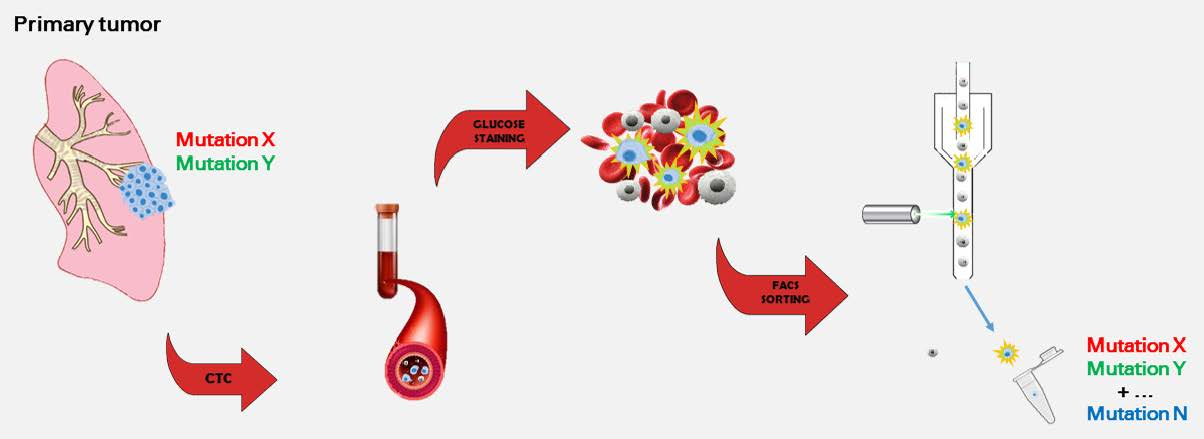
Assessment of the Mutational Status of NSCLC Using Hypermetabolic Circulating Tumor Cells

 , , , ,
, , , ,
Abstract
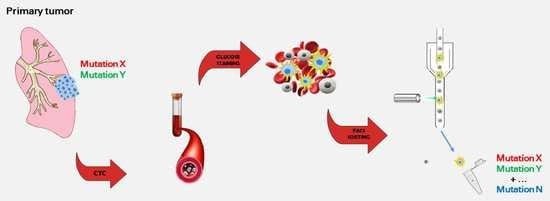
:
1. Introduction
2. Results
2.1. Feasibility Study
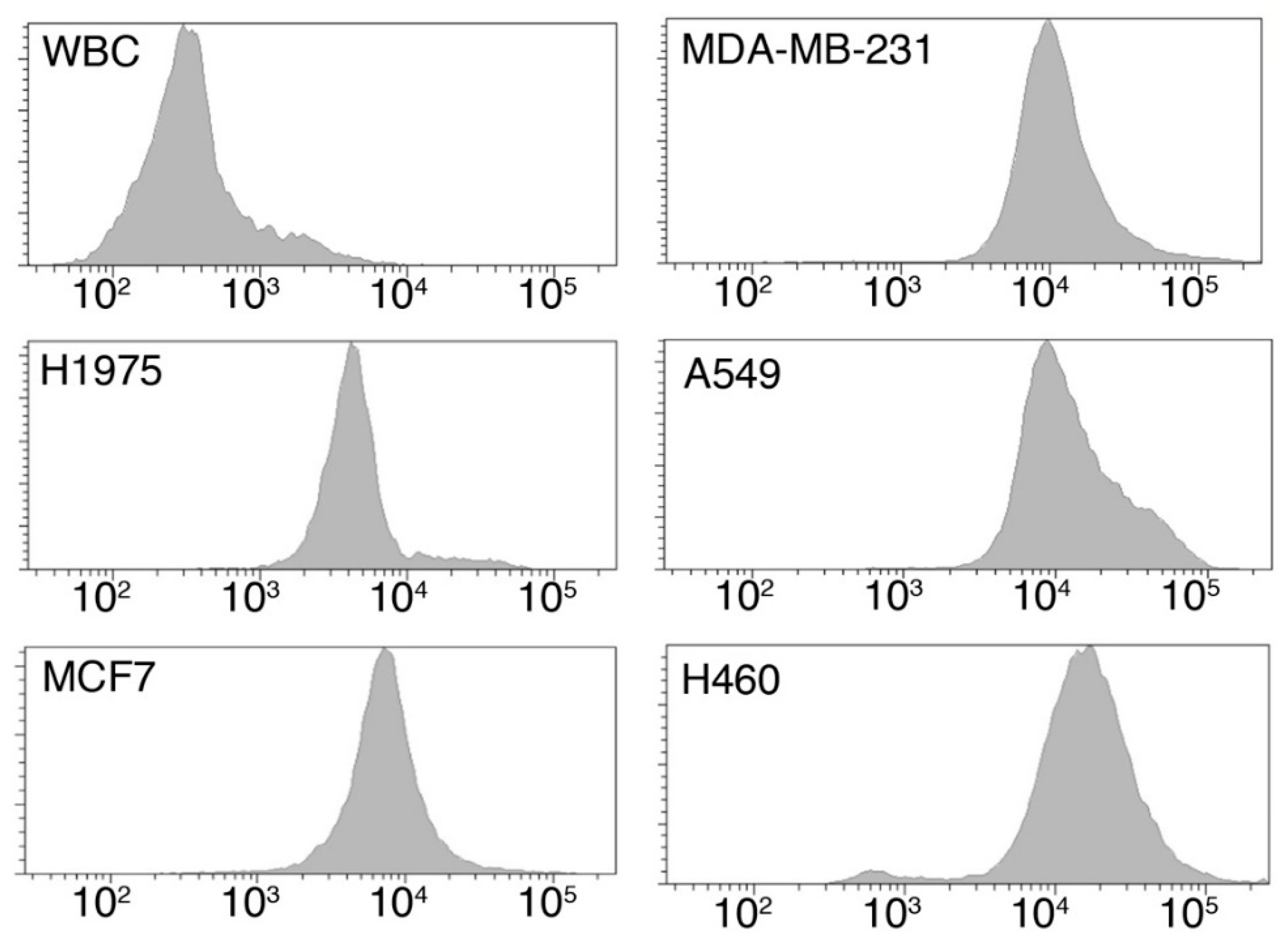
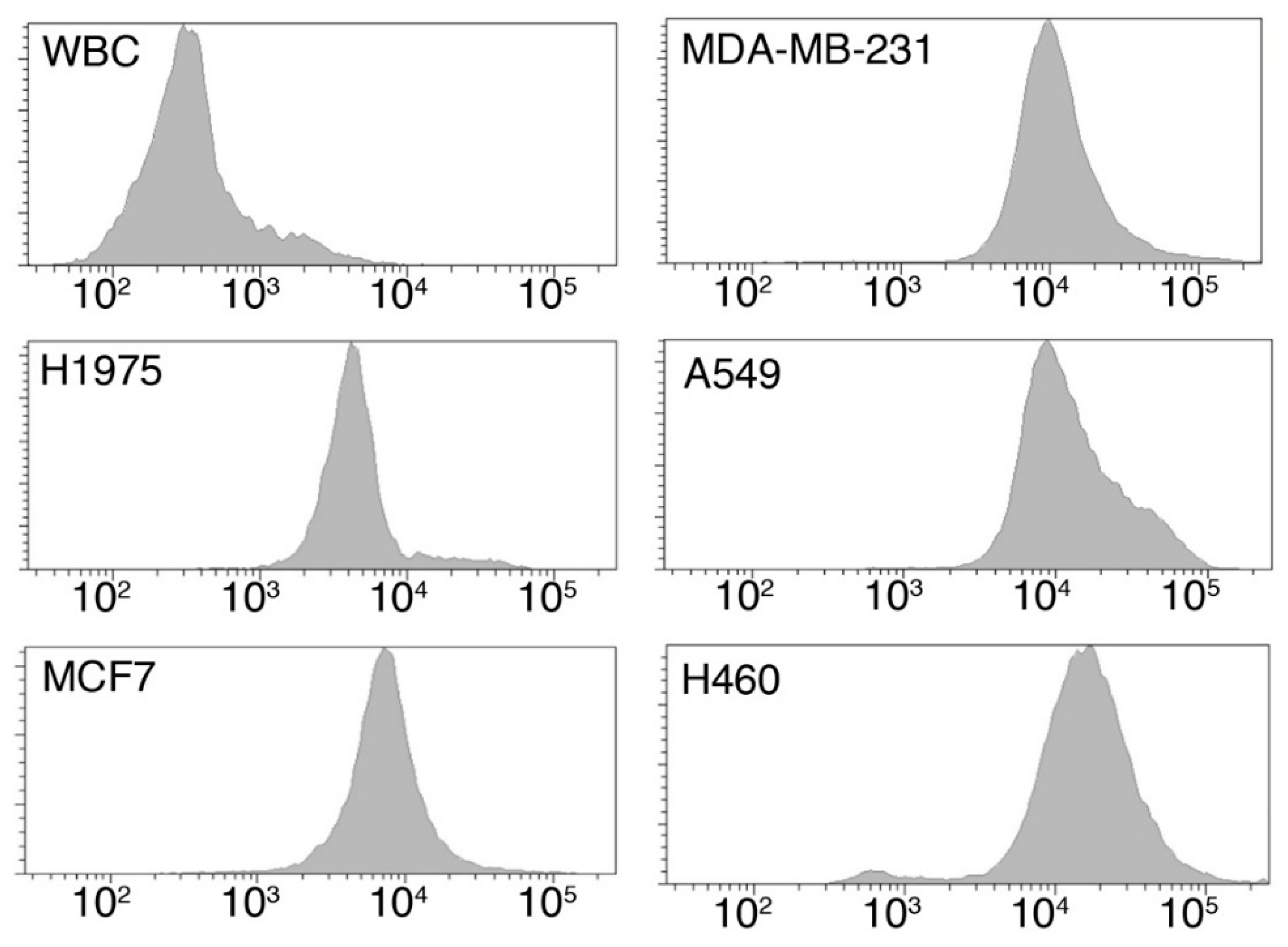
2.1.1. Tumor Cells Are Characterized by an Increased Glucose Uptake
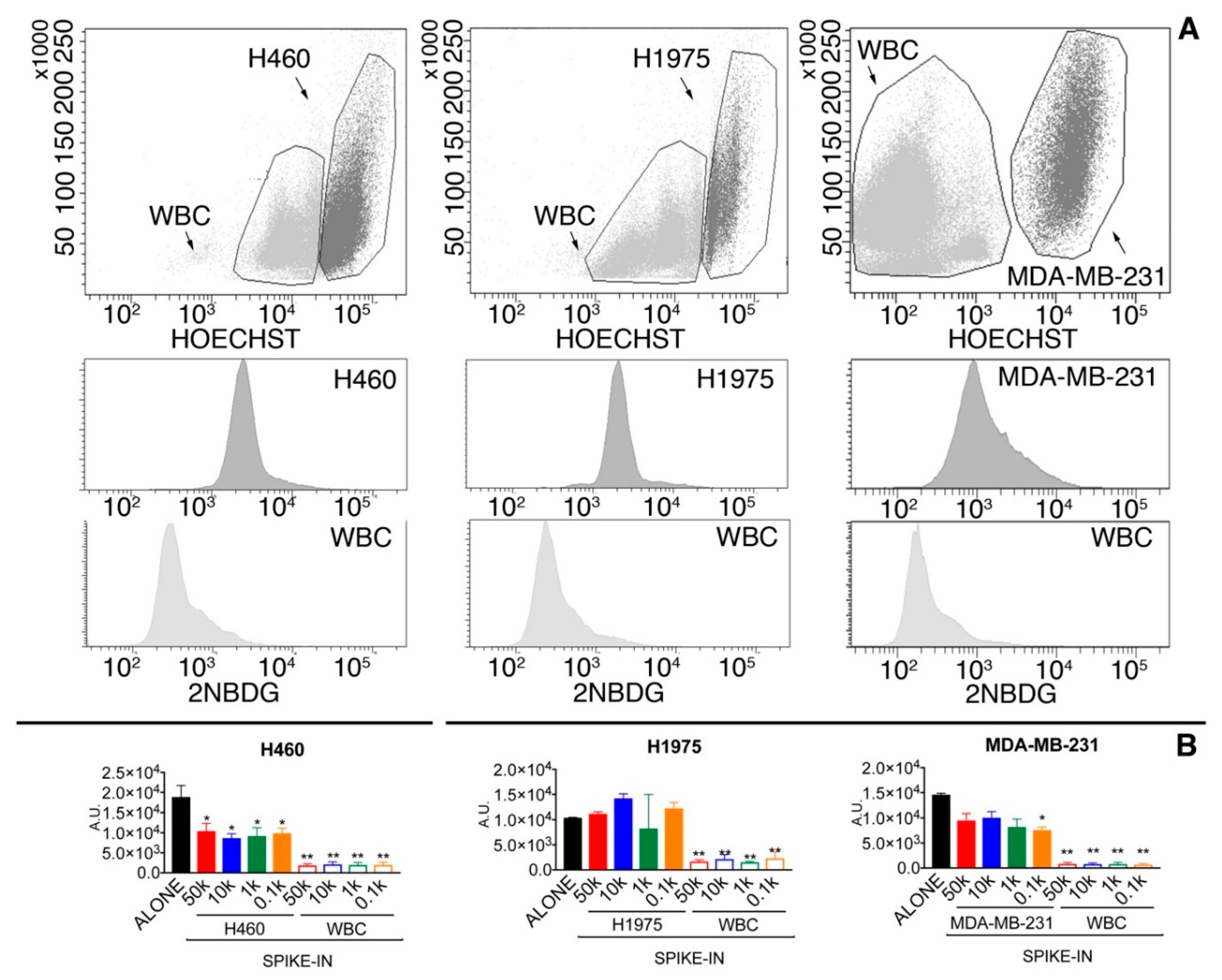
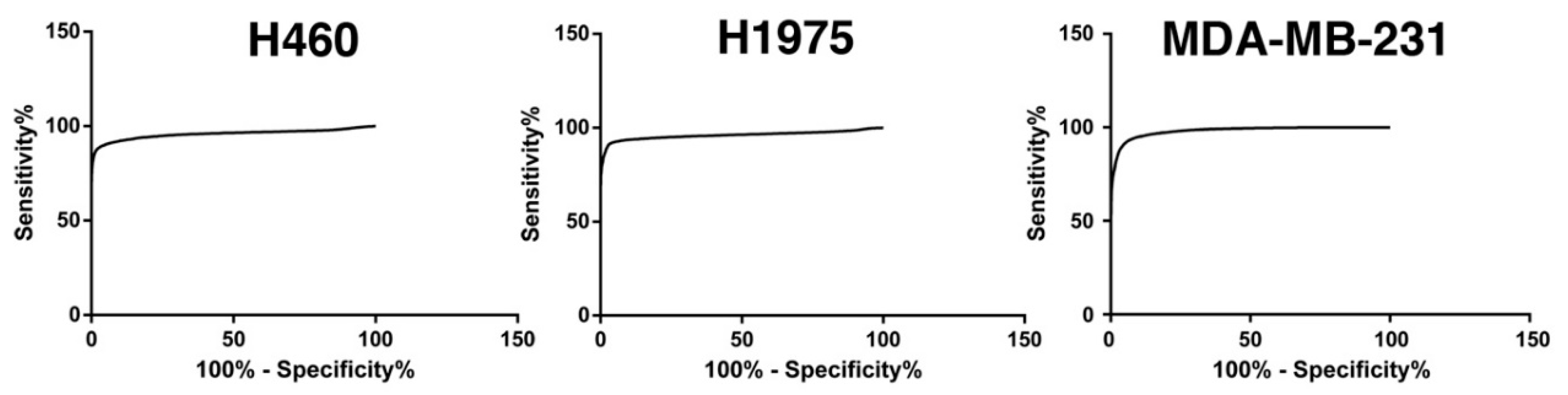
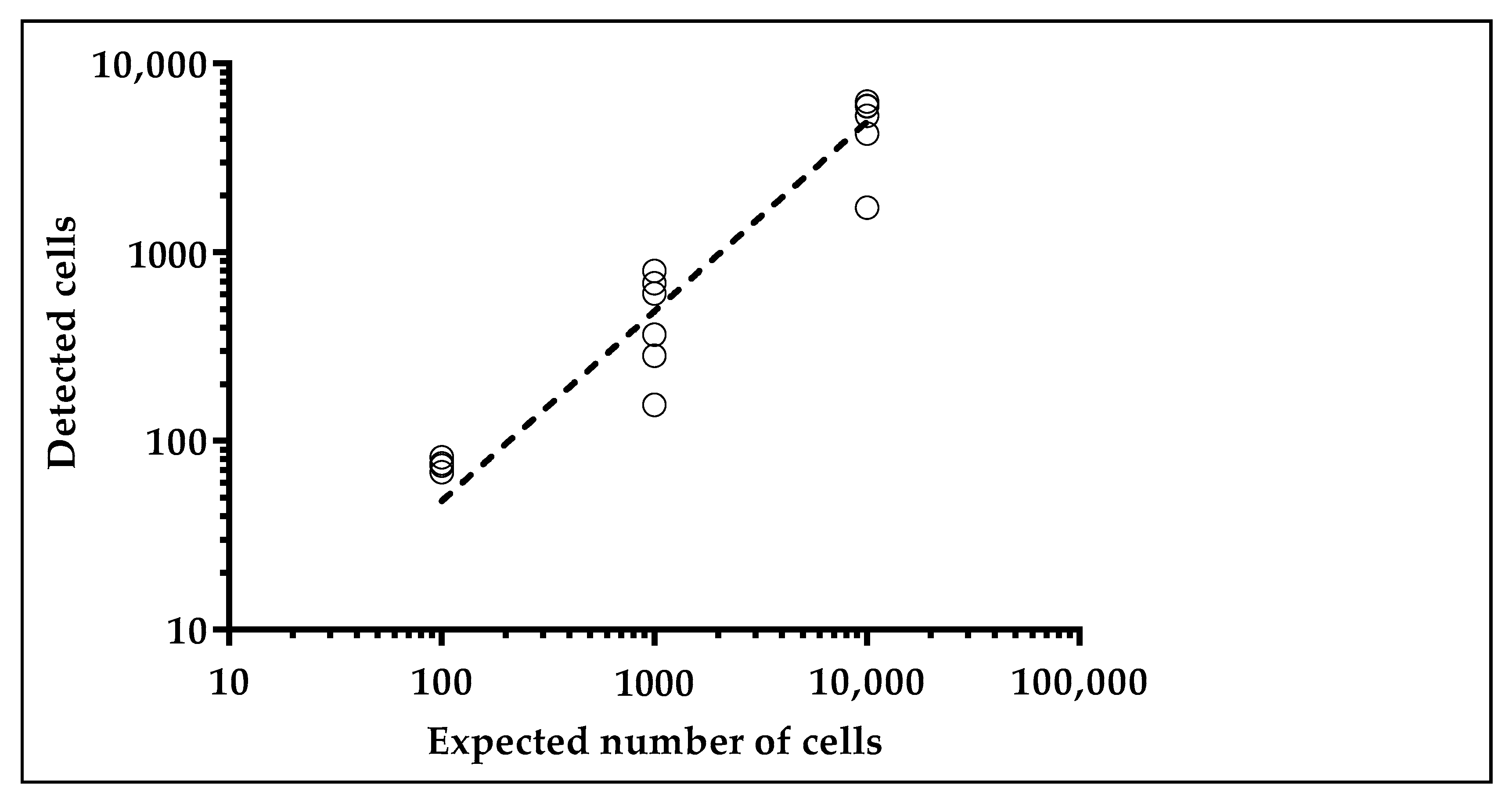
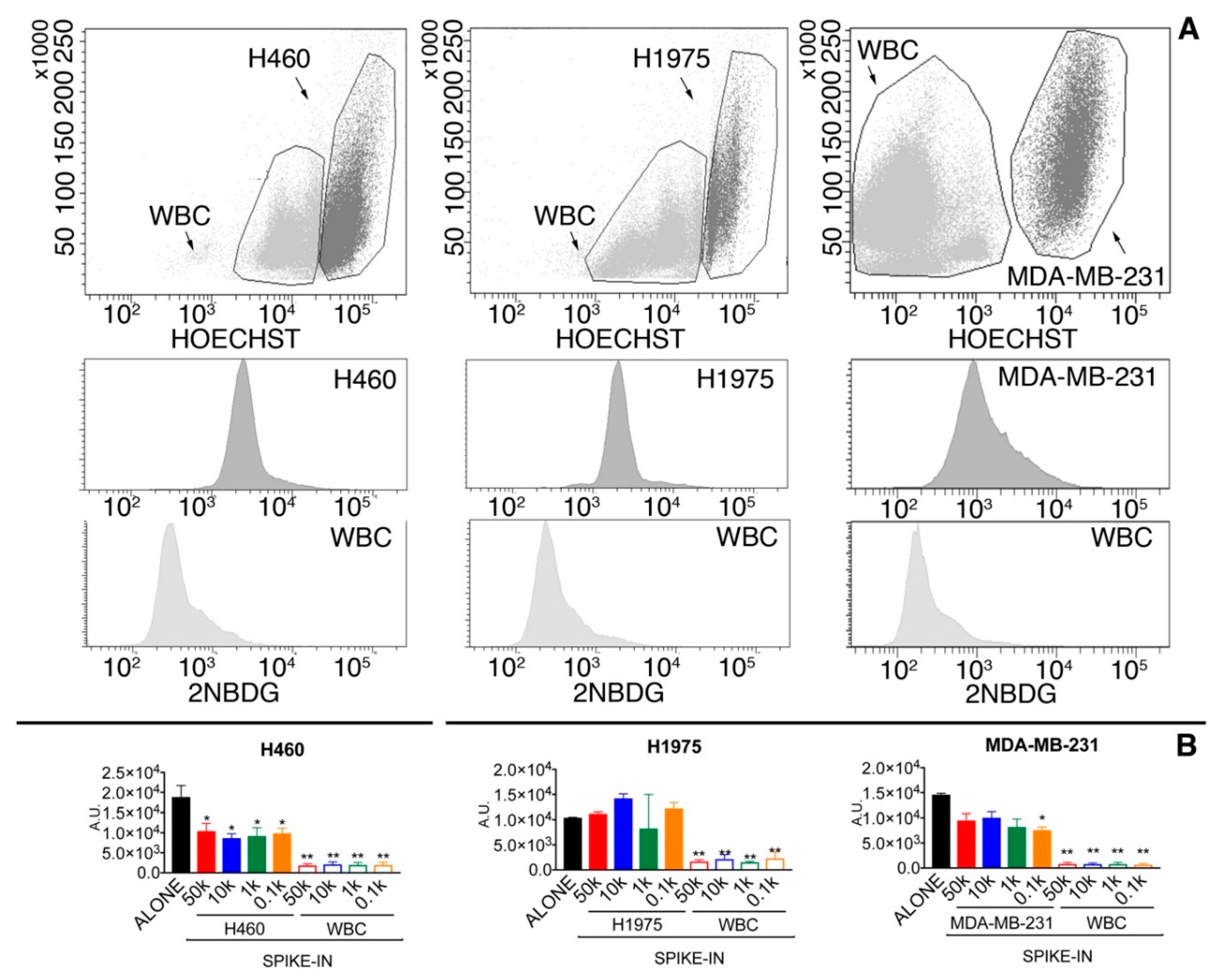
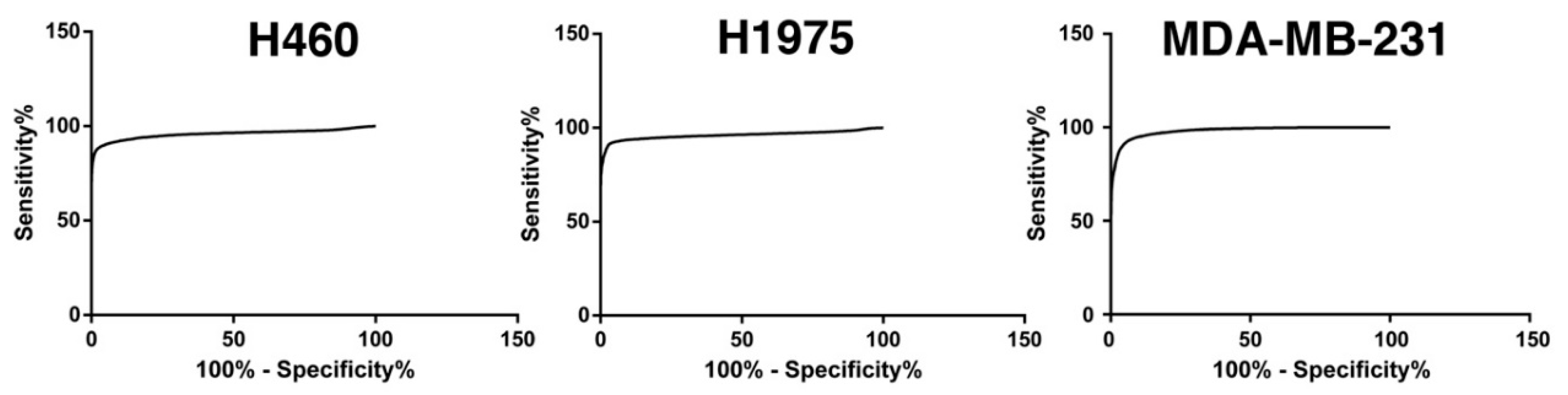
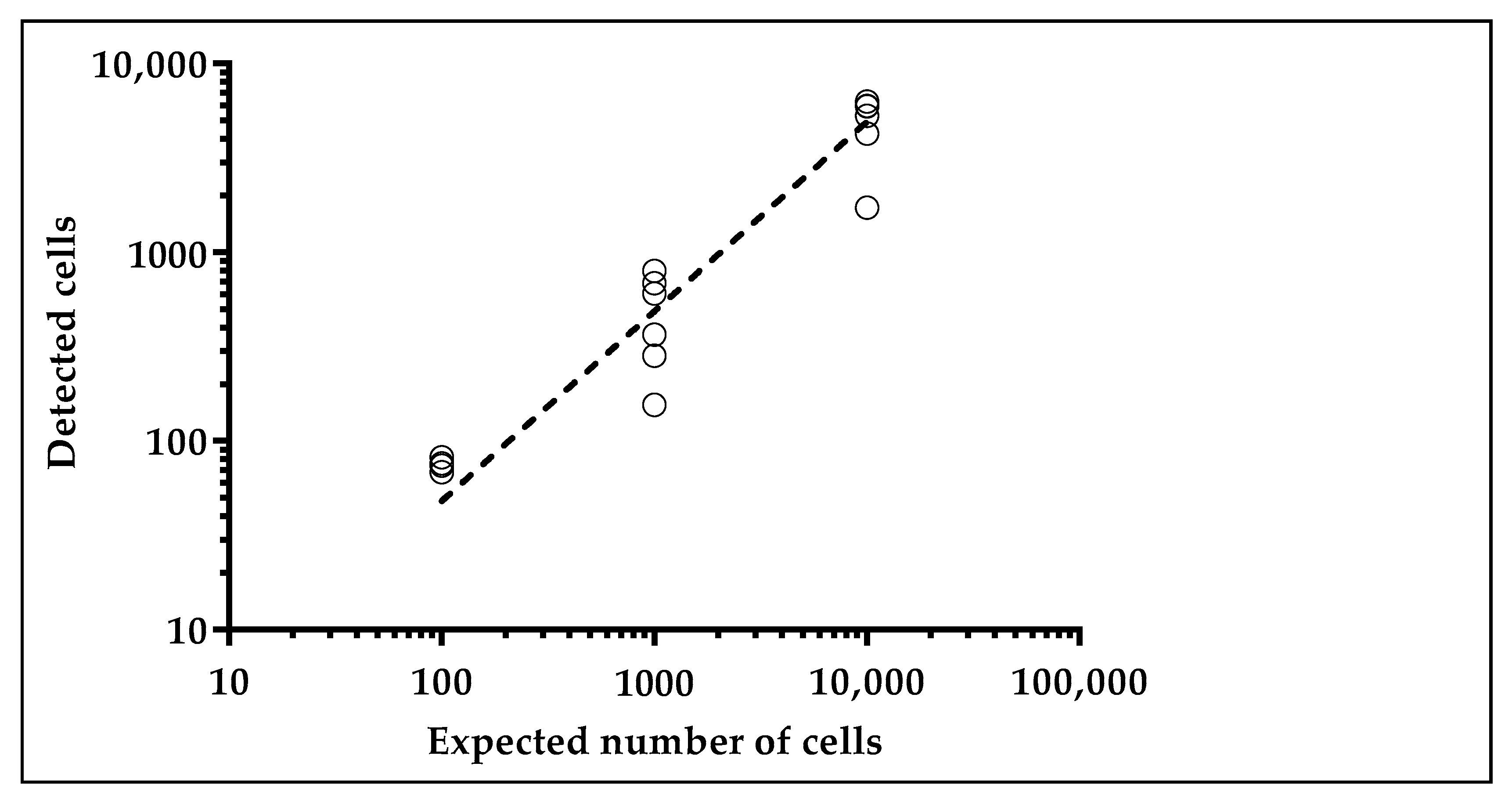
2.1.2. Glucose Uptake Can Be Used to Recover Tumor Cells from Spike-in Samples
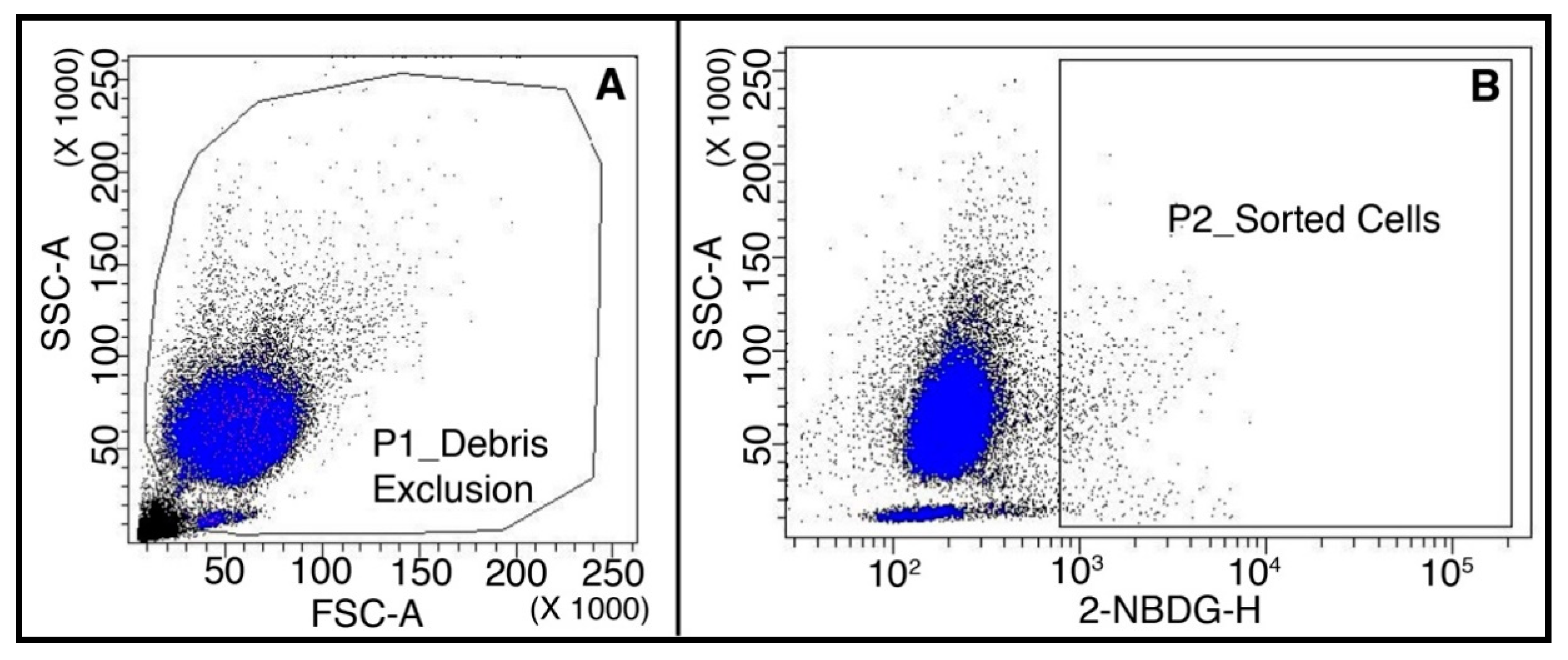
2.1.3. Highly Metabolic Cells Sorted from Spike-in Samples by FACS Are Suitable for ddPCR Analysis
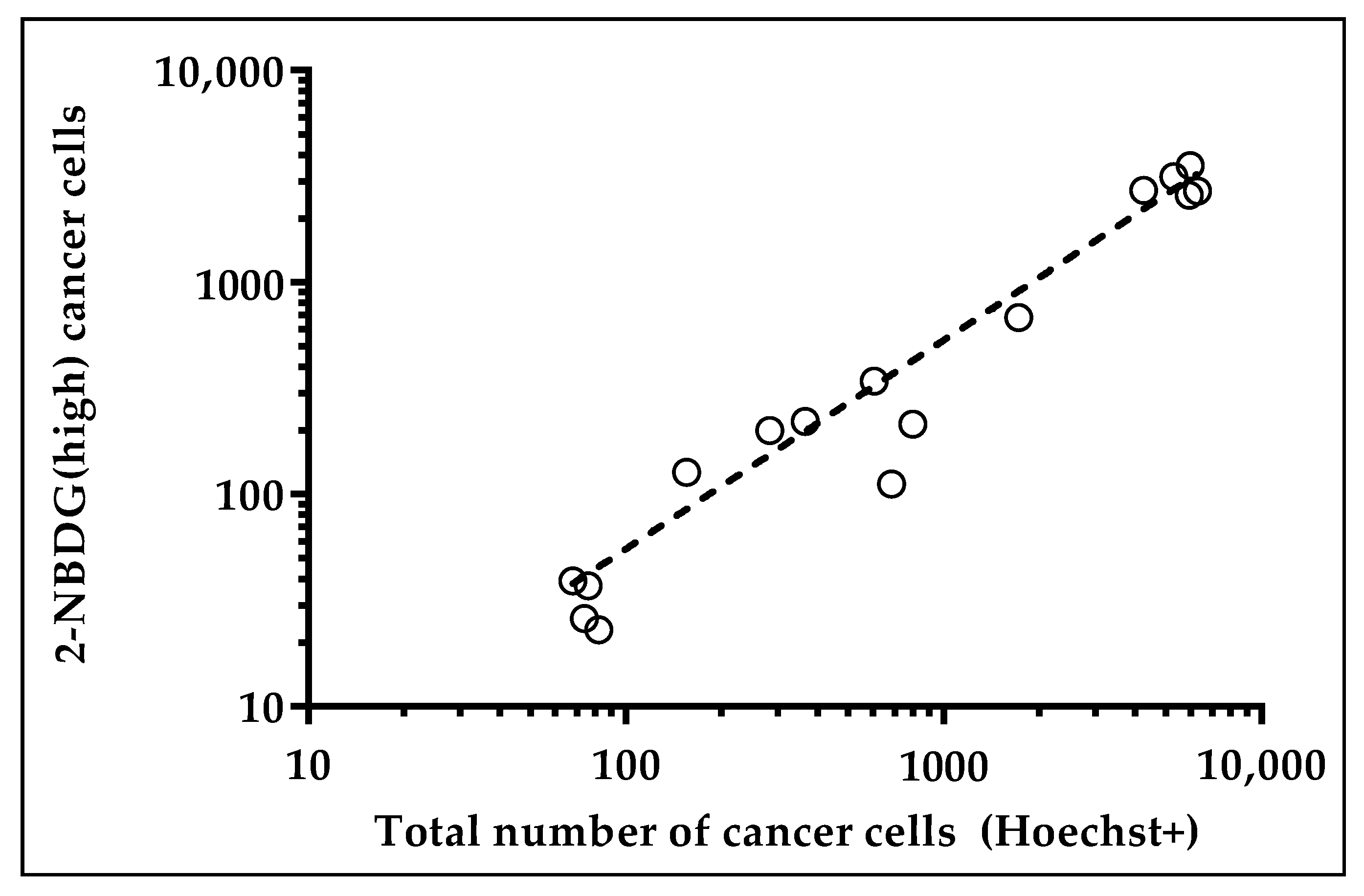
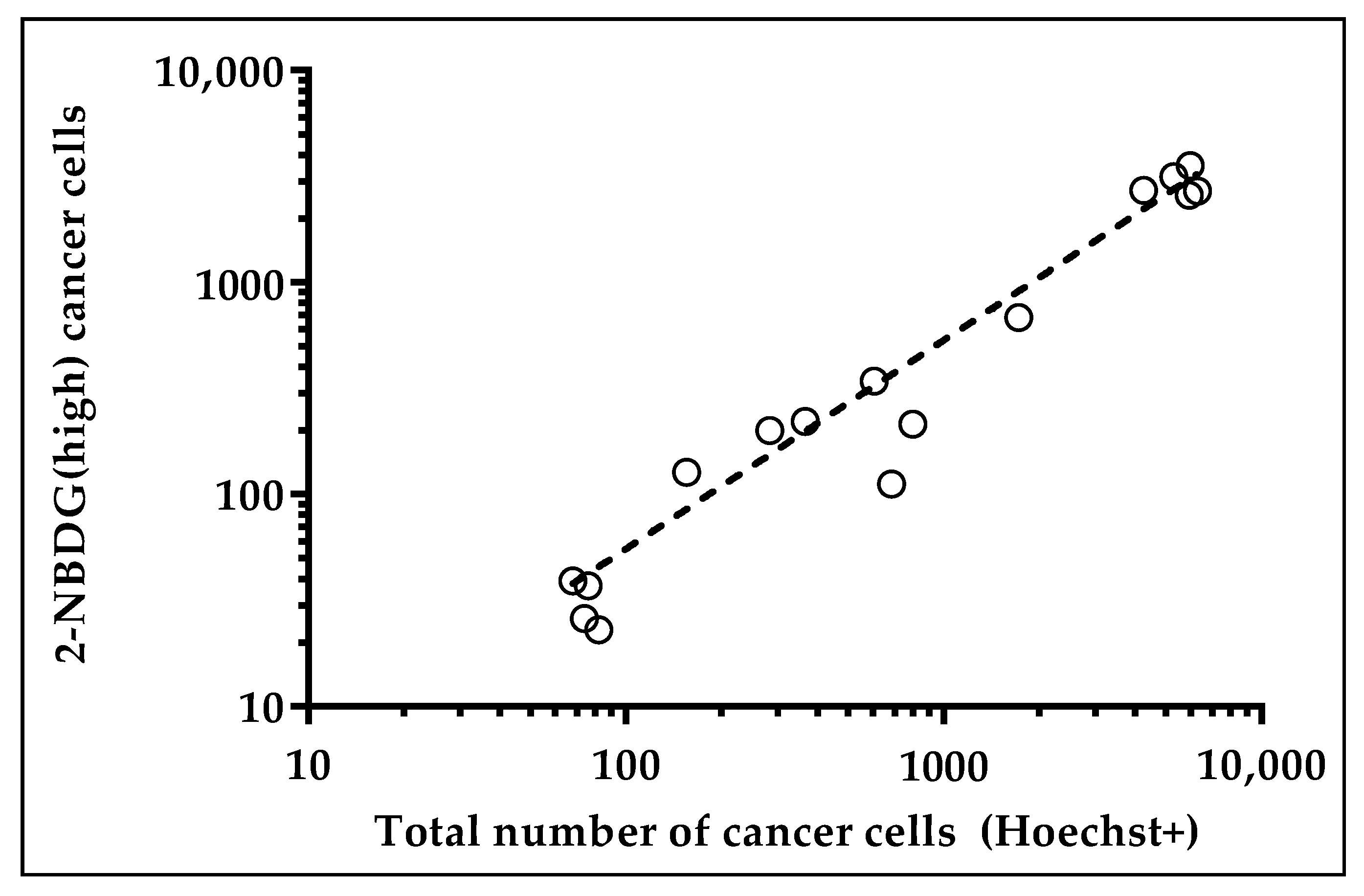
2.2. Harvesting and Molecular Analysis of Hypermetabolic Fraction in NSCLC Patients
3. Discussion
4. Materials and Methods
4.1. 2-NBDG Uptake by White Blood Cells and Cancer Cell Lines
4.2. Spike-in Assay
4.3. Patient Samples, Healthy Donors and Clinical Data
4.4. Patient Sample Preparation and Staining
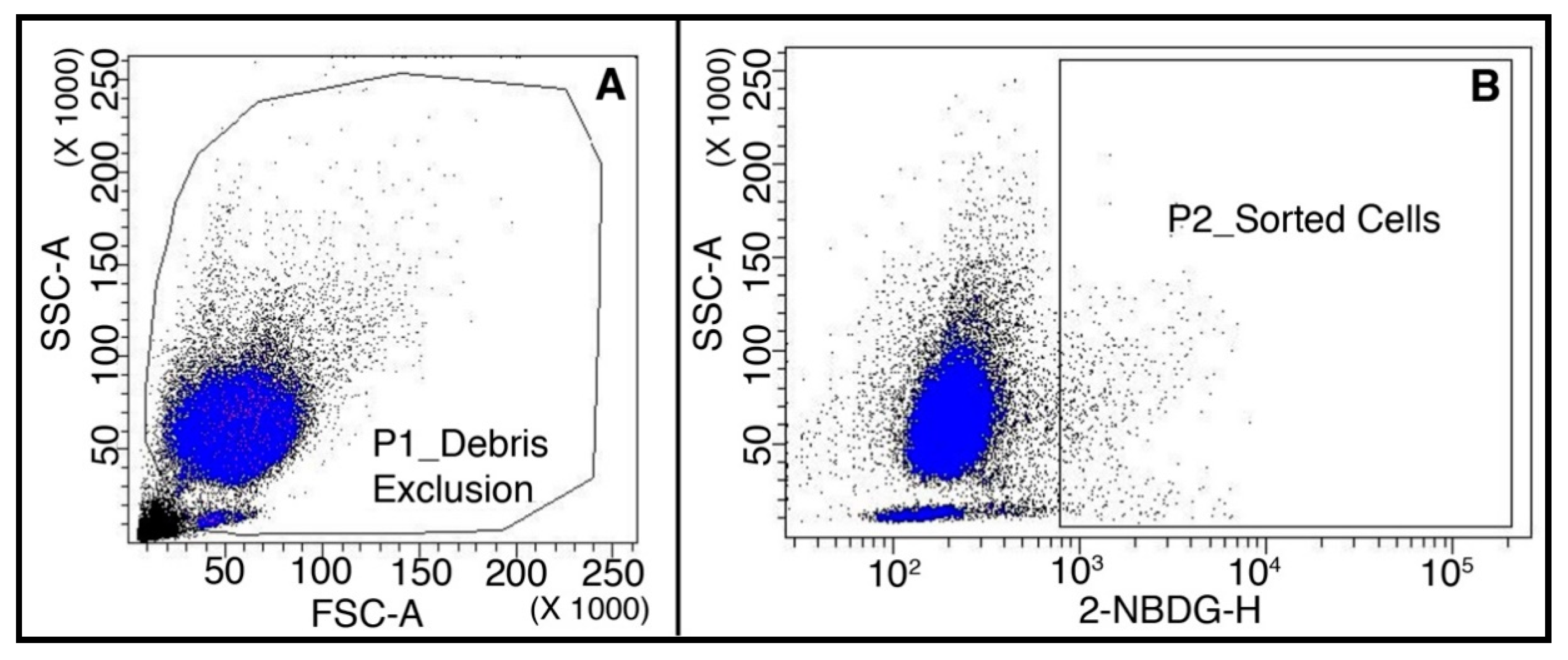
4.5. Flow Cytometry and Cell Sorting
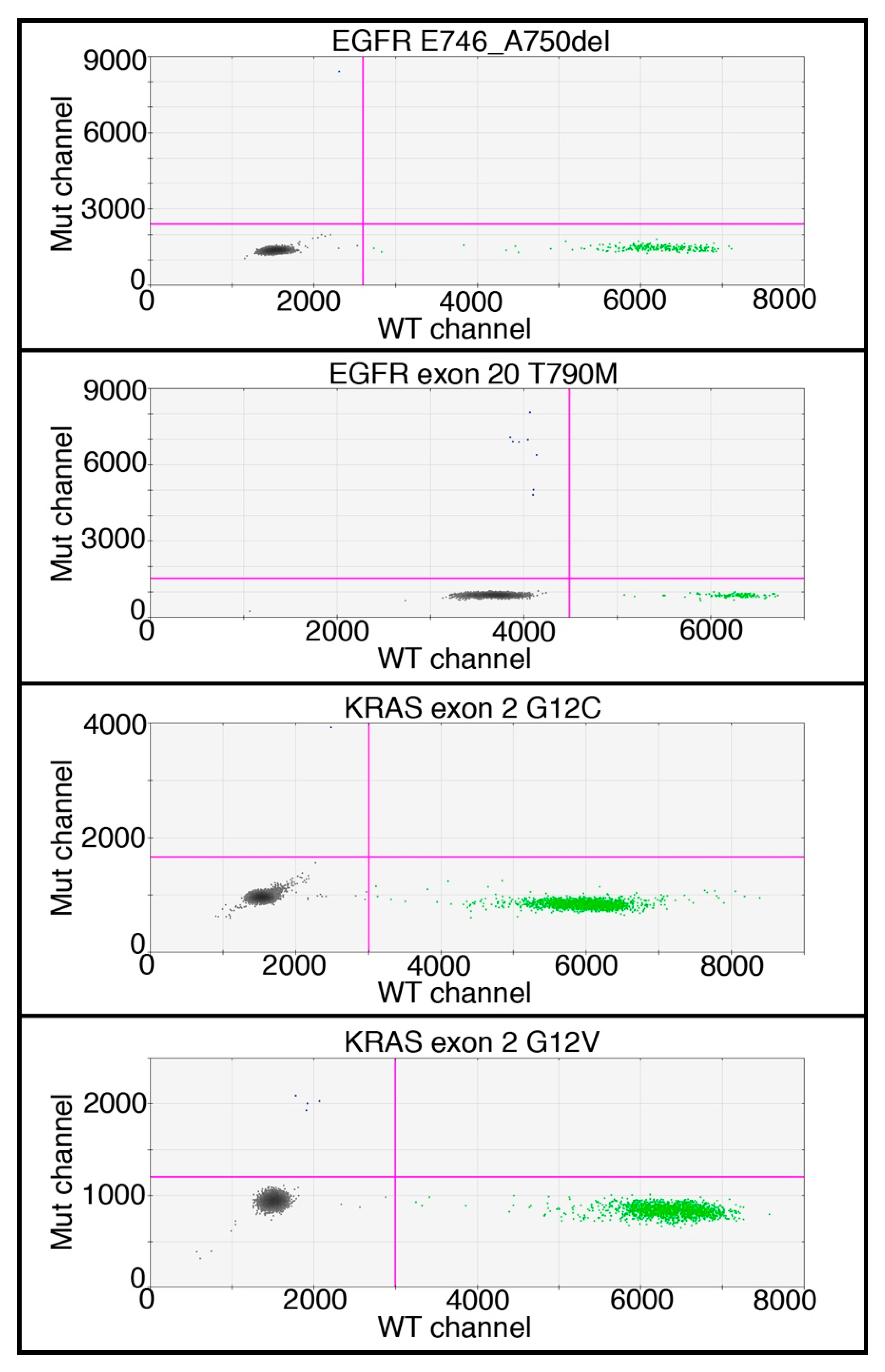
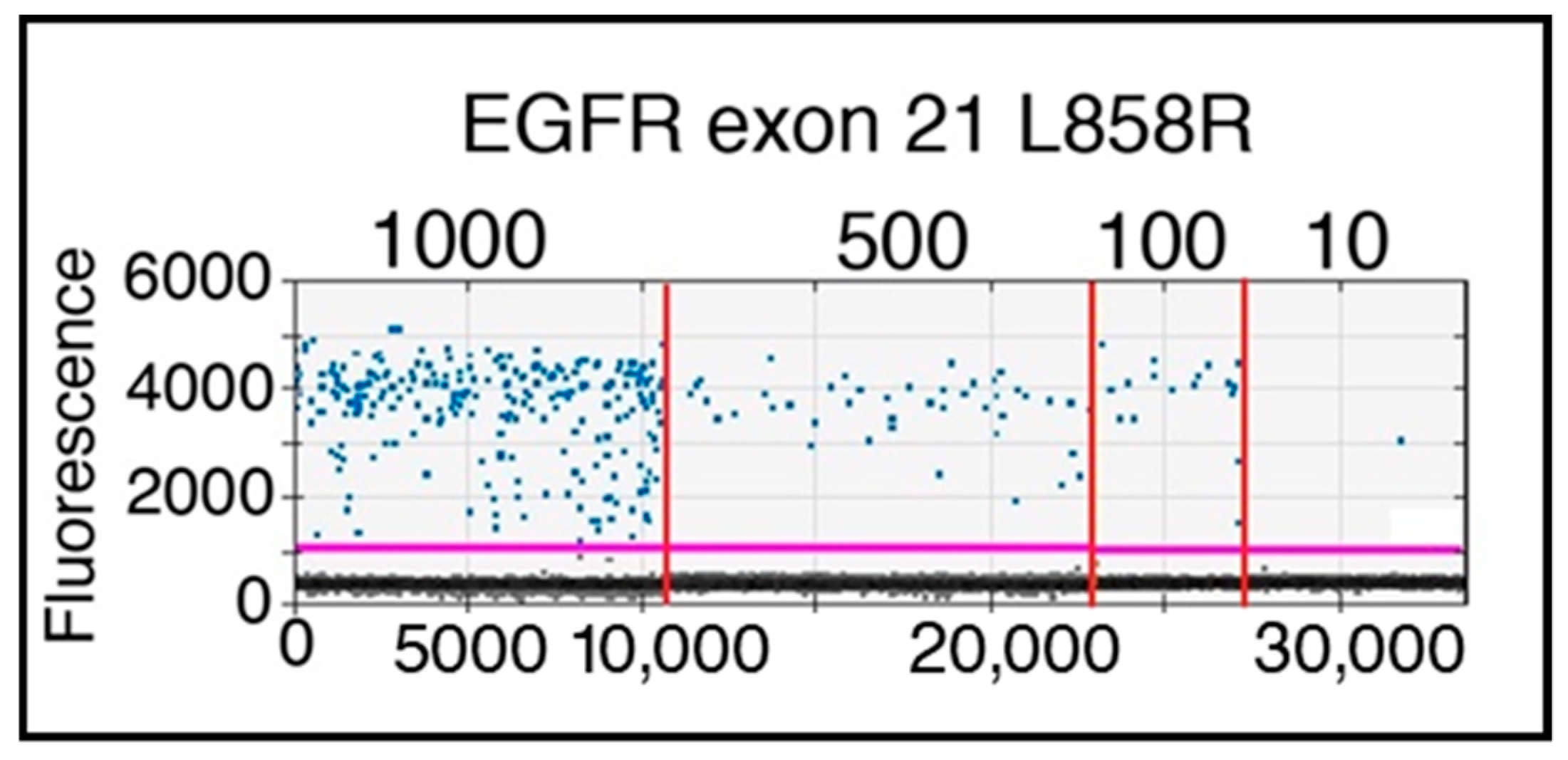
4.6. DNA Mutation Detection
- EGFR: dHsaCP000039 p.E746_A750del WT: dHsaCP2000040
- EGFR: dHsaCP000019 p.T790M WT: dHsaCP2000020
- EGFR: dHsaCP000021 p.L858R WT: dHsaCP2000022
- KRAS: dHsaCP000007 p.G12C WT: dHsaCP2500585
- KRAS: dHsaCP2500592 p.G12V WT: dHsaCP2500593
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Hughes, B.G.M. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Sequist, L.V.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Schuler, M.; Mok, T.; et al. EGFR mutation detection in circulating cell-free DNA of lung adenocarcinoma patients: Analysis of LUX-Lung 3 and 6. Br. J. Cancer 2017, 116, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Bulfoni, M.; Turetta, M.; Del Ben, F.; Di Loreto, C.; Beltrami, A.; Cesselli, D. Dissecting the Heterogeneity of Circulating Tumor Cells in Metastatic Breast Cancer: Going Far Beyond the Needle in the Haystack. Int. J. Mol. Sci. 2016, 17, 1775. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. Semin. Oncol. 2006, 33 (Suppl. 9), 9–14. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.S.-J.; Tsui, D.W.D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.O.M.; Chin, S.F.S.-F.; Dunning, M.J.M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; Kinzler, K.W.; Vogelstein, B.; Diaz, L.A. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.G.; Sloane, R.; Priest, L.; Lancashire, L.; Hou, J.-M.; Greystoke, A.; Ward, T.H.; Ferraldeschi, R.; Hughes, A.; Clack, G.; et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Marrinucci, D.; Bethel, K.; Kolatkar, A.; Luttgen, M.S.; Malchiodi, M.; Baehring, F.; Voigt, K.; Lazar, D.; Nieva, J.; Bazhenova, L.; et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys. Biol. 2012, 9, 016003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Veridex, L. Veridex CellSearch—Intended Use, Section 3; Menarini Silicon Biosystems: Bologna, Italy, 2008. [Google Scholar]
- Marchetti, A.; Del Grammastro, M.; Felicioni, L.; Malatesta, S.; Filice, G.; Centi, I.; De Pas, T.; Santoro, A.; Chella, A.; Brandes, A.A.; et al. Assessment of EGFR mutations in circulating tumor cell preparations from NSCLC patients by next generation sequencing: Toward a real-time liquid biopsy for treatment. PLoS ONE 2014, 9, e103883. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, A.; Wagner, J.; Gorges, T.M.; Taenzer, A.; Uzunoglu, F.G.; Driemel, C.; Stoecklein, N.H.; Knoefel, W.T.; Angenendt, S.; Hauch, S.; et al. Characterization of different CTC subpopulations in non-small cell lung cancer. Sci. Rep. 2016, 6, 28010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, J.F.; Kao, G.D.; MacArthur, K.M.; Ju, M.; Steinmetz, D.; Wileyto, E.P.; Simone, C.B.; Hahn, S.M. Tracking viable circulating tumor cells (CTCs) in the peripheral blood of non-small cell lung cancer (NSCLC) patients undergoing definitive radiation therapy: Pilot study results. Cancer 2015, 121, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Nel, I.; Jehn, U.; Gauler, T.; Hoffmann, A.-C. Individual profiling of circulating tumor cell composition in patients with non-small cell lung cancer receiving platinum based treatment. Transl. Lung Cancer Res. 2014, 3, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, F.; Turetta, M.; Celetti, G.; Piruska, A.; Bulfoni, M.; Cesselli, D.; Huck, W.T.S.; Scoles, G.; Del Ben, F.; Turetta, M.; et al. A Method for Detecting Circulating Tumor Cells Based on the Measurement of Single-Cell Metabolism in Droplet-Based Microfluidics. Angew. Chem. Int. Ed. 2016, 55, 8581–8584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Wang, Z.; Li, Z.; Kim, J.; Deng, Y.; Li, Y.; Heath, J.R.; Wei, W.; Lu, S.; Shi, Q. High-throughput screening of rare metabolically active tumor cells in pleural effusion and peripheral blood of lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 2544–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Peng, F. 2-NBDG fluorescence imaging of hypermetabolic circulating tumor cells in mouse xenograft model of breast cancer. J. Fluoresc. 2013, 23, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Lechago, L.V.; Somoano, J.R.; Mosharaf, M.; Lechago, J. Wide expression of the human erythrocyte glucose transporter Glut1 in human cancers. Cancer Res. 1996, 56, 1164–1167. [Google Scholar] [PubMed]
- Ooi, A.T.; Gower, A.C.; Zhang, K.X.; Vick, J.L.; Hong, L.; Nagao, B.; Wallace, W.D.; Elashoff, D.A.; Walser, T.C.; Dubinett, S.M.; et al. Molecular profiling of premalignant lesions in lung squamous cell carcinomas identifies mechanisms involved in stepwise carcinogenesis. Cancer Prev. Res. (Philadelphia PA) 2014, 7, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.W.; Lee, S.J.; Park, S.Y. Differential expression and prognostic significance of GLUT1 according to histologic type of non-small-cell lung cancer and its association with volume-dependent parameters. Lung Cancer 2017, 104, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Yang, C.; Zhang, X.; Zheng, P.; Shen, W. Expression of glucose transporter 1 and prognosis in non-small cell lung cancer: A pooled analysis of 1665 patients. Oncotarget 2017, 8, 60954–60961. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Sivridis, E.; Arelaki, S.; Koukourakis, M.I. Expression of enzymes related to glucose metabolism in non-small cell lung cancer and prognosis. Exp. Lung Res. 2017, 43, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.W.H.; Schuurbiers, O.C.J.; Kaanders, J.H.A.M.; Looijen-Salamon, M.G.; de Geus-Oei, L.-F.; Verhagen, A.F.T.M.; Lok, J.; van der Heijden, H.F.M.; Rademakers, S.E.; Span, P.N.; et al. Differences in metabolism between adeno- and squamous cell non-small cell lung carcinomas: Spatial distribution and prognostic value of GLUT1 and MCT4. Lung Cancer 2012, 76, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Schrevens, L.; Lorent, N.; Dooms, C.; Vansteenkiste, J. The role of PET scan in diagnosis, staging, and management of non-small cell lung cancer. Oncologist 2004, 9, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.F. PET scan in the staging of non-small cell lung cancer. Lung Cancer (Amsterdam Netherlands) 2003, 42 (Suppl. 1), 27–37. [Google Scholar] [CrossRef]
- Venturelli, L.; Nappini, S.; Bulfoni, M.; Gianfranceschi, G.; Dal Zilio, S.; Coceano, G.; Del Ben, F.; Turetta, M.; Scoles, G.; Vaccari, L.; et al. Glucose is a key driver for GLUT1-mediated nanoparticles internalization in breast cancer cells. Sci. Rep. 2016, 6, 21629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulfoni, M.; Gerratana, L.; Del Ben, F.; Marzinotto, S.; Sorrentino, M.; Turetta, M.; Scoles, G.; Toffoletto, B.; Isola, M.; Beltrami, C.A.; et al. In patients with metastatic breast cancer the identification of circulating tumor cells in epithelial-to-mesenchymal transition is associated with a poor prognosis. Breast Cancer Res. BCR 2016, 18, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Kularatne, S.A.; Kalli, K.R.; Prendergast, F.G.; Amato, R.J.; Klee, G.G.; Hartmann, L.C.; Low, P.S. Quantitation of circulating tumor cells in blood samples from ovarian and prostate cancer patients using tumor-specific fluorescent ligands. Int. J. Cancer 2008, 123, 1968–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, Y.; Humberg, V.; Erlmeier, F.; Steffens, S.; Steinestel, J.; Bögemann, M.; Schrader, A.J.; Bernemann, C. Comparison of isolation platforms for detection of circulating renal cell carcinoma cells. Oncotarget 2017, 8, 87710–87717. [Google Scholar] [CrossRef] [PubMed]
- Kallergi, G.; Politaki, E.; Alkahtani, S.; Stournaras, C.; Georgoulias, V. Evaluation of Isolation Methods for Circulating Tumor Cells (CTCs). Cell. Physiol. Biochem. 2016, 40, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Droplet Digital™ PCR Applications Guide. Bio-Rad Laboratories. Available online: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf (accessed on 12 August 2018).
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.R.; Pantel, K.; Riethdorf, S. Heterogeneity of Epidermal Growth Factor Receptor Status and Mutations of KRAS/PIK3CA in Circulating Tumor Cells of Patients with Colorectal Cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Mostert, B.; Jiang, Y.; Sieuwerts, A.M.; Wang, H.; Bolt-De Vries, J.; Biermann, K.; Kraan, J.; Lalmahomed, Z.; Van Galen, A.; De Weerd, V.; et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int. J. Cancer 2013, 133, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Saito, M.; Oh, K.-B.; Nemoto, Y.; Matsuoka, H.; Natsume, M.; Abe, H. Intracellular Fate of 2-NBDG, a Fluorescent Probe for Glucose Uptake Activity, in Escherichia coli Cells. Biosci. Biotechnol. Biochem. 1996, 60, 1899–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanein, M.; Weidow, B.; Koehler, E.; Bakane, N.; Garbett, S.; Shyr, Y.; Quaranta, V. Development of high-throughput quantitative assays for glucose uptake in cancer cell lines. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2011, 13, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, X.; Peng, Y.; Chai, H.; Xu, Y.; Wei, J.; Ren, X.; Wang, X.; Liu, W.; Chen, M.; et al. Comparison of the sorting efficiency and influence on cell function between the sterile flow cytometry and immunomagnetic bead purification methods. Prep. Biochem. Biotechnol. 2013, 43, 197–206. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
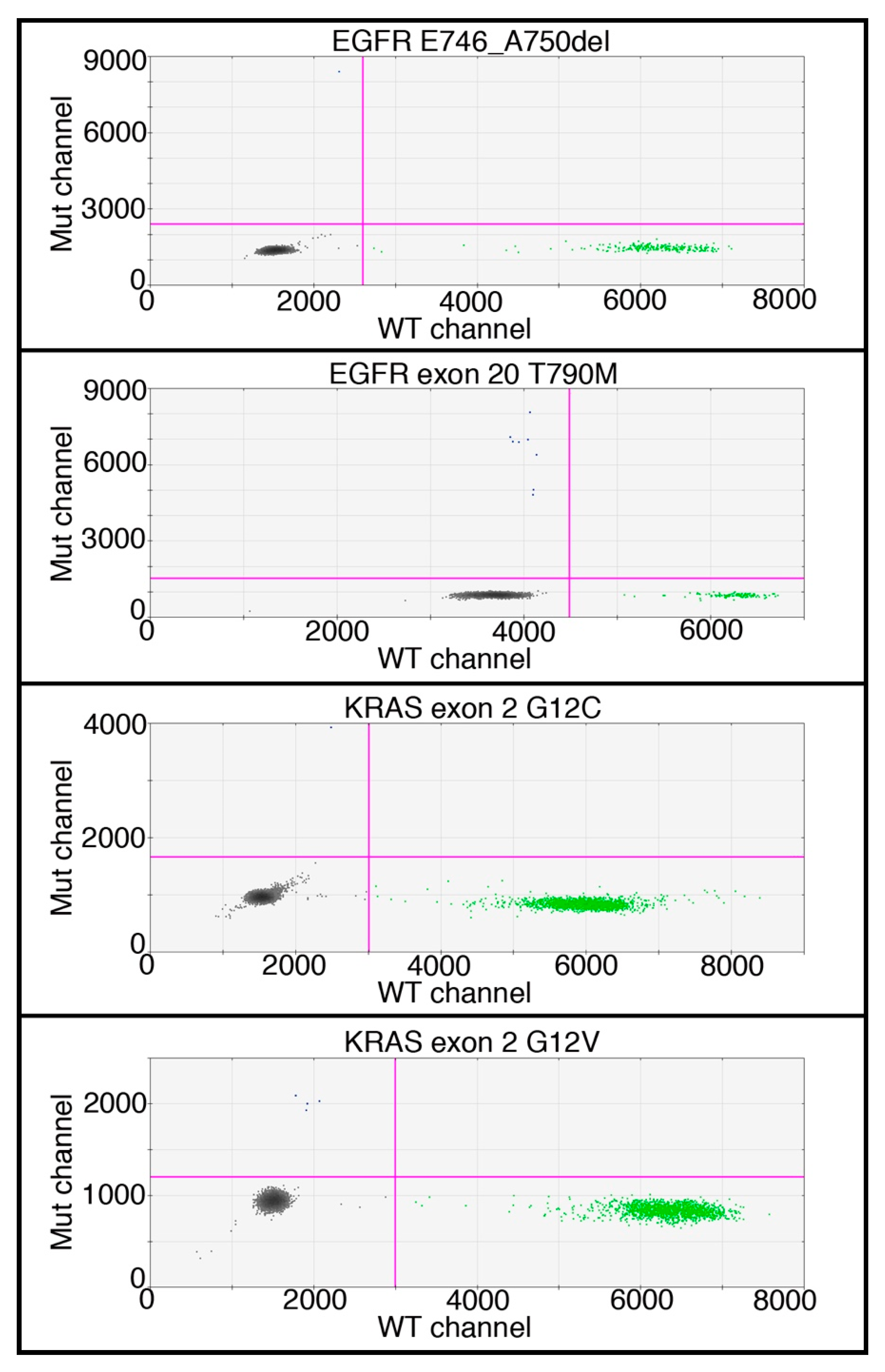{kind=link}
| Cell Type | Origin | Median Fluorescence Intensity (A.U.) | p-Value * |
|---|---|---|---|
| WBC | Healthy donor | 291 | - |
| H1975 | Lung Cancer | 4758 | 0.0238 |
| MDA-MB-231 | Breast Cancer | 8338 | 0.0043 |
| MCF-7 | Breast Cancer | 6983 | 0.0238 |
| A549 | Lung Cancer | 9977 | 0.0238 |
| H460 | Lung Cancer | 19,764 | 0.0095 |
| Cut-Off Level | Tumor Cell Recovery | WBC Contaminants |
|---|---|---|
| 3-fold WBC average | 74.9% | 9341 |
| 5-fold WBC average | 46.5% | 3052 |
| 7-fold WBC average | 32.1% | 1412 |
| WBC average + 2.5-folds SD | 59.8% | 4473 |
| Primary Tumor | 2-NBDG(high) | Matching | |
|---|---|---|---|
| PATIENT 1 | KRAS p.G12C | KRAS p.G12C | Perfect |
| PATIENT 2 | KRAS p.G12C | KRAS p.G12C | Perfect |
| PATIENT 3 | KRAS p.G12C | KRAS p.G12C | Perfect |
| PATIENT 4 * | KRAS p.G12D | KRAS p.G12C | n.a. |
| PATIENT 5 | EGFR p.L858R | EGFR p.L858R | Perfect |
| PATIENT 6 | EGFR p.E746_A750del and EGFR p.T790M | EGFR p.E746_A750del and EGFR p.T790M | Perfect |
| PATIENT 7 | EGFR p.E746_A750del | EGFR p.E746_A750del | Perfect |
| PATIENT 8 | EGFR p.L858R | EGFR p.L858R | Perfect |
| PATIENT 9 | EGFR p.L858R and EGFR p.T790M | EGFR p.Leu858Arg and EGFR p.T790M | Perfect |
| PATIENT 10 | EGFR p.L858R | EGFR p.L858R | Perfect |
| PATIENT 11 | EGFR p.E746_A750del | EGFR p.E746_A750del | Perfect |
| PATIENT 12 | EGFR p.E746_A750del | EGFR p.E746_A750del | Perfect |
| PATIENT 13 | EGFR p.L858R | EGFR p.L858R | Perfect |
| PATIENT 14 | EGFR p.E746_A750del | EGFR p.E746_A750del | Perfect |
| PATIENT 15 | EGFR p.E746_A750del | EGFR p.E746_A750del | Perfect |
| PATIENT 16 | EGFR p.E746_A750del | EGFR p.E746_A750del | Perfect |
| PATIENT 17 ** | KRAS exon 2 [not specified] | KRAS p.G12C and KRAS p.G12V | n.a. |
| PATIENT 18 | EGFR p.E746_A750del | EGFR p.T790M | Different |
| PATIENT 19 | KRAS p.G12C | WT | NEG |
| PATIENT 20 | KRAS p.G12C | WT | NEG |
| PATIENT 23 | EGFR p.L858R and EGFR p.T790M | EGFR p.T790M | Partial |
| PATIENT 25 | EGFR p.E746_A750del | EGFR p.T790M | Different |
| PATIENT 26 ** | KRAS p.G12C | KRAS p.G12C and KRAS G12V | Additional |
| PATIENT 27 *** | EGFR p.G719C and EGFR p.T790M | EGFR p.T790M | n.a. |
| PATIENT 29 | KRAS p.G12V | WT | Negative |
| PATIENT 30 | KRAS p.G12C | WT | Negative |
| PATIENT 21 | WT | WT | Perfect |
| PATIENT 22 | WT | EGFR p.E746_A750del | New mutation |
| PATIENT 24 | WT | KRAS p.G12V | New mutation |
| PATIENT 28 | WT | WT | Perfect |
| Characteristic | Years, mean (range) |
| Age | 68 (51–87) |
| Characteristic | Number of patients (% on total) |
| Gender (n of patients (%)) | |
| Male | 16 (53.3%) |
| Female | 14 (46.7%) |
| Histological subtype (n of patients (%)) | |
| Adenocarcinoma (ADC) | 29 (96.7%) |
| Unknown | 1 (3.3%) |
| Mutation (n of patients (%)) | |
| Mutation of the primary tumor: | 26 (86.7%) |
| EGFR | 16 (61.5%) |
| p.E746_A750del | 9 (34.6%) |
| p.L858R | 6 (23.1%) |
| p.T790M | 4 (15.4%) |
| KRAS | 10 (38.5%) |
| p.G12C | 8 (30.8%) |
| p.G12V | 1 (3.8%) |
| Unknown | 1 (3.8%) |
| Wild-type primary tumor | 4 (13.3%) |
| ECOG Performance Status (n of patients (%)) | |
| 0 | 18 (60%) |
| 1 | 9 (30%) |
| 2 | 2 (6.7%) |
| Unknown | 1 (3.3%) |
| Stage (n of patients (%)) | |
| IV | 29 (96.7%) |
| Unknown | 1 (3.3%) |
| Metastatic status (n of patients (%)) | |
| Mx | 1 (3.3%) |
| M1 | 29 (96.7%) |
| Metastatic sites (n of patients (%)) | |
| Lung | 21 (70%) |
| Pleural effusion | 11 (36.7%) |
| Lymph nodes | 3 (10%) |
| Bone | 10 (33.3%) |
| CNS | 6 (20%) |
| Adrenal gland | 2 (6.7%) |
| Liver | 3 (10%) |
| Kidney | 1 (3.3%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turetta, M.; Bulfoni, M.; Brisotto, G.; Fasola, G.; Zanello, A.; Biscontin, E.; Mariuzzi, L.; Steffan, A.; Di Loreto, C.; Cesselli, D.; et al. Assessment of the Mutational Status of NSCLC Using Hypermetabolic Circulating Tumor Cells. Cancers 2018, 10, 270. https://doi.org/10.3390/cancers10080270
Turetta M, Bulfoni M, Brisotto G, Fasola G, Zanello A, Biscontin E, Mariuzzi L, Steffan A, Di Loreto C, Cesselli D, et al. Assessment of the Mutational Status of NSCLC Using Hypermetabolic Circulating Tumor Cells. Cancers. 2018; 10(8):270. https://doi.org/10.3390/cancers10080270
Chicago/Turabian StyleTuretta, Matteo, Michela Bulfoni, Giulia Brisotto, Gianpiero Fasola, Andrea Zanello, Eva Biscontin, Laura Mariuzzi, Agostino Steffan, Carla Di Loreto, Daniela Cesselli, and et al. 2018. "Assessment of the Mutational Status of NSCLC Using Hypermetabolic Circulating Tumor Cells" Cancers 10, no. 8: 270. https://doi.org/10.3390/cancers10080270