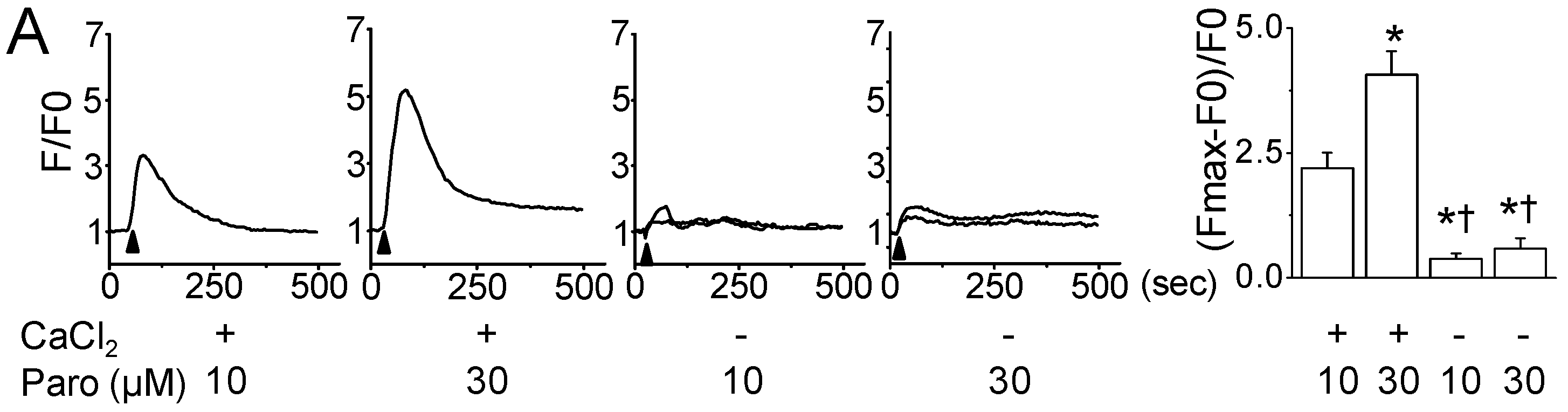
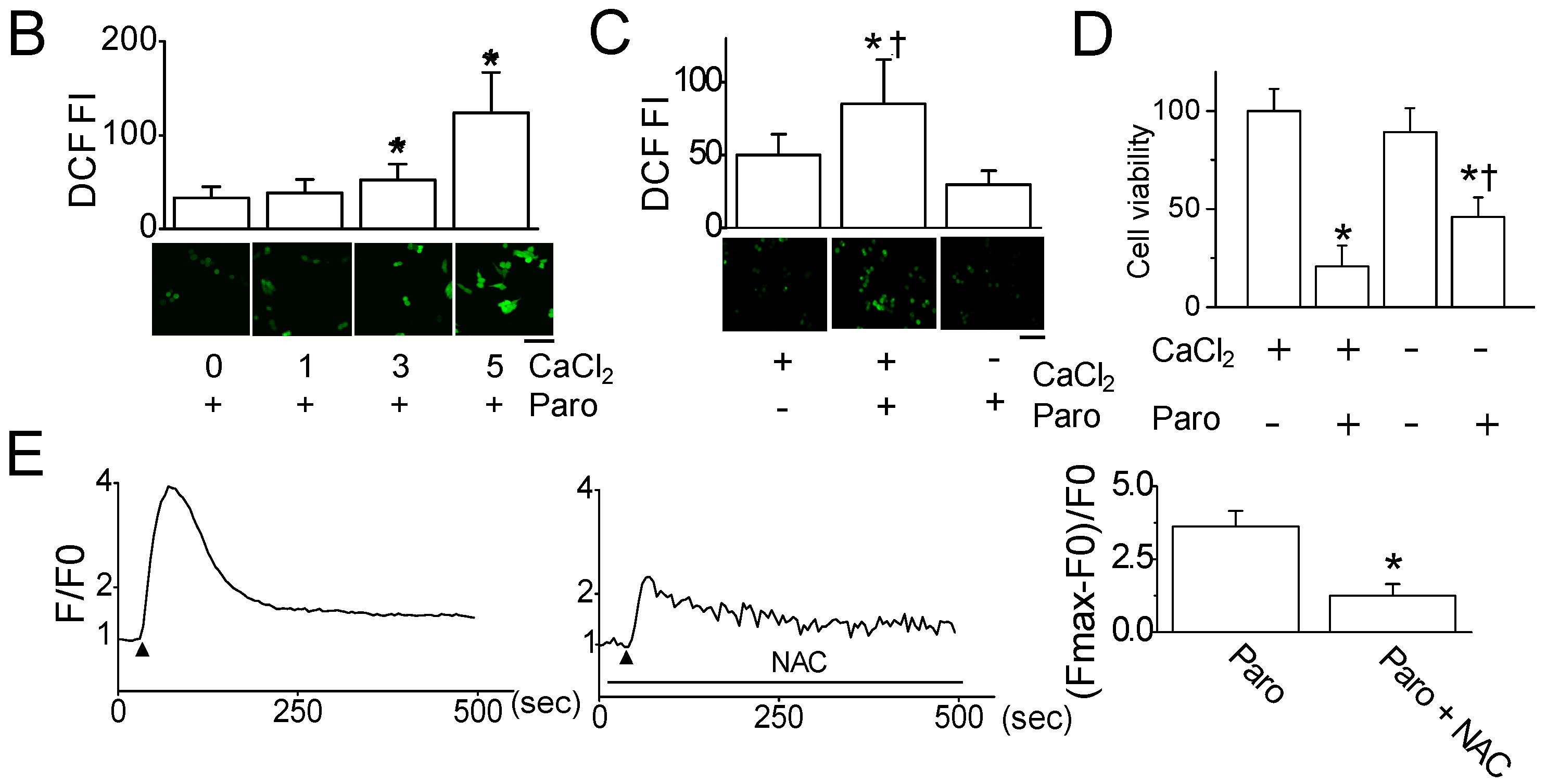
2.2. Paroxetine-Induced ROS Generation and Increase in Intracellular Ca2+ Levels
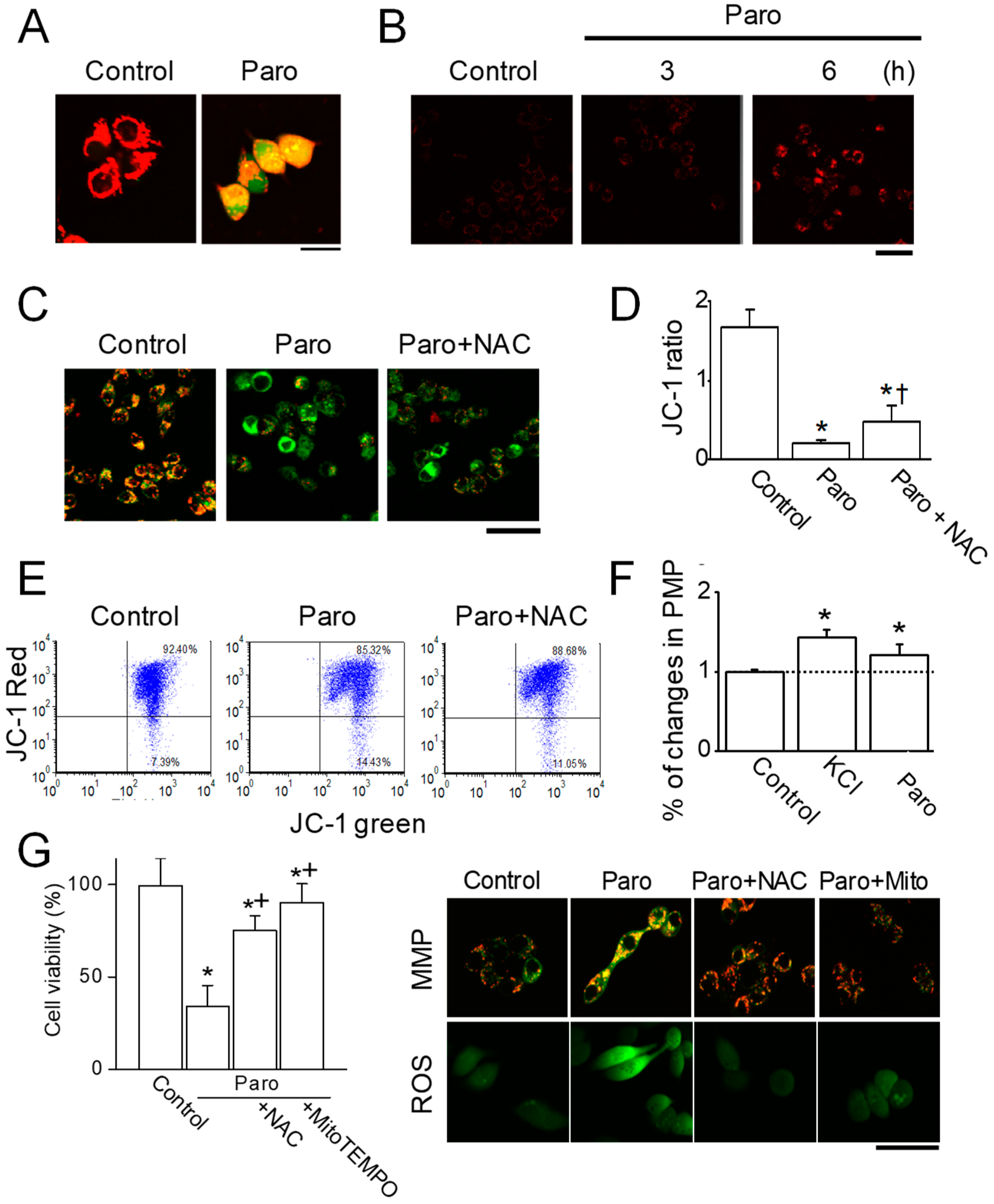
2.3. Paroxetine-Induced Mitochondrial Dysfunction in MCF-7 Cells
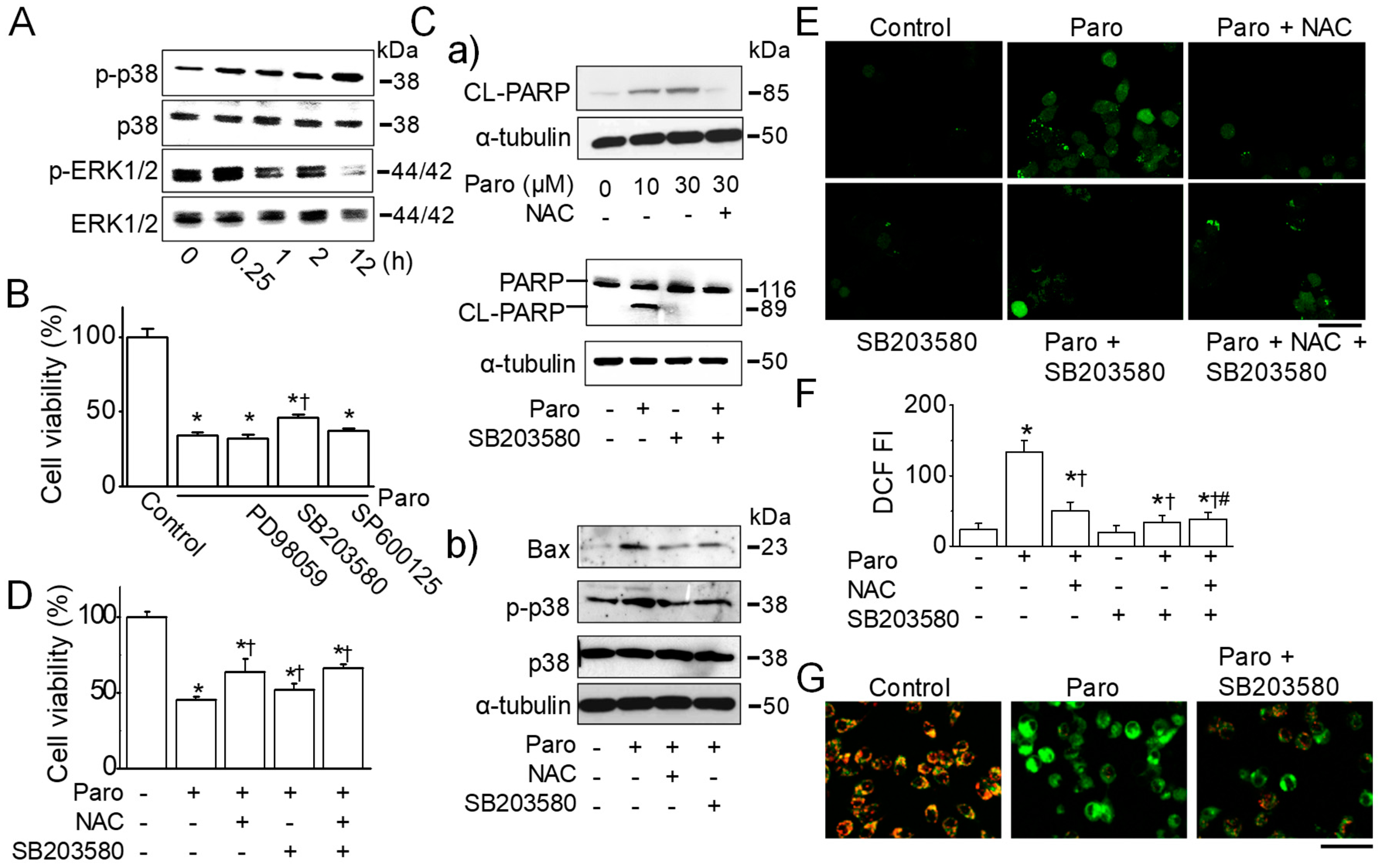
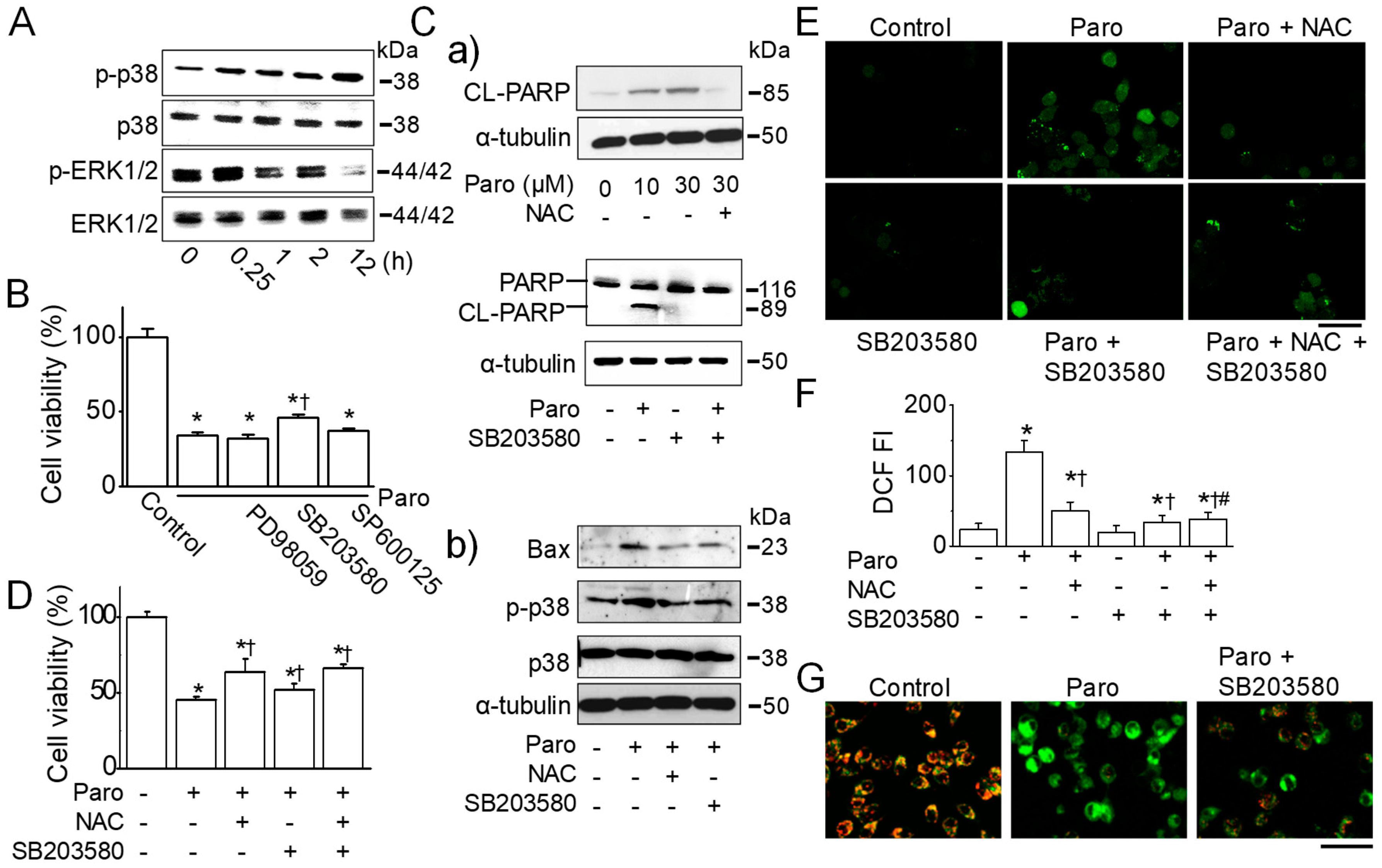
2.4. Paroxetine-Induced ROS Generation Is Due to the Activation of p38 MAPK
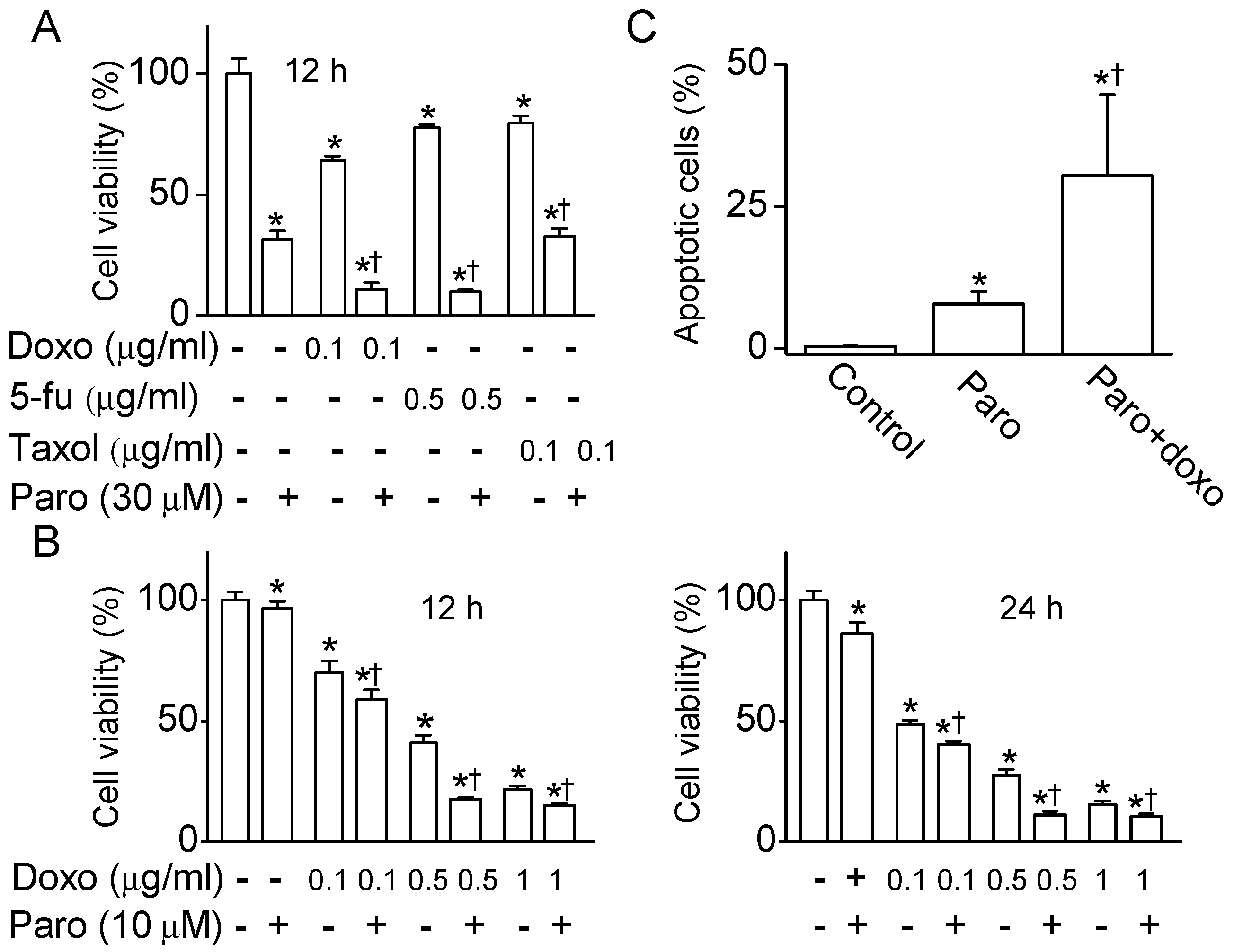
2.5. Synergistic Effect of Paroxetine on the MCF-7 Cell Death Induced by Anticancer Drugs
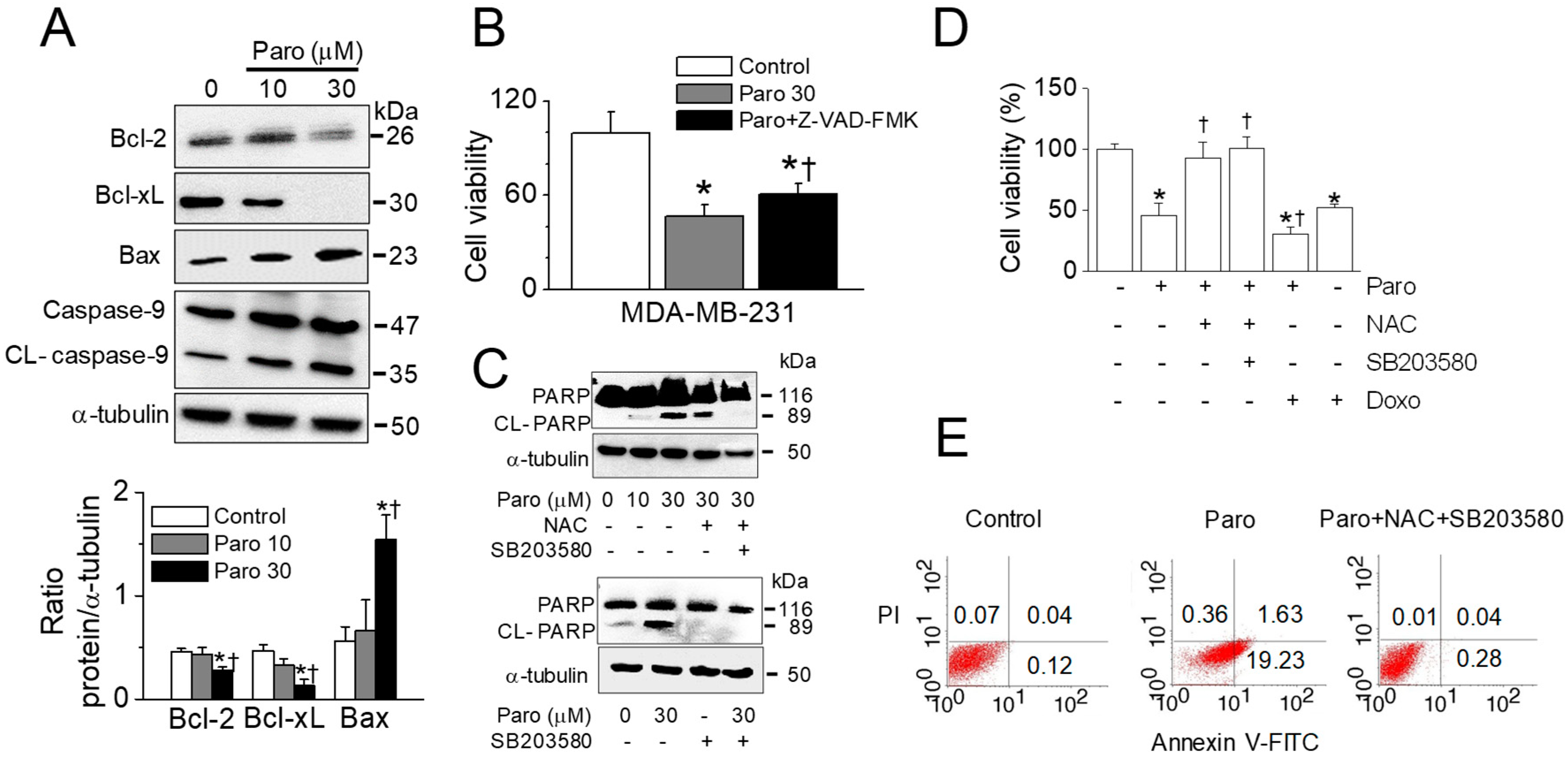
2.6. Apoptotic Death of Breast Cancer MDA-MB-231 Cells Induced by Paroxetine Treatment
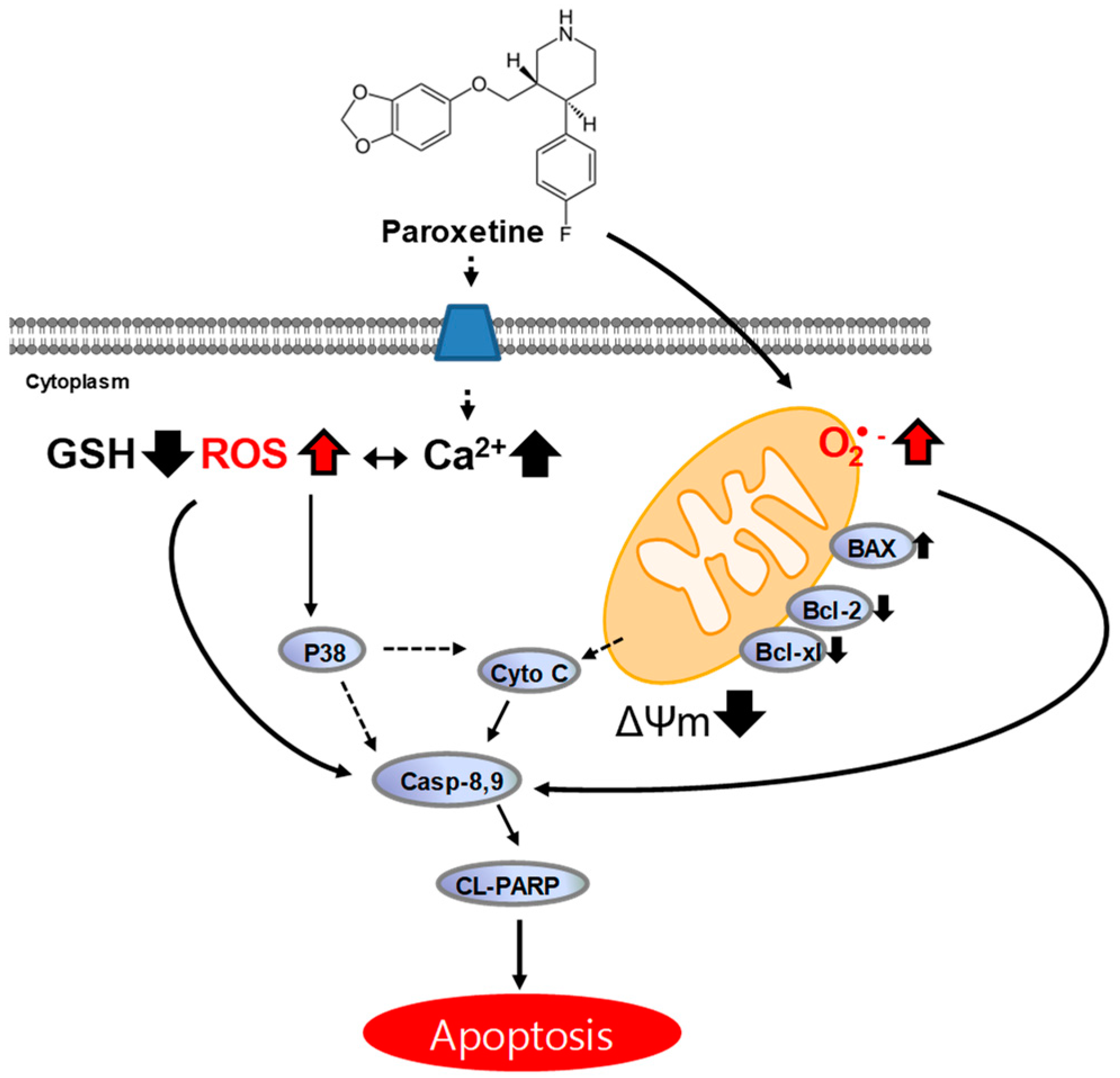
3. Discussion
3.1. Cytotoxic Effect of Paroxetine on Breast Cancer Cells
3.2. Potential Role of Paroxetine in Breast Cancer
4. Materials and Methods
4.1. Chemicals and Stock Solutions
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Flow Cytometry Analysis
4.5. Measurement of Caspase Activity
4.6. Measurement of Intracellular Ca2+ Concentration
4.7. Measurement of Intracellular Reactive Oxygen Species (ROS) Levels
4.8. Measurement of Glutathione (GSH) Concentration
4.9. Measurement of Mitochondrial ROS
4.10. Measurement of Mitochondrial Membrane Potential (MMP) and Plasma Membrane Potential
4.11. Western blot Analysis
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mincey, B.A.; Perez, E.A. Advances in screening, diagnosis, and treatment of breast cancer. Mayo Clin. Proc. 2004, 79, 810–816. [Google Scholar] [CrossRef]
- Massie, M.J. Prevalence of depression in patients with cancer. J. Natl. Cancer Inst. Monogr. 2004, 32, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Pitman, A.; Suleman, S.; Hyde, N.; Hodgkiss, A. Depression and anxiety in patients with cancer. BMJ 2018, 361, k1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostuzzi, G.; Matcham, F.; Dauchy, S.; Barbui, C.; Hotopf, M. Antidepressants for the treatment of depression in people with cancer. Cochrane Database Syst. Rev. 2018, 4, CD011006. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, D.; Venkateswaran, C.; Nayar, K.; Unnikrishnan, U.G. Prevalence of Depression in Breast Cancer Patients and its Association with their Quality of Life: A Cross-sectional Observational Study. Indian J. Palliat. Care 2017, 23, 268–273. [Google Scholar] [PubMed]
- Anxiety, Fear, and Depression. Available online: https://www.cancer.org/treatment/treatments-and-side-effects/emotional-side-effects/anxiety-fear-depression.html (accessed on 9 January 2019).
- Grassi, L.; Nanni, M.G.; Rodin, G.; Li, M.; Caruso, R. The use of antidepressants in oncology: A review and practical tips for oncologists. Ann. Oncol. 2018, 29, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Frick, L.R.; Rapanelli, M. Antidepressants: Influence on cancer and immunity? Life Sci. 2013, 92, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, L.; Shi, L.; Creasey, D.E.; Anaya-McKivergan, M.; Myers, J.A.; Huybrechts, K.F. Antidepressants and breast and ovarian cancer risk: A review of the literature and researchers’ financial associations with industry. PLoS ONE 2011, 6, e18210. [Google Scholar] [CrossRef] [PubMed]
- Janicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat. 2009, 117, 219–221. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Nicco, C.; Batteux, F. ROS Modulator Molecules with Therapeutic Potential in Cancers Treatments. Molecules 2017, 23, 84. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Trebak, M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium 2017, 63, 70–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scanlon, J.M.; Reynolds, I.J. Effects of oxidants and glutamate receptor activation on mitochondrial membrane potential in rat forebrain neurons. J. Neurochem. 1998, 71, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Liao, X.; Mo, L.; Yang, C.; Yang, Z.; Wang, X.; Hu, F.; Chen, P.; Feng, J.; Zheng, D.; et al. Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS ONE 2011, 6, e25921. [Google Scholar] [CrossRef] [PubMed]
- Abdul, M.; Logothetis, C.J.; Hoosein, N.M. Growth-inhibitory effects of serotonin uptake inhibitors on human prostate carcinoma cell lines. J. Urol. 1995, 154, 247–250. [Google Scholar] [CrossRef]
- Rosetti, M.; Frasnelli, M.; Tesei, A.; Zoli, W.; Conti, M. Cytotoxicity of different selective serotonin reuptake inhibitors (SSRIs) against cancer cells. J. Exp. Ther. Oncol. 2006, 6, 23–29. [Google Scholar] [PubMed]
- Schuster, C.; Fernbach, N.; Rix, U.; Superti-Furga, G.; Holy, M.; Freissmuth, M.; Sitte, H.H.; Sexl, V. Selective serotonin reuptake inhibitors—A new modality for the treatment of lymphoma/leukaemia? Biochem. Pharmacol. 2007, 74, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, J.; Yamada, T.; Egashira, N.; Ueda, M.; Zukeyama, N.; Ushio, S.; Masuda, S. Comparison of the Anti-tumor Effects of Selective Serotonin Reuptake Inhibitors as Well as Serotonin and Norepinephrine Reuptake Inhibitors in Human Hepatocellular Carcinoma Cells. Biol. Pharm. Bull. 2015, 38, 1410–1414. [Google Scholar] [CrossRef] [Green Version]
- Gil-Ad, I.; Zolokov, A.; Lomnitski, L.; Taler, M.; Bar, M.; Luria, D.; Ram, E.; Weizman, A. Evaluation of the potential anti-cancer activity of the antidepressant sertraline in human colon cancer cell lines and in colorectal cancer-xenografted mice. Int. J. Oncol. 2008, 33, 277–286. [Google Scholar] [CrossRef]
- Levkovitz, Y.; Gil-Ad, I.; Zeldich, E.; Dayag, M.; Weizman, A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: Evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. J. Mol. Neurosci. 2005, 27, 29–42. [Google Scholar] [CrossRef]
- Chou, C.T.; He, S.; Jan, C.R. Paroxetine-induced apoptosis in human osteosarcoma cells: Activation of p38 MAP kinase and caspase-3 pathways without involvement of [Ca2+]i elevation. Toxicol. Appl. Pharmacol. 2007, 218, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, F.; Singh, S.V.; Wang, Q.J. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res. 2008, 68, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.C.; Chou, C.T.; Pan, C.C.; Hsieh, Y.D.; Liang, W.Z.; Chao, D.; Tsai, J.Y.; Liao, W.C.; Kuo, D.H.; Shieh, P.; et al. Paroxetine-induced Ca2+ movement and death in OC2 human oral cancer cells. Chin. J. Physiol. 2011, 54, 310–317. [Google Scholar] [PubMed]
- Then, C.K.; Liu, K.H.; Liao, M.H.; Chung, K.H.; Wang, J.Y.; Shen, S.C. Antidepressants, sertraline and paroxetine, increase calcium influx and induce mitochondrial damage-mediated apoptosis of astrocytes. Oncotarget 2017, 8, 115490–115502. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef]
- Yermolaieva, O.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Hoshi, T. Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Voeikov, V. Reactive oxygen species, water, photons and life. Riv. Biol. 2001, 94, 237–258. [Google Scholar]
- Kang, K.R.; Cho, Y.W.; Ryu, J.H.; Kim, E.J.; Han, J.; Kang, D. Paroxetine induces apoptosis in human breast cancer MCF-7 cells through mitochondrial dysfunction and modulation of K+ channels. In Proceedings of the AACR 107th Annual Meeting 2016, New Orleans, LA, USA, 16–20 April 2016. [Google Scholar]
- Annunziato, L.; Amoroso, S.; Pannaccione, A.; Cataldi, M.; Pignataro, G.; D’Alessio, A.; Sirabella, R.; Secondo, A.; Sibaud, L.; Di Renzo, G.F. Apoptosis induced in neuronal cells by oxidative stress: Role played by caspases and intracellular calcium ions. Toxicol. Lett. 2003, 139, 125–133. [Google Scholar] [CrossRef]
- Kumar, D.; Jugdutt, B.I. Apoptosis and oxidants in the heart. J. Lab. Clin. Med. 2003, 142, 288–297. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cano, J.; Roche, O.; Cimas, F.J.; Pascual-Serra, R.; Ortega-Muelas, M.; Fernandez-Aroca, D.M.; Sanchez-Prieto, R. p38MAPK and Chemotherapy: We Always Need to Hear Both Sides of the Story. Front. Cell Dev. Biol. 2016, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.Y.; Park, J.H.; Paik, H.D.; Nah, S.Y.; Kim, D.S.; Han, Y.S. Acacetin-induced apoptosis of human breast cancer MCF-7 cells involves caspase cascade, mitochondria-mediated death signaling and SAPK/JNK1/2-c-Jun activation. Mol. Cells 2007, 24, 95–104. [Google Scholar]
- Arkin, M. Protein-protein interactions and cancer: Small molecules going in for the kill. Curr. Opin. Chem. Biol. 2005, 9, 317–324. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; Cheer, S.M.; Matheson, A.J.; Ormrod, D.; Goa, K.L. Paroxetine: An update of its use in psychiatric disorders in adults. Drugs 2002, 62, 655–703. [Google Scholar] [CrossRef]
- Nevels, R.M.; Gontkovsky, S.T.; Williams, B.E. Paroxetine-The Antidepressant from Hell? Probably Not, But Caution Required. Psychopharmacol. Bull. 2016, 46, 77–104. [Google Scholar] [PubMed]
- Carrasco, J.L.; Sandner, C. Clinical effects of pharmacological variations in selective serotonin reuptake inhibitors: An overview. Int. J. Clin. Pract. 2005, 59, 1428–1434. [Google Scholar] [CrossRef]
- Negroni, M.P.; Fiszman, G.L.; Azar, M.E.; Morgado, C.C.; Espanol, A.J.; Pelegrina, L.T.; de la Torre, E.; Sales, M.E. Immunoglobulin G from breast cancer patients in stage I stimulates muscarinic acetylcholine receptors in MCF7 cells and induces proliferation. Participation of nitric oxide synthase-derived nitric oxide. J. Clin. Immunol. 2010, 30, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Haque, R.; Shi, J.; Schottinger, J.E.; Ahmed, S.A.; Cheetham, T.C.; Chung, J.; Avila, C.; Kleinman, K.; Habel, L.A.; Fletcher, S.W.; et al. Tamoxifen and Antidepressant Drug Interaction in a Cohort of 16,887 Breast Cancer Survivors. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Roy-Byrne, P. Antidepressants Do Not Impair the Anticancer Effects of Tamoxifen. NEJM J. Watch 2016. Available online: https://www.jwatch.org/na39889/2016/01/04/antidepressants-do-not-impair-anticancer-effects-tamoxifen (accessed on 9 January 2019).
- Donneyong, M.M.; Bykov, K.; Bosco-Levy, P.; Dong, Y.H.; Levin, R.; Gagne, J.J. Risk of mortality with concomitant use of tamoxifen and selective serotonin reuptake inhibitors: Multi-database cohort study. BMJ 2016, 354, i5014. [Google Scholar] [CrossRef]
- Tomita, T.; Yasui-Furukori, N.; Nakagami, T.; Tsuchimine, S.; Ishioka, M.; Kaneda, A.; Nakamura, K.; Kaneko, S. Therapeutic reference range for plasma concentrations of paroxetine in patients with major depressive disorders. Ther. Drug Monit. 2014, 36, 480–485. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, Y.-W.; Kim, E.-J.; Nyiramana, M.M.; Shin, E.-J.; Jin, H.; Ryu, J.H.; Kang, K.R.; Lee, G.-W.; Kim, H.J.; Han, J.; et al. Paroxetine Induces Apoptosis of Human Breast Cancer MCF-7 Cells through Ca2+-and p38 MAP Kinase-Dependent ROS Generation. Cancers 2019, 11, 64. https://doi.org/10.3390/cancers11010064
Cho Y-W, Kim E-J, Nyiramana MM, Shin E-J, Jin H, Ryu JH, Kang KR, Lee G-W, Kim HJ, Han J, et al. Paroxetine Induces Apoptosis of Human Breast Cancer MCF-7 Cells through Ca2+-and p38 MAP Kinase-Dependent ROS Generation. Cancers. 2019; 11(1):64. https://doi.org/10.3390/cancers11010064
Chicago/Turabian StyleCho, Young-Woo, Eun-Jin Kim, Marie Merci Nyiramana, Eui-Jung Shin, Hana Jin, Ji Hyeon Ryu, Kee Ryeon Kang, Gyeong-Won Lee, Hye Jung Kim, Jaehee Han, and et al. 2019. "Paroxetine Induces Apoptosis of Human Breast Cancer MCF-7 Cells through Ca2+-and p38 MAP Kinase-Dependent ROS Generation" Cancers 11, no. 1: 64. https://doi.org/10.3390/cancers11010064