Proliferative and Invasive Colorectal Tumors in Pet Dogs Provide Unique Insights into Human Colorectal Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
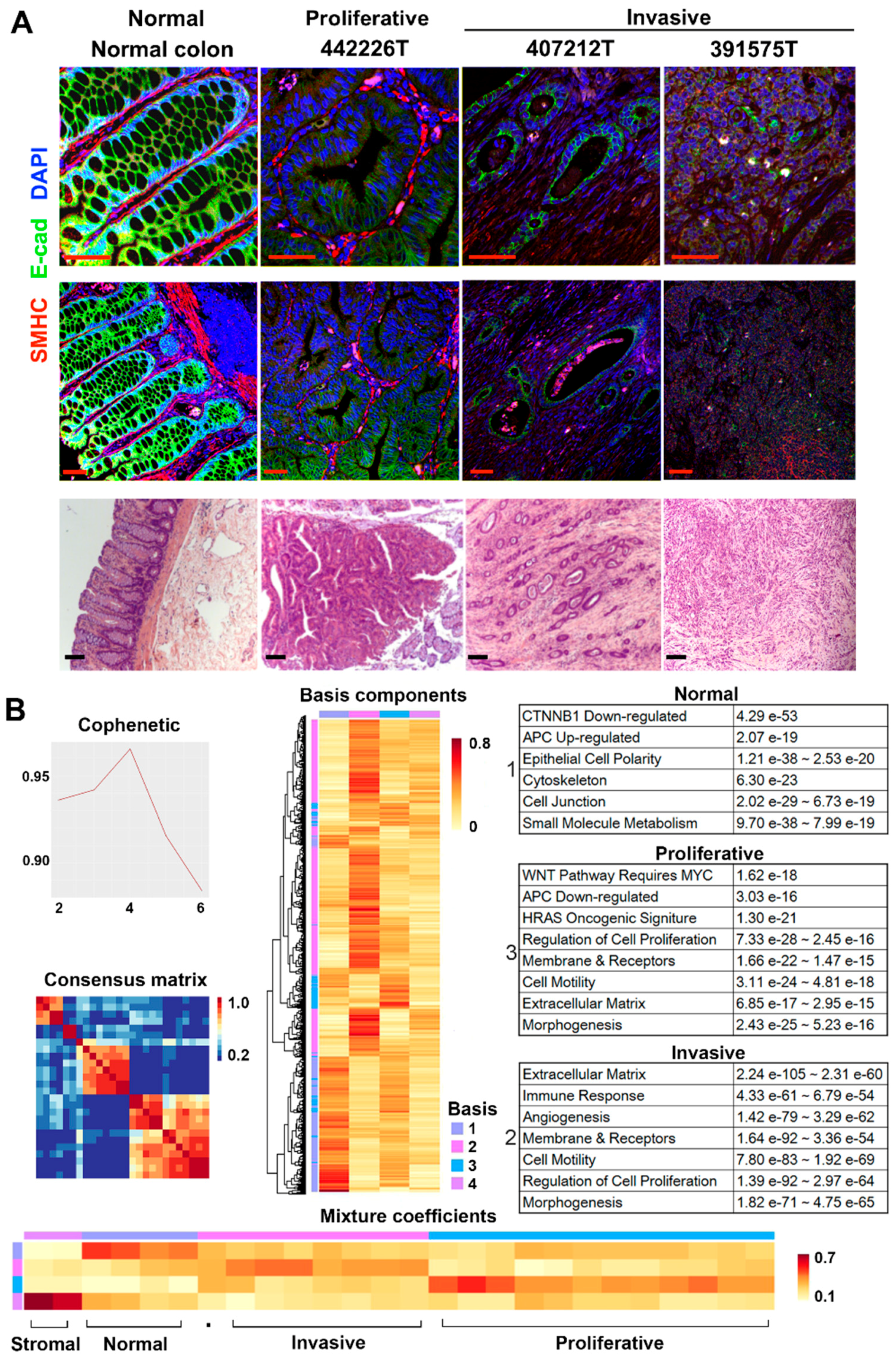
2.1. RNA-Seq Analysis Clusters the Tumors into Two Major Groups
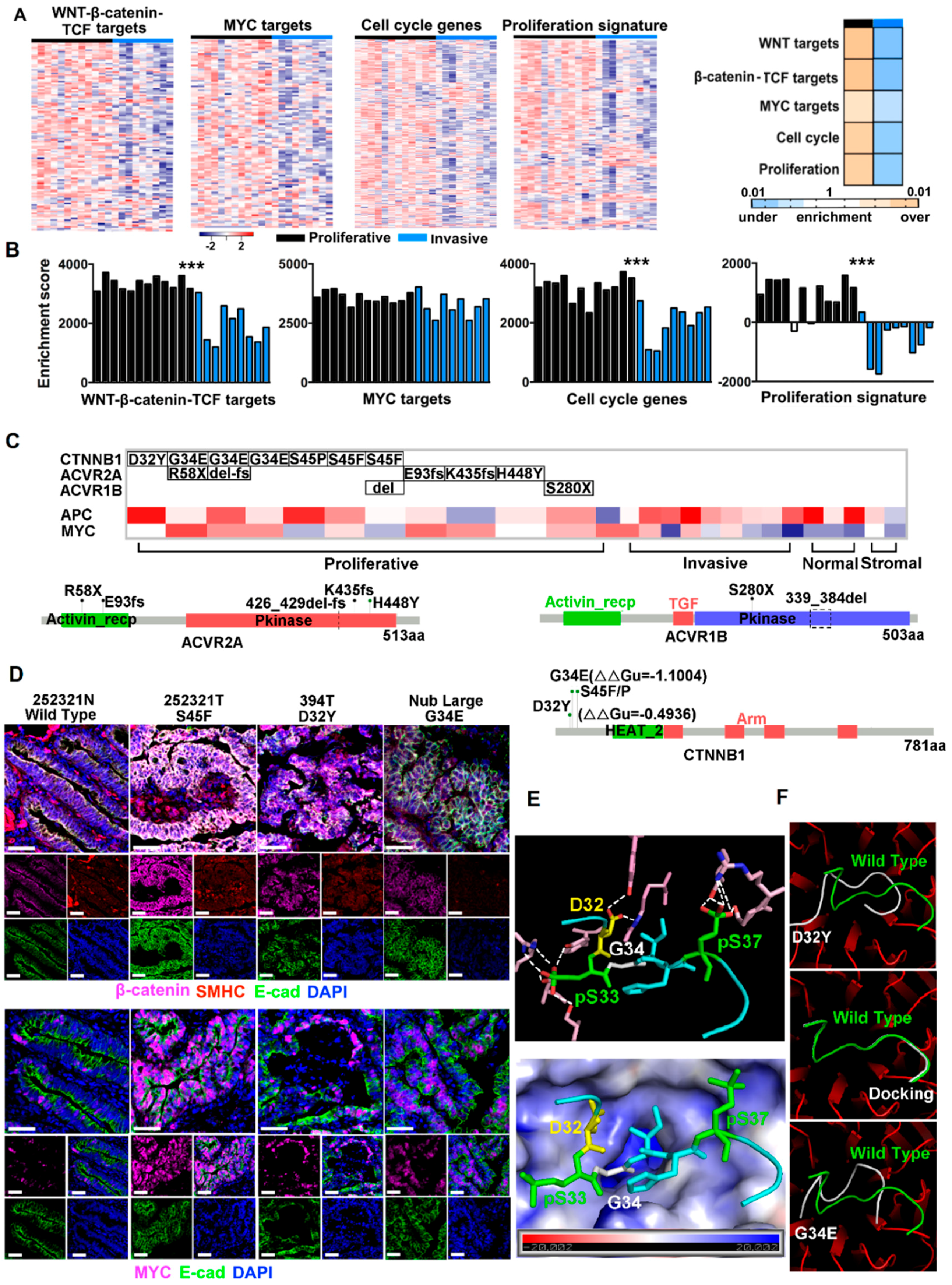
2.2. Canonical CRC Pathways Are Activated in Proliferative Tumors
2.3. CTNNB1 and TGF-β Signaling Genes Were Recurrently Mutated in Proliferative Tumors
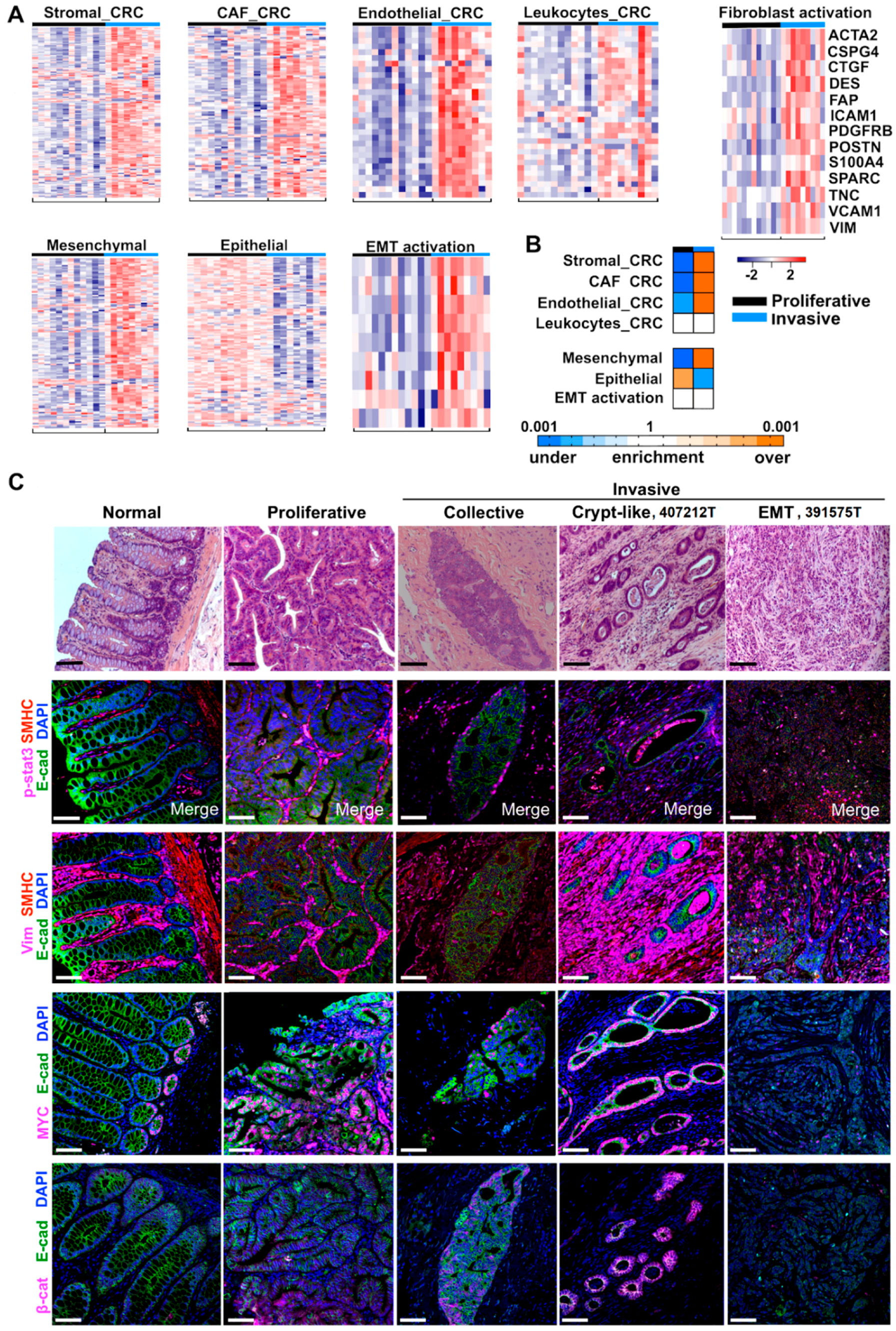
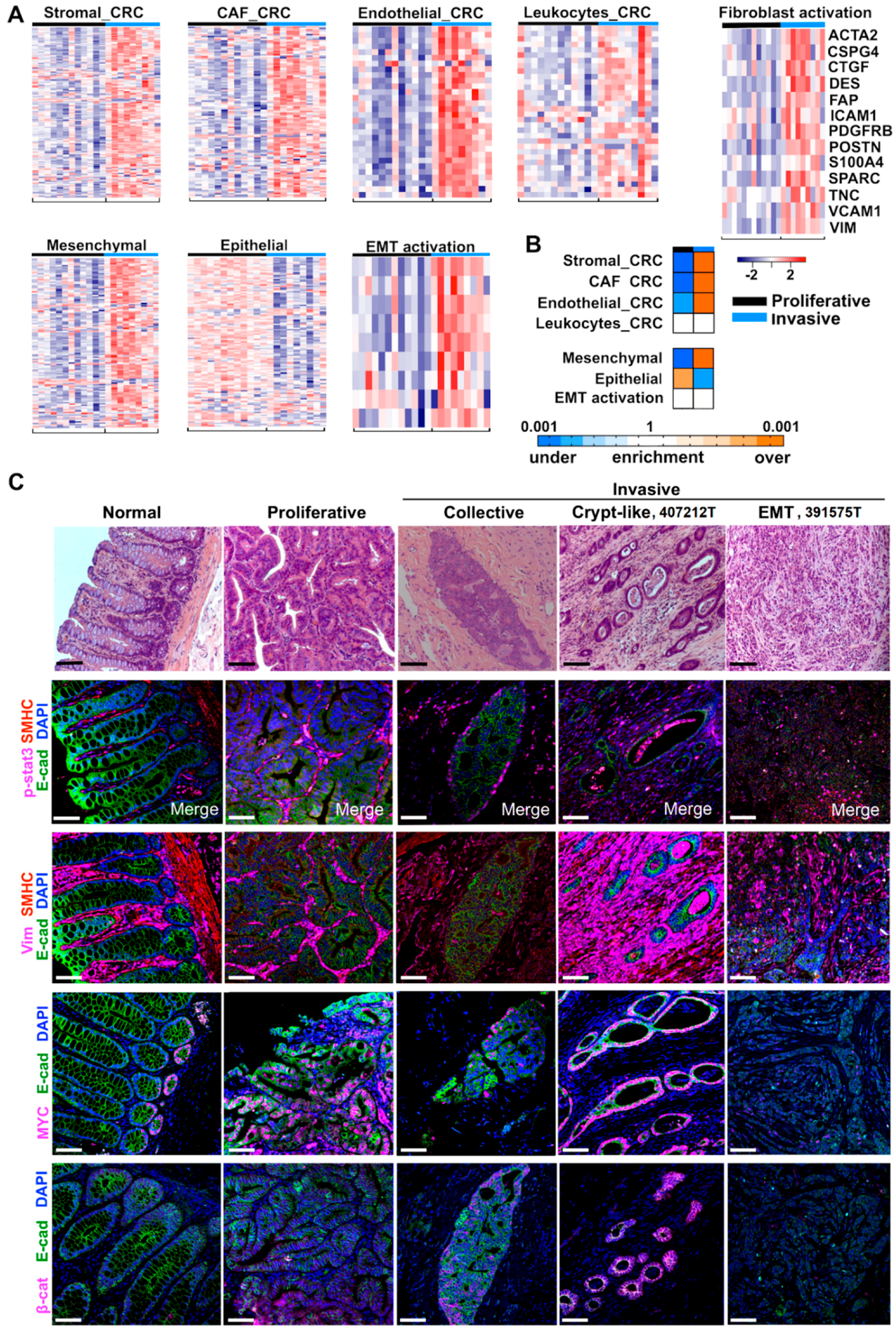
2.4. Cancer-Associated Fibroblast (CAF) and Stromal Signatures Are Activated in Invasive Tumors
2.5. Three Modes of Cancer Cell Invasion Were Observed
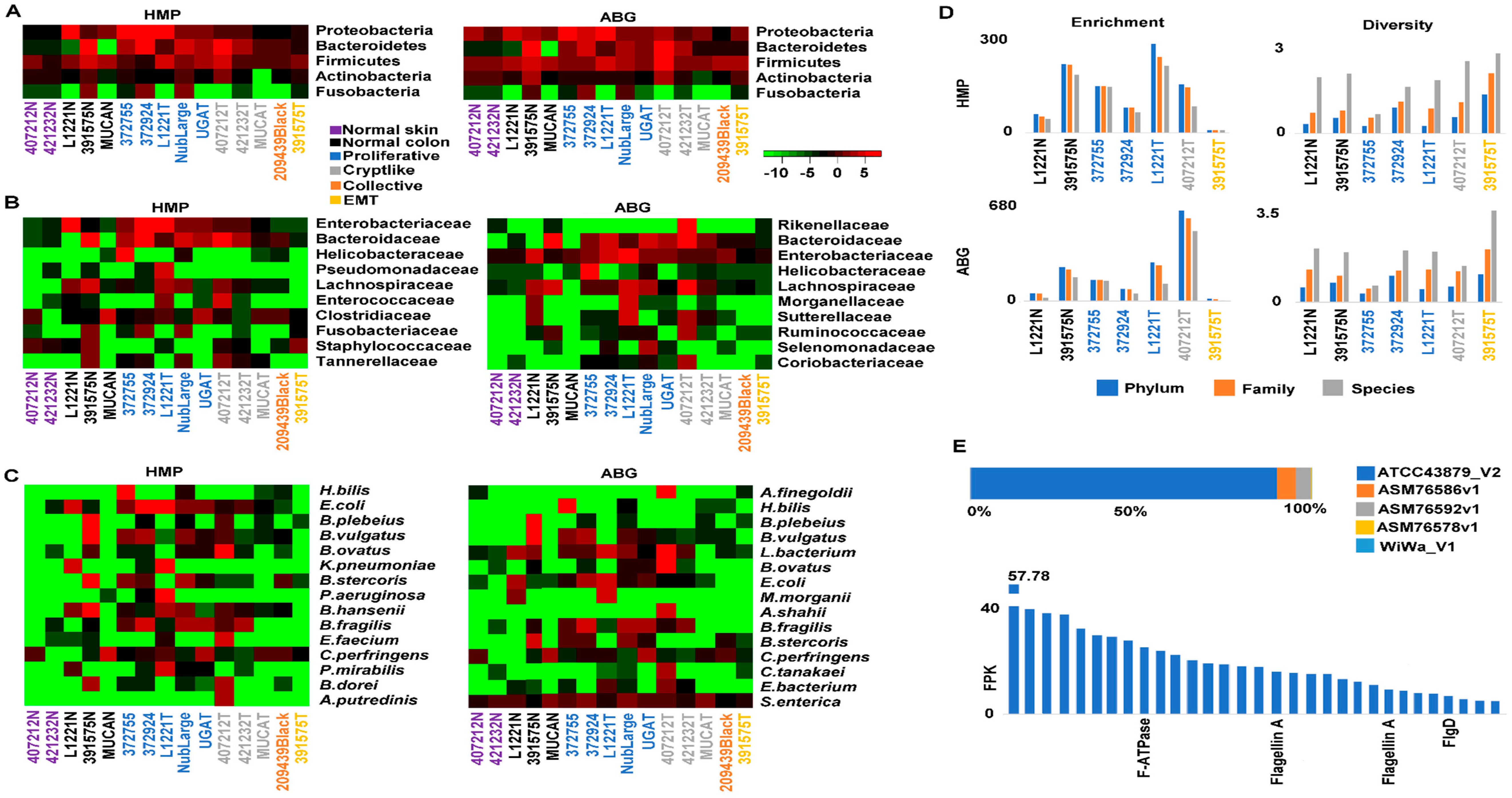
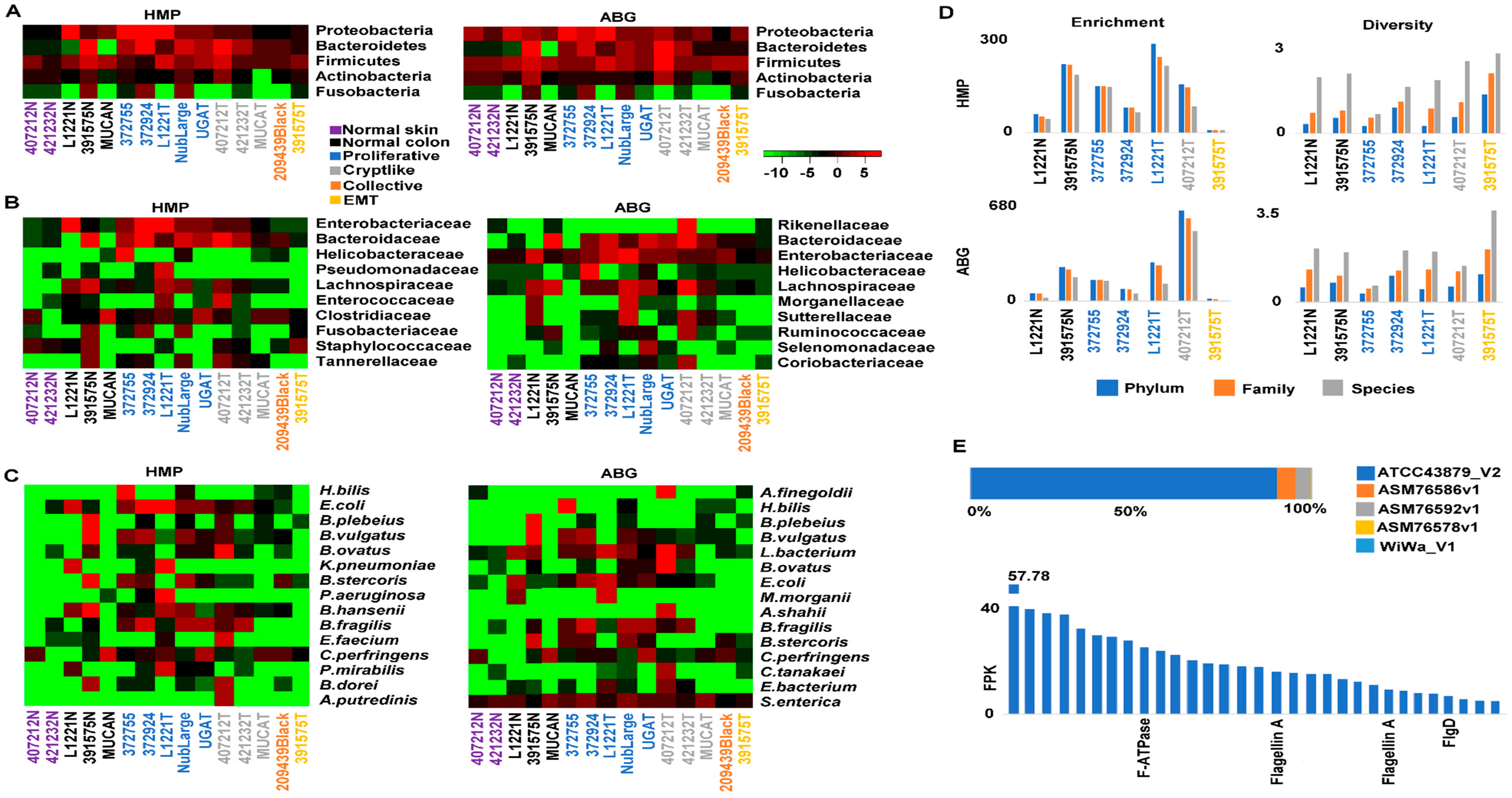
2.6. Crypt-Like Invasion Tumor Harbors Mucosa-Like Microbiome
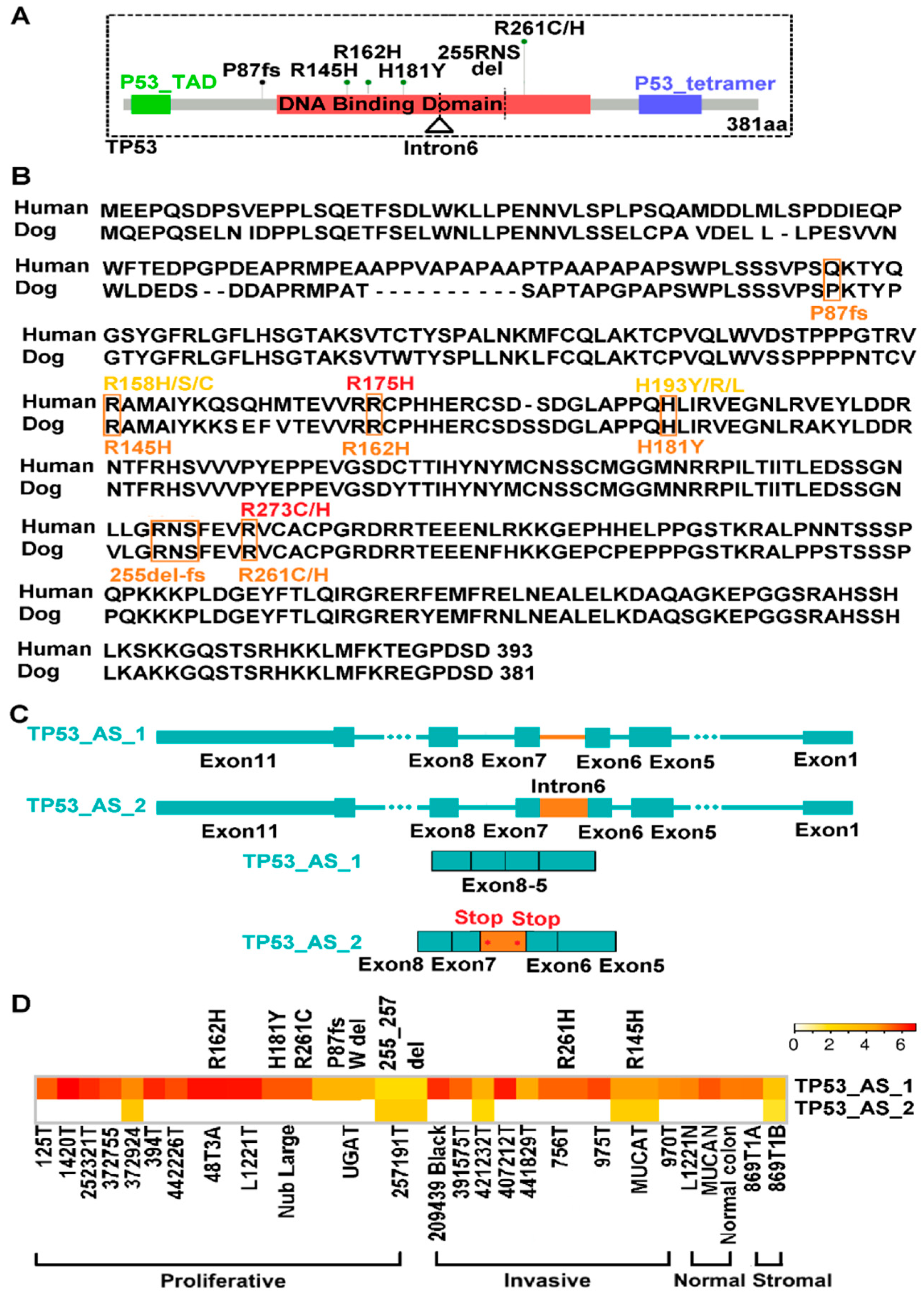
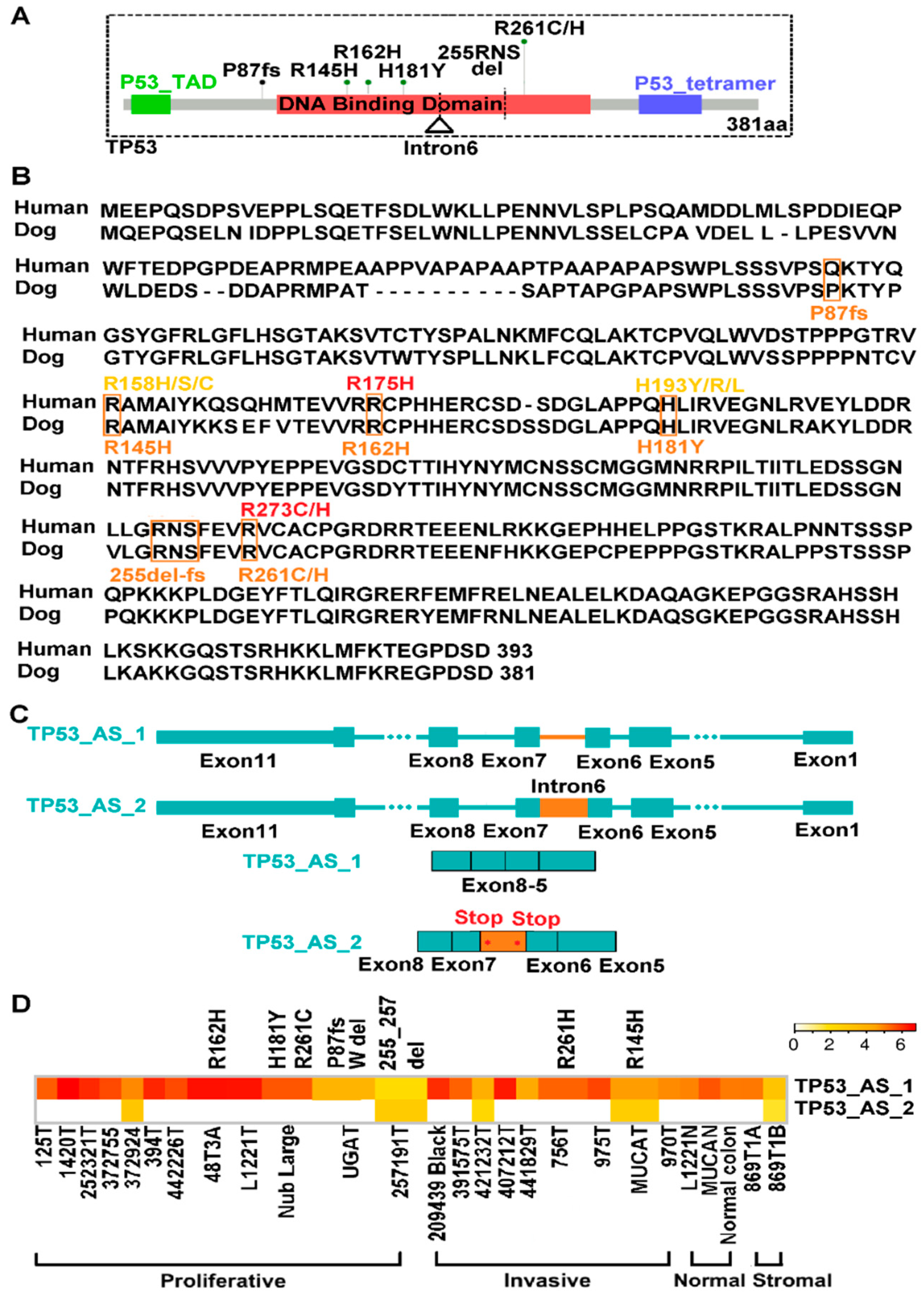
2.7. TP53 Is Recurrently Altered in Both Proliferative and Invasive Tumors
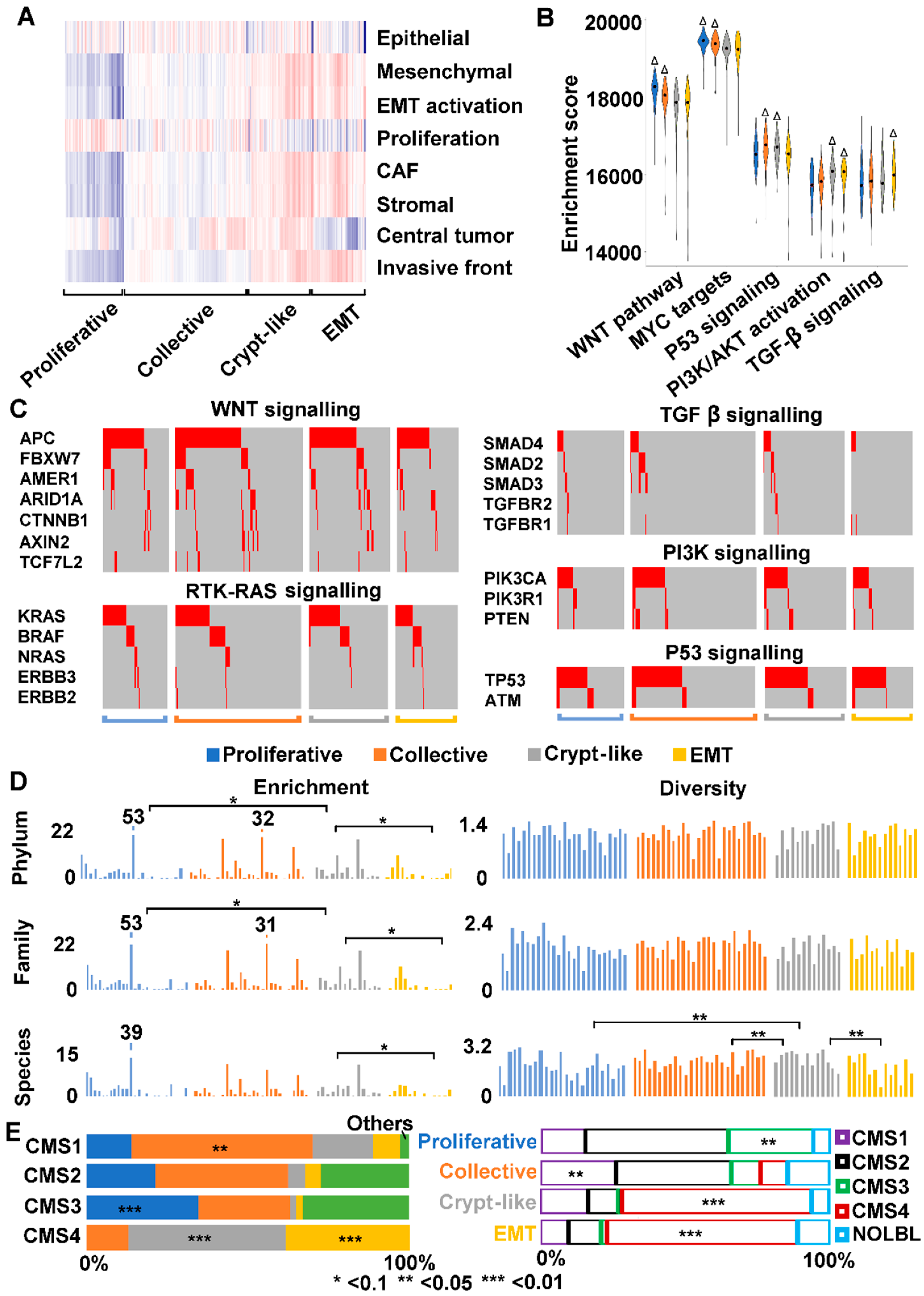
2.8. We Identified Three Types of Invasion in Human Colon Cancers
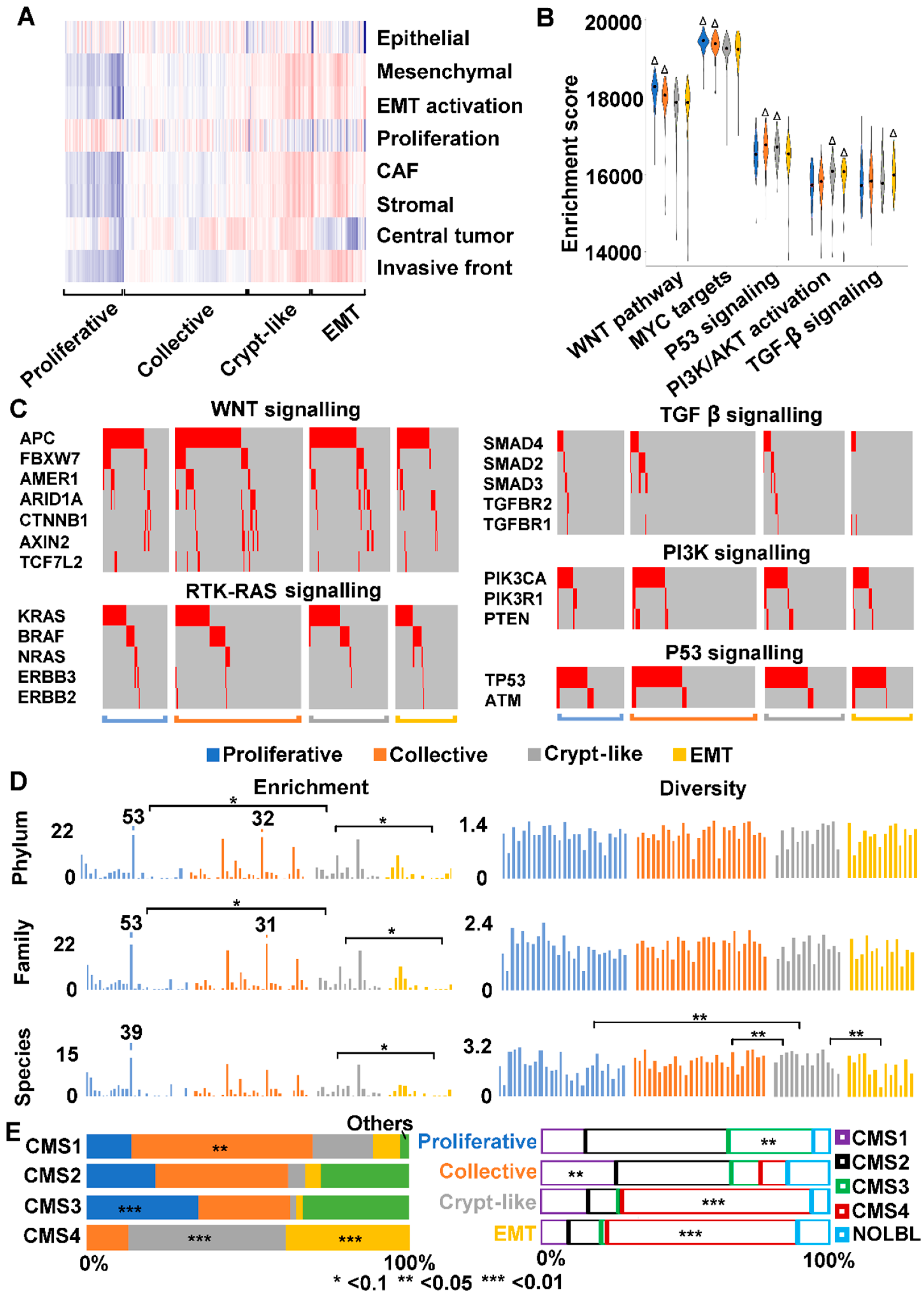
2.9. We Classified Consensus Molecular Subtype 4 (CMS4) into Crypt-Like and EMT Invasions
3. Discussion
3.1. Canine Colorectal Tumors Follow Canonical Pathogenic Pathways of Human CRC
3.2. We Have Detected Three Invasion Modes of Canine Cancer Cells
3.3. Human CMS4 Colon Cancers Consist of Crypt-Like and EMT Invasion Subtypes that Differ in TGF-β Signaling
3.4. Dog-Human Comparison Could Be Effective for Driver-Passenger Discrimination for Missense Mutations
4. Materials and Methods
4.1. Canine Samples
4.2. Tissue Dissection, DNA and RNA Extraction, and Quality Control
4.3. Immunohistochemical (IHC) Analysis
4.4. Paired-End WGS and RNA-Seq
4.5. Sequence Data Analyses
4.6. Microbiome Analysis
- (1)
- Simpson’s Diversity:
- (2)
- Shannon-Wiener Diversity:
4.7. TCGA Data Analysis
4.8. Data Access
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Paoloni, M.; Khanna, C. Translation of new cancer treatments from pet dogs to humans. Nat. Rev. Cancer 2008, 8, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.; Paoloni, M.; Mazcko, C.; Khanna, C. The Comparative Oncology Trials Consortium: Using spontaneously occurring cancers in dogs to inform the cancer drug development pathway. PLoS Med. 2009, 6, e1000161. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyko, A.R. The domestic dog: Man’s best friend in the genomic era. Genome Biol. 2011, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, H.G.; Shearin, A.L.; Ostrander, E.A. Man’s best friend becomes biology’s best in show: Genome analyses in the domestic dog. Annu. Rev. Genet. 2010, 44, 309–336. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, Y.; Lyon, K.; Camps, J.; Dalton, S.; Ried, T.; Zhao, S. Cancer driver-passenger distinction via sporadic human and dog cancer comparison: A proof-of-principle study with colorectal cancer. Oncogene 2014, 33, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xiong, H.; Ellis, A.E.; Northrup, N.C.; Dobbin, K.K.; Shin, D.M.; Zhao, S. Canine spontaneous head and neck squamous cell carcinomas represent their human counterparts at the molecular level. PLoS Genet. 2015, 11, e1005277. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xiong, H.; Ellis, A.E.; Northrup, N.C.; Rodriguez, C.O., Jr.; O’Regan, R.M.; Dalton, S.; Zhao, S. Molecular homology and difference between spontaneous canine mammary cancer and human breast cancer. Cancer Res. 2014, 74, 5045–5056. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Le, S.; Sun, L.; Yan, X.; Zhang, M.; Macleod, J.; Leroy, B.; Northrup, N.; Ellis, A.; Yeatman, T.J.; et al. Copy number abnormalities in sporadic canine colorectal cancers. Genome Res. 2010, 20, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, T.; Bishop, M.A.; Edwards, J.F.; Yin, H.; Dalton, S.; Bryan, L.K.; Zhao, S. Collaborating genomic, transcriptomic and microbiomic alterations lead to canine extreme intestinal polyposis. Oncotarget 2018, 9, 29162–29179. [Google Scholar] [CrossRef] [PubMed]
- Nasir, L.; Devlin, P.; McKevitt, T.; Rutteman, G.; Argyle, D.J. Telomere lengths and telomerase activity in dog tissues: A potential model system to study human telomere and telomerase biology. Neoplasia 2001, 3, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Sun, J.; Huang, Z.; Hou, H., Jr.; Arcilla, M.; Rakhilin, N.; Joe, D.J.; Choi, J.; Gadamsetty, P.; Milsom, J.; et al. Comprehensive models of human primary and metastatic colorectal tumors in immunodeficient and immunocompetent mice by chemokine targeting. Nat. Biotechnol. 2015, 33, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cecilia, S.; Zhang, F.; Sancho, A.; Li, S.D.; Aguilo, F.; Sun, Y.F.; Rengasamy, M.; Zhang, W.J.; Del Vecchio, L.; Salvatore, F.; et al. RBM5-AS1 Is Critical for Self-Renewal of Colon Cancer Stem-like Cells. Cancer Res. 2016, 76, 5615–5627. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Dihlmann, S.; Doeberitz, M.V.K. Wnt/beta-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int. J. Cancer 2005, 113, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers 2018, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Herring, E.; Kanaoka, S.; Tremblay, E.; Beaulieu, J.F. A Stool Multitarget mRNA Assay for the Detection of Colorectal Neoplasms. Methods Mol. Biol. 2018, 1765, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Xiong, H.; Ma, Z.; Wang, Z.; Kipreos, E.T.; Dalton, S.; Zhao, S. Cancer driver candidate genes AVL9, DENND5A and NUPL1 contribute to MDCK cystogenesis. Oncoscience 2014, 1, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.M.H.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; Di Narzo, A.F.; Yan, P.; Hodgson, J.G.; Weinrich, S.; et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int. J. Cancer 2014, 134, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.G.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, P.D.; McArt, D.G.; Bradley, C.A.; O’Reilly, P.G.; Barrett, H.L.; Cummins, R.; O’Grady, T.; Arthur, K.; Loughrey, M.B.; Allen, W.L.; et al. Challenging the Cancer Molecular Stratification Dogma: Intratumoral Heterogeneity Undermines Consensus Molecular Subtypes and Potential Diagnostic Value in Colorectal Cancer. Clin. Cancer Res. 2016, 22, 4095–4104. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, C.; Li, V.S.W.; Chan, A.S.Y.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, L.G.; Sabates-Bellver, J.; Oving, I.; Haegebarth, A.; De Palo, M.; Anti, M.; Van Gijn, M.E.; Suijkerbuijk, S.; Van de Wetering, M.; Marra, G.; et al. The Intestinal Wnt/TCF Signature. Gastroenterology 2007, 132, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Nebozhyn, M.V.; Watters, J.W.; Buser, C.A.; Shaw, P.M.; Huang, P.S.; Van’t Veer, L.; Tollenaar, R.A.; Jackson, D.B.; Agrawal, D.; et al. EMT is the dominant program in human colon cancer. BMC. Med Genom. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Wu, G.; Xu, G.; Schulman, B.A.; Jeffrey, P.D.; Harper, J.W.; Pavletich, N.P. Structure of a beta-TrCP1-Skp1-beta-catenin complex: Destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol. Cell 2003, 11, 1445–1456. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.; Suchodolski, J. Understanding the canine intestinal microbiota and its modification by pro-, pre-and synbiotics-what is the evidence? Vet. Med. Sci. 2016, 2, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Fenner, L.; Roux, V.; Ananian, P.; Raoult, D. Alistipes finegoldii in blood cultures from colon cancer patients. Emerg. Infect. Dis. 2007, 13, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Shomer, N.H.; Dangler, C.A.; Schrenzel, M.D.; Fox, J.G. Helicobacter bilis-induced inflammatory bowel disease in scid mice with defined flora. Infect. Immun. 1997, 65, 4858–4864. [Google Scholar] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Castro-Piedras, I.; Simmons, G.E., Jr.; Pruitt, K. Dishevelled: A masterful conductor of complex Wnt signals. Cell. Signal. 2018, 47, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Rajaram, M.; Li, J.; Egeblad, M.; Powers, R.S. System-wide analysis reveals a complex network of tumor-fibroblast interactions involved in tumorigenicity. PLoS Genet. 2013, 9, e1003789. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Tabassum, D.P.; Janiszewska, M.; Place, A.E.; Trinh, A.; Rozhok, A.I.; Pyne, S.; Guerriero, J.L.; Shu, S.; Ekram, M.; et al. Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical Outcomes. Cancer Res. 2016, 76, 6495–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, T.; Sun, Y.; Feng, Y.; Kisseberth, W.C.; Henry, C.J.; Mok, I.; Lana, S.E.; Dobbin, K.; Northrup, N.; et al. Proliferative and Invasive Colorectal Tumors in Pet Dogs Provide Unique Insights into Human Colorectal Cancer. Cancers 2018, 10, 330. https://doi.org/10.3390/cancers10090330
Wang J, Wang T, Sun Y, Feng Y, Kisseberth WC, Henry CJ, Mok I, Lana SE, Dobbin K, Northrup N, et al. Proliferative and Invasive Colorectal Tumors in Pet Dogs Provide Unique Insights into Human Colorectal Cancer. Cancers. 2018; 10(9):330. https://doi.org/10.3390/cancers10090330
Chicago/Turabian StyleWang, Jin, Tianfang Wang, Yanfang Sun, Yuan Feng, William C. Kisseberth, Carolyn J. Henry, Irene Mok, Susan E. Lana, Kevin Dobbin, Nicole Northrup, and et al. 2018. "Proliferative and Invasive Colorectal Tumors in Pet Dogs Provide Unique Insights into Human Colorectal Cancer" Cancers 10, no. 9: 330. https://doi.org/10.3390/cancers10090330