Epithelioid Hemangioendothelioma as a Model of YAP/TAZ-Driven Cancer: Insights from a Rare Fusion Sarcoma



{kind=link}
Abstract
:1. Introduction
2. The EHE Phenotype
2.1. Clinical Features
2.2. Diagnosis
3. Molecular Pathology
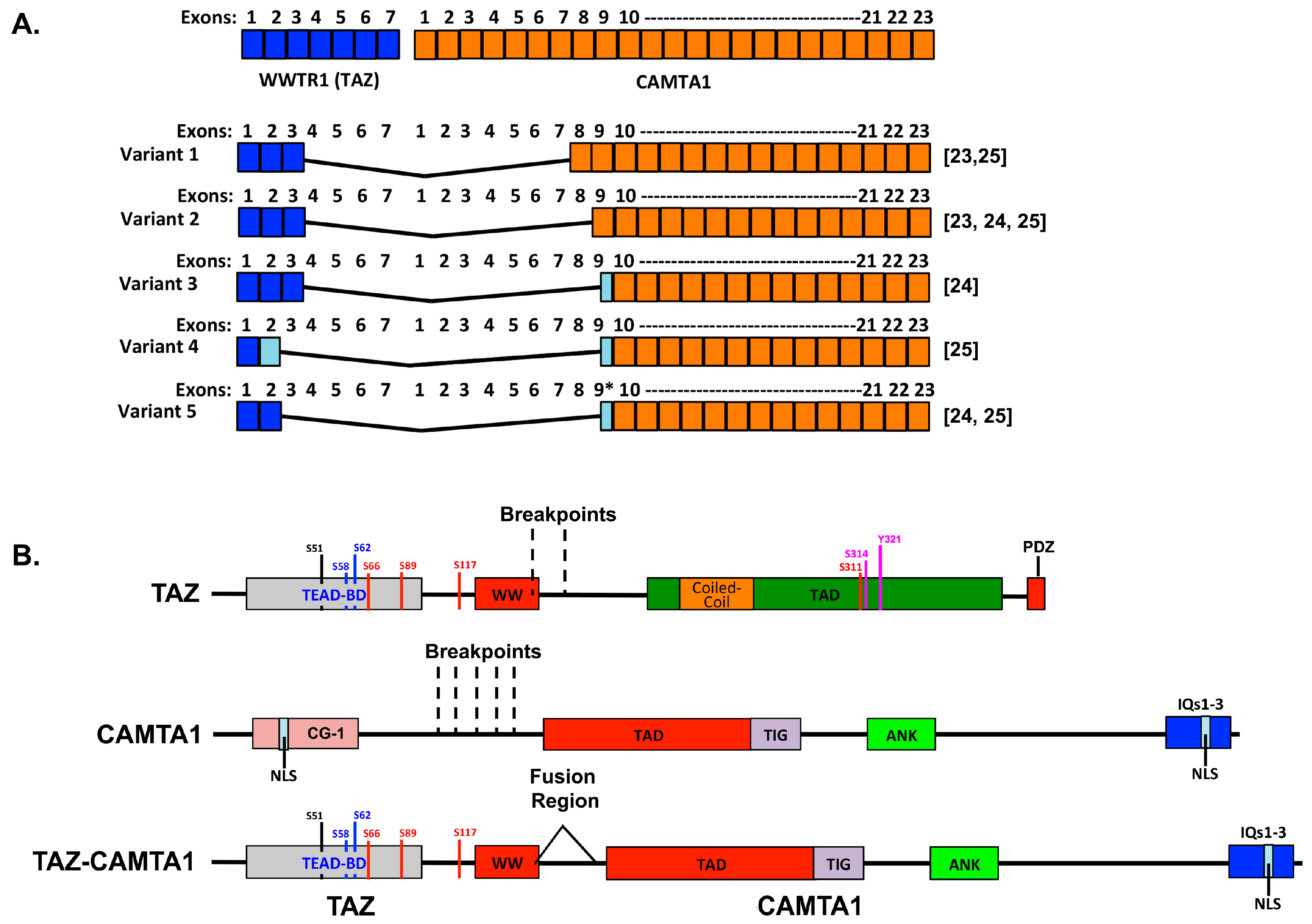
3.1. The WWTR1–CAMTA1 Fusion and Its Role in EHE
3.2. Overlap among Clinical Features of EHE and TAZ-Driven Cell Phenotypes
4. Treatment
4.1. Orthodox Approaches to Treatment
4.2. Untested Potential Treatments
4.3. Strategies to Target the WWTR1–CAMTA1 Fusion to Treat EHE
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| CAMTA | Calmodulin Binding Transcription Activator 1 |
| CT | Computed Tomography |
| CTGF | Connective Tissue Growth Factor |
| EHE | Epithelioid Hemangioendothelioma |
| ERG | ETS related gene |
| GSK-3 | Glycogen synthase kinase-3 |
| LATS | Large Tumor Suppressor Homolog |
| MRI | Magnetic resonance imaging |
| NLS | nuclear localization signal |
| PECAM 1 | Platelet endothelial cell adhesion molecule 1 |
| PI3K | Phosphatidylinositol 4,5-bisphosphate 3-kinase |
| qPCR | quantitative polymerase chain reaction |
| TAD | Transactivation Domain |
| TAZ | Transcriptional Co-activator with PDZ-binding Motif |
| TEAD | TEA Domain Family Member |
| TFE3 | Transcription factor E3 |
| UTR | untranslated region |
| WWTR1 | WW Domain Containing Transcription Regulator 1 |
| YAP | Yes Associated Protein |
References
- Taleb, N.N. The Black Swan: The Impact of the Highly Improbable; Random House: New York, NY, USA, 2007; p. 400. [Google Scholar]
- Garrod, A.E. The incidence of alkaptonuria: A study in chemical individuality. 1902 [classical article]. Yale J. Biol. Med. 2002, 75, 221–231. [Google Scholar] [PubMed]
- Antonescu, C.R.; Le Loarer, F.; Mosquera, J.M.; Sboner, A.; Zhang, L.; Chen, C.L.; Chen, H.W.; Pathan, N.; Krausz, T.; Dickson, B.C.; et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013, 52, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Tanas, M.R.; Ma, S.; Jadaan, F.O.; Ng, C.K.; Weigelt, B.; Reis-Filho, J.S.; Rubin, B.P. Mechanism of action of a WWTR1(TAZ)-CAMTA1 fusion oncoprotein. Oncogene 2016, 35, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.S.A.; Xiao, Y.; Lamar, J.M. YAP/TAZ activation as a target for treating metastatic cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Janse van Rensburg, H.J.; Yang, X. The roles of the hippo pathway in cancer metastasis. Cell Signal. 2016, 28, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Massad, M.; Pollak, C.; Rubin, C.; Yeh, J.; Wang, J.; Edelman, G.; Yeh, J.; Prasad, S.; Weinberg, G. Clinical patterns and outcome in epithelioid hemangioendothelioma with or without pulmonary involvement: Insights from an internet registry in the study of a rare cancer. Chest 2011, 140, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Mallory, F.B. The results of the application of special histological methods to the study of tumors. J. Exp. Med. 1908, 10, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Sardaro, A.; Bardoscia, L.; Petruzzelli, M.F.; Portaluri, M. Epithelioid hemangioendothelioma: An overview and update on a rare vascular tumor. Oncol. Rev. 2014, 8, 259. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.W.; Enzinger, F.M. Epithelioid hemangioendothelioma: A vascular tumor often mistaken for a carcinoma. Cancer 1982, 50, 970–981. [Google Scholar] [CrossRef]
- Corrin, B.; Manners, B.; Millard, M.; Weaver, L. Histogenesis of the so-called “intravascular bronchioloalveolar tumour”. J. Pathol. 1979, 128, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Weldon-Linne, C.M.; Victor, T.A.; Christ, M.L.; Fry, W.A. Angiogenic nature of the “intravascular bronchioloalveolar tumor” of the lung: An electron microscopic study. Arch. Pathol. Lab. Med. 1981, 105, 174–179. [Google Scholar] [PubMed]
- Makhlouf, H.R.; Ishak, K.G.; Goodman, Z.D. Epithelioid hemangioendothelioma of the liver: A clinicopathologic study of 137 cases. Cancer 1999, 85, 562–582. [Google Scholar] [CrossRef]
- Deyrup, A.T.; Tighiouart, M.; Montag, A.G.; Weiss, S.W. Epithelioid hemangioendothelioma of soft tissue: A proposal for risk stratification based on 49 cases. Am. J. Surg. Pathol. 2008, 32, 924–927. [Google Scholar] [CrossRef] [PubMed]
- Groeschl, R.T.; Miura, J.T.; Oshima, K.; Gamblin, T.C.; Turaga, K.K. Does histology predict outcome for malignant vascular tumors of the liver? J. Surg. Oncol. 2014, 109, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C. Malignant vascular tumors—An update. Mod. Pathol. 2014, 27 (Suppl. 1), S30–S38. [Google Scholar] [CrossRef] [PubMed]
- Flucke, U.; Vogels, R.J.; de Saint Aubain Somerhausen, N.; Creytens, D.H.; Riedl, R.G.; van Gorp, J.M.; Milne, A.N.; Huysentruyt, C.J.; Verdijk, M.A.; van Asseldonk, M.M.; et al. Epithelioid hemangioendothelioma: Clinicopathologic, immunhistochemical, and molecular genetic analysis of 39 cases. Diagn. Pathol. 2014, 9, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.A.; Fletcher, C.D.; Hornick, J.L. Nuclear expression of camta1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am. J. Surg. Pathol. 2016, 40, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, R.; Matsuyama, A.; Shiba, E.; Harada, H.; Yabuki, K.; Hisaoka, M. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology 2015, 67, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Mendlick, M.R.; Nelson, M.; Pickering, D.; Johansson, S.L.; Seemayer, T.A.; Neff, J.R.; Vergara, G.; Rosenthal, H.; Bridge, J.A. Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am. J. Surg. Pathol. 2001, 25, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Errani, C.; Zhang, L.; Sung, Y.S.; Hajdu, M.; Singer, S.; Maki, R.G.; Healey, J.H.; Antonescu, C.R. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 2011, 50, 644–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanas, M.R.; Sboner, A.; Oliveira, A.M.; Erickson-Johnson, M.R.; Hespelt, J.; Hanwright, P.J.; Flanagan, J.; Luo, Y.; Fenwick, K.; Natrajan, R.; et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci. Transl. Med. 2011, 3, 98ra82. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.R.; Salim, A.A.; Sayeed, H.; Sarabia, S.F.; Hollingsworth, F.; Warren, M.; Jakacky, J.; Tanas, M.; Oliveira, A.M.; Rubin, B.P.; et al. Molecular characterization of epithelioid haemangioendotheliomas identifies novel WWTR1-CAMTA1 fusion variants. Histopathology 2015, 67, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.; Zhang, L.; Hameed, M.; Rusch, V.; Travis, W.D.; Antonescu, C.R. Thoracic epithelioid malignant vascular tumors: A clinicopathologic study of 52 cases with emphasis on pathologic grading and molecular studies of WWTR1-CAMTA1 fusions. Am. J. Surg. Pathol. 2015, 39, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X. The hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Fu, V.; Plouffe, S.W.; Guan, K.L. The hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell Biol. 2017, 49, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Barbashina, V.; Salazar, P.; Holland, E.C.; Rosenblum, M.K.; Ladanyi, M. Allelic losses at 1p36 and 19q13 in gliomas: Correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin. Cancer Res. 2005, 11, 1119–1128. [Google Scholar] [PubMed]
- Henrich, K.O.; Claas, A.; Praml, C.; Benner, A.; Mollenhauer, J.; Poustka, A.; Schwab, M.; Westermann, F. Allelic variants of CAMTA1 and FLJ10737 within a commonly deleted region at 1p36 in neuroblastoma. Eur. J. Cancer 2007, 43, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Henrich, K.O.; Fischer, M.; Mertens, D.; Benner, A.; Wiedemeyer, R.; Brors, B.; Oberthuer, A.; Berthold, F.; Wei, J.S.; Khan, J.; et al. Reduced expression of camta1 correlates with adverse outcome in neuroblastoma patients. Clin. Cancer Res. 2006, 12, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Schraivogel, D.; Weinmann, L.; Beier, D.; Tabatabai, G.; Eichner, A.; Zhu, J.Y.; Anton, M.; Sixt, M.; Weller, M.; Beier, C.P.; et al. CAMTA1 is a novel tumour suppressor regulated by miR-9/9* in glioblastoma stem cells. EMBO J. 2011, 30, 4309–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bas-Orth, C.; Tan, Y.W.; Oliveira, A.M.; Bengtson, C.P.; Bading, H. The calmodulin-binding transcription activator camta1 is required for long-term memory formation in mice. Learn. Mem. 2016, 23, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Thevenon, J.; Lopez, E.; Keren, B.; Heron, D.; Mignot, C.; Altuzarra, C.; Beri-Dexheimer, M.; Bonnet, C.; Magnin, E.; Burglen, L.; et al. Intragenic camta1 rearrangements cause non-progressive congenital ataxia with or without intellectual disability. J. Med. Genet. 2012, 49, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Huentelman, M.J.; Papassotiropoulos, A.; Craig, D.W.; Hoerndli, F.J.; Pearson, J.V.; Huynh, K.D.; Corneveaux, J.; Hanggi, J.; Mondadori, C.R.; Buchmann, A.; et al. Calmodulin-binding transcription activator 1 (CAMTA1) alleles predispose human episodic memory performance. Hum. Mol. Genet. 2007, 16, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Shinawi, M.; Coorg, R.; Shimony, J.S.; Grange, D.K.; Al-Kateb, H. Intragenic camta1 deletions are associated with a spectrum of neurobehavioral phenotypes. Clin. Genet. 2015, 87, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Grueter, C.E.; Song, K.; Qin, S.; Qi, X.; Kong, Y.M.; Shelton, J.M.; Richardson, J.A.; Zhang, C.L.; Bassel-Duby, R.; et al. Ataxia and purkinje cell degeneration in mice lacking the CAMTA1 transcription factor. Proc. Natl. Acad. Sci. USA 2014, 111, 11521–11526. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Han, J.; Reddig, K.; Li, H.S. A potential dimerization region of dcamta is critical for termination of fly visual response. J. Biol. Chem. 2007, 282, 21253–21258. [Google Scholar] [CrossRef] [PubMed]
- Muller-Borer, B.; Esch, G.; Aldina, R.; Woon, W.; Fox, R.; Bursac, N.; Hiller, S.; Maeda, N.; Shepherd, N.; Jin, J.P.; et al. Calcium dependent camta1 in adult stem cell commitment to a myocardial lineage. PLoS ONE 2012, 7, e38454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, K.O.; Bauer, T.; Schulte, J.; Ehemann, V.; Deubzer, H.; Gogolin, S.; Muth, D.; Fischer, M.; Benner, A.; Konig, R.; et al. CAMTA1, a 1p36 tumor suppressor candidate, inhibits growth and activates differentiation programs in neuroblastoma cells. Cancer Res. 2011, 71, 3142–3151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, C.Y.; Zha, Z.Y.; Zhao, B.; Yao, J.; Zhao, S.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. Tead transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 2009, 284, 13355–13362. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. Taz: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.D.; Tremblay, A.M.; Murray, G.I.; Wackerhage, H. The hippo signal transduction pathway in soft tissue sarcomas. Biochim. Biophys. Acta 2015, 1856, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Eisinger-Mathason, T.S. Targeting the hippo pathway: Clinical implications and therapeutics. Pharmacol. Res. 2015, 103, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Xu, S.; Guo, F. TEAD1 mediates the oncogenic activities of Hippo-YAP1 signaling in osteosarcoma. Biochem. Biophys. Res. Commun. 2017, 488, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Huang, K.; Ma, Y.; Zhou, M.; Fan, S. The TAZ-miR-224-SMAD4 axis promotes tumorigenesis in osteosarcoma. Cell Death Dis. 2017, 8, e2539. [Google Scholar] [CrossRef] [PubMed]
- Slemmons, K.K.; Crose, L.E.S.; Riedel, S.; Sushnitha, M.; Belyea, B.C.; Linardic, C.M. A novel notch-YAP circuit drives stemness and tumorigenesis in embryonal rhabdomyosarcoma. Mol. Cancer Res. 2017, 15, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Crose, L.E.; Galindo, K.A.; Kephart, J.G.; Chen, C.; Fitamant, J.; Bardeesy, N.; Bentley, R.C.; Galindo, R.L.; Chi, J.T.; Linardic, C.M. Alveolar rhabdomyosarcoma-associated PAX3-FOXO1 promotes tumorigenesis via hippo pathway suppression. J. Clin. Investig. 2014, 124, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Slemmons, K.K.; Crose, L.E.; Rudzinski, E.; Bentley, R.C.; Linardic, C.M. Role of the YAP Oncoprotein in Priming Ras-Driven Rhabdomyosarcoma. PLoS ONE 2015, 10, e0140781. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The hippo transducer yap1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Sun, C.; De Mello, V.; Selfe, J.; Missiaglia, E.; Shipley, J.; Murray, G.I.; Zammit, P.S.; Wackerhage, H. The hippo effector TAZ (WWTR1) transforms myoblasts and its abundance is associated with reduced survival in embryonal rhabdomyosarcoma. J. Pathol. 2016, 240, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Deel, M.D.; Slemmons, K.K.; Hinson, A.R.; Genadry, K.C.; Burgess, B.A.; Crose, L.E.S.; Kuprasertkul, N.; Oristian, K.M.; Bentley, R.C.; Linardic, C.M. The transcriptional coactivator taz is a potent mediator of alveolar rhabdomyosarcoma tumorigenesis. Clin. Cancer Res. 2018, 24, 2616–2630. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.; Mucaj, V.; Biju, K.M.; Nakazawa, M.S.; Gohil, M.; Cash, T.P.; Yoon, S.S.; Skuli, N.; Park, K.M.; Gerecht, S.; et al. Deregulation of the hippo pathway in soft-tissue sarcoma promotes foxm1 expression and tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E3402–E3411. [Google Scholar] [CrossRef] [PubMed]
- Deel, M.D.; Li, J.J.; Crose, L.E.; Linardic, C.M. A review: Molecular aberrations within hippo signaling in bone and soft-tissue sarcomas. Front. Oncol. 2015, 5, 190. [Google Scholar] [CrossRef] [PubMed]
- Fullenkamp, C.A.; Hall, S.L.; Jaber, O.I.; Pakalniskis, B.L.; Savage, E.C.; Savage, J.M.; Ofori-Amanfo, G.K.; Lambertz, A.M.; Ivins, S.D.; Stipp, C.S.; et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget 2016, 7, 30094–30108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, T.; Cottrill, K.A.; Annis, S.; Bhat, B.; Gochuico, B.R.; Osorio, J.C.; Rosas, I.; Haley, K.J.; Corey, K.E.; Chung, R.T.; et al. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci. Rep. 2015, 5, 18277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Yu, M.; Xia, R.; Song, K.; Wang, J.; Luo, J.; Chen, G.; Cheng, J. YAP/TAZ deletion in Gli(+) cell-derived myofibroblasts attenuates fibrosis. J. Am. Soc. Nephrol. 2017, 28, 3278–3290. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Mikami, Y.; Urushiyama, H.; Horie, M.; Matsuzaki, H.; Takeshima, H.; Makita, K.; Miyashita, N.; Mitani, A.; et al. Taz contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci. Rep. 2017, 7, 42595. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Looney, A.P.; Baker, B.M.; Stawski, L.; Haines, P.; Simms, R.; Szymaniak, A.D.; Varelas, X.; Trojanowska, M. Therapeutic targeting of TAZ and YAP by dimethyl fumarate in systemic sclerosis fibrosis. J. Investig. Dermatol. 2018, 138, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ are mechanoregulators of TGF-Beta-Smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.Z.; Bialik, J.F.; Speight, P.; Dan, Q.; Yeung, T.; Szaszi, K.; Pedersen, S.F.; Kapus, A. TGF-Beta1 regulates the expression and transcriptional activity of TAZ protein via a SMAD3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017, 292, 14902–14920. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, A.J.; Choi, K.M.; Sicard, D.; Smith, K.M.; Hiemer, S.E.; Varelas, X.; Tschumperlin, D.J. Taz activation drives fibroblast spheroid growth, expression of profibrotic paracrine signals, and context-dependent ecm gene expression. Am. J. Physiol. Cell Physiol. 2017, 312, C277–C285. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Guo, S.; Zhang, Y.; Yin, J.; Yin, W.; Tao, S.; Wang, Y.; Zhang, C. Proton-sensing gpcr-yap signalling promotes cancer-associated fibroblast activation of mesenchymal stem cells. Int. J. Biol. Sci. 2016, 12, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Y.; Chang, C.C.; Prakash, E.; Kuo, M.L. Connective tissue growth factor (CTGF) and cancer progression. J. Biomed. Sci. 2008, 15, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: Lessons from transgenic animals. J. Cell Commun. Signal. 2010, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem. Cell Biol. 2003, 81, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Haberal Reyhan, N. Liver transplant for nonhepatocellular carcinoma malignancy. Exp. Clin. Transplant. 2017, 15, 69–73. [Google Scholar] [PubMed]
- Eghtesad, B.; Aucejo, F. Liver transplantation for malignancies. J. Gastrointest. Cancer 2014, 45, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Bufton, S.; Haydon, G.; Neil, D. Liver transplantation for hepatic epithelioid hemangioendothelioma: A case series. Prog. Transplant. 2007, 17, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Mehrabi, A.; Kashfi, A.; Fonouni, H.; Schemmer, P.; Schmied, B.M.; Hallscheidt, P.; Schirmacher, P.; Weitz, J.; Friess, H.; Buchler, M.W.; et al. Primary malignant hepatic epithelioid hemangioendothelioma: A comprehensive review of the literature with emphasis on the surgical therapy. Cancer 2006, 107, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Feys, E.; Karam, V.; Adam, R.; Klempnauer, J.; Oliverius, M.; Mazzaferro, V.; Pascher, A.; Remiszewski, P.; Isoniemi, H.; et al. Hepatic epithelioid hemangioendothelioma and adult liver transplantation: Proposal for a prognostic score based on the analysis of the ELTR-ELITA registry. Transplantation 2017, 101, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.; Agulnik, M. Epithelioid hemangioendothelioma: Update on diagnosis and treatment. Curr. Treat. Options Oncol. 2018, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Idilman, R.; Dokmeci, A.; Beyler, A.R.; Bastemir, M.; Ormeci, N.; Aras, N.; Ekinci, C.; Uzunalimoglu, O.; De Maria, N.; Van Thiel, D.H. Successful medical treatment of an epithelioid hemangioendothelioma of liver. Oncology 1997, 54, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Li, W.; Feng, J.; Shi, J.X.; Chen, Y.; Han, B.H. Treatment of pulmonary epithelioid hemangioendothelioma with combination chemotherapy: Report of three cases and review of the literature. Oncol. Lett. 2013, 5, 1491–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Malangone, S.; Green, M.; Badari, A.; Clarke, K.; Elquza, E. Combination capecitabine and bevacizumab in the treatment of metastatic hepatic epithelioid hemangioendothelioma. Ther. Adv. Med. Oncol. 2015, 7, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinet, C.; Magnan, A.; Garbe, L.; Payan, M.J.; Vervloet, D. Aggressive form of pleural epithelioid haemangioendothelioma: Complete response after chemotherapy. Eur. Respir. J. 1999, 14, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, A.; Fuhrer, G.; Malekiani, C.; McKay, S.; Thurber, J. Primary pleural epithelioid hemangioendothelioma (EHE)—Two cases and review of the literature. Clin. Respir. J. 2011, 5, e1–e5. [Google Scholar] [CrossRef] [PubMed]
- Semenisty, V.; Naroditsky, I.; Keidar, Z.; Bar-Sela, G. Pazopanib for metastatic pulmonary epithelioid hemangioendothelioma—A suitable treatment option: Case report and review of anti-angiogenic treatment options. BMC Cancer 2015, 15, 402. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Shimamura, T.; Tokuhisa, M.; Goto, A.; Ichikawa, Y. Sorafenib monotherapy in a patient with unresectable hepatic epithelioid hemangioendothelioma. Case Rep. Oncol. 2016, 9, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Gaur, S.; Torabi, A.; O’Neill, T.J. Activity of angiogenesis inhibitors in metastatic epithelioid hemangioendothelioma: A case report. Cancer Biol. Med. 2012, 9, 133–136. [Google Scholar] [PubMed]
- Agulnik, M.; Yarber, J.L.; Okuno, S.H.; von Mehren, M.; Jovanovic, B.D.; Brockstein, B.E.; Evens, A.M.; Benjamin, R.S. An open-label, multicenter, phase ii study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann. Oncol. 2013, 24, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wang, H.; Jiang, H.; Chen, E.; Zhang, J.; Xie, X. Apatinib for the treatment of pulmonary epithelioid hemangioendothelioma: A case report and literature review. Medicine 2017, 96, e8507. [Google Scholar] [CrossRef] [PubMed]
- Raphael, C.; Hudson, E.; Williams, L.; Lester, J.F.; Savage, P.M. Successful treatment of metastatic hepatic epithelioid hemangioendothelioma with thalidomide: A case report. J. Med. Case Rep. 2010, 4, 413. [Google Scholar] [CrossRef] [PubMed]
- Salech, F.; Valderrama, S.; Nervi, B.; Rodriguez, J.C.; Oksenberg, D.; Koch, A.; Smok, G.; Duarte, I.; Perez-Ayuso, R.M.; Jarufe, N.; et al. Thalidomide for the treatment of metastatic hepatic epithelioid hemangioendothelioma: A case report with a long term follow-up. Ann. Hepatol. 2011, 10, 99–102. [Google Scholar] [PubMed]
- Kanemura, S.; Kuribayashi, K.; Moriya, Y.; Shimizu, S.; Tsujimura, T.; Nakano, T. Pemetrexed for epithelioid haemangioendothelioma of the pleura. Respirol. Case Rep. 2016, 4, e00191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacchiotti, S.; Provenzano, S.; Dagrada, G.; Negri, T.; Brich, S.; Basso, U.; Brunello, A.; Grosso, F.; Galli, L.; Palassini, E.; et al. Sirolimus in advanced epithelioid hemangioendothelioma: A retrospective case-series analysis from the italian rare cancer network database. Ann. Surg. Oncol. 2016, 23, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.G.; Ng, Y.L.; Lam, W.L.; Plouffe, S.W.; Guan, K.L. The hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to MTORC1. Cell Res. 2015, 25, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibault, F.; Corvaisier, M.; Bailly, F.; Huet, G.; Melnyk, P.; Cotelle, P. Non-photoinduced biological properties of verteporfin. Curr. Med. Chem. 2016, 23, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Q.; Dai, Y.Y.; Hsu, P.C.; Wang, H.; Cheng, L.; Yang, Y.L.; Wang, Y.C.; Xu, Z.D.; Liu, S.; Chan, G.; et al. Targeting YAP in malignant pleural mesothelioma. J. Cell. Mol. Med. 2017, 21, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Gou, J.; Jia, J.; Yi, T.; Cui, T.; Li, Z. Verteporfin, a suppressor of yap-tead complex, presents promising antitumor properties on ovarian cancer. OncoTargets Ther. 2016, 9, 5371–5381. [Google Scholar]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Cao, L.; Rahma, O.E.; Makarova-Rusher, O.V.; Kleiner, D.E.; Fioravanti, S.; Walker, M.; Carey, S.; Yu, Y.; et al. A phase ii study of TRC105 in patients with hepatocellular carcinoma who have progressed on sorafenib. United Eur. Gastroenterol. J. 2015, 3, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Apolo, A.B.; Karzai, F.H.; Trepel, J.B.; Alarcon, S.; Lee, S.; Lee, M.J.; Tomita, Y.; Cao, L.; Yu, Y.; Merino, M.J.; et al. A phase ii clinical trial of TRC105 (anti-endoglin antibody) in adults with advanced/metastatic urothelial carcinoma. Clin. Genitourin. Cancer 2017, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Brian, R.; (Cleveland, OH, USA). Personal Communication, 2018.
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The hippo pathway component TAZ promotes immune evasion in human cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-induced PD-L1 expression drives immune evasion in brafi-resistant melanoma. Cancer Immunol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodaka, M.; Hata, Y. The mammalian hippo pathway: Regulation and function of YAP1 and TAZ. Cell. Mol. Life Sci. 2015, 72, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lv, X.; Liu, C.; Zha, Z.; Zhang, H.; Jiang, Y.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. The n-terminal phosphodegron targets TAZ/WWTR1 protein for SCFBeta-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J. Biol. Chem. 2012, 287, 26245–26253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, N.; Li, X.; Tran, M.K.; Han, X.; Chen, J. Tankyrase inhibitors target YAP by stabilizing angiomotin family proteins. Cell Rep. 2015, 13, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Troilo, A.; Benson, E.K.; Esposito, D.; Garibsingh, R.A.; Reddy, E.P.; Mungamuri, S.K.; Aaronson, S.A. Angiomotin stabilization by tankyrase inhibitors antagonizes constitutive tead-dependent transcription and proliferation of human tumor cells with hippo pathway core component mutations. Oncotarget 2016, 7, 28765–28782. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, B.; Castillo, J.; Zhang, Y.; Yang, Z.; McAllister, G.; Lindeman, A.; Reece-Hoyes, J.; Tallarico, J.; Russ, C.; et al. Tankyrase inhibitor sensitizes lung cancer cells to endothelial growth factor receptor (EGFR) inhibition via stabilizing angiomotins and inhibiting YAP signaling. J. Biol. Chem. 2016, 291, 15256–15266. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Qiao, Y.; Pilo, M.G.; Cigliano, A.; Liu, X.; Shao, Z.; Calvisi, D.F.; Chen, X. Tankyrase inhibitors suppress hepatocellular carcinoma cell growth via modulating the hippo cascade. PLoS ONE 2017, 12, e0184068. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Miyoshi, Y.; Takahata, C.; Irahara, N.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Down-regulation of LATS1 and LATS2 mrna expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin. Cancer Res. 2005, 11, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schagdarsurengin, U.; Blumke, K.; Wurl, P.; Pfeifer, G.P.; Hauptmann, S.; Taubert, H.; Dammann, R. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol. Carcinog. 2007, 46, 865–871. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamar, J.M.; Motilal Nehru, V.; Weinberg, G. Epithelioid Hemangioendothelioma as a Model of YAP/TAZ-Driven Cancer: Insights from a Rare Fusion Sarcoma. Cancers 2018, 10, 229. https://doi.org/10.3390/cancers10070229
Lamar JM, Motilal Nehru V, Weinberg G. Epithelioid Hemangioendothelioma as a Model of YAP/TAZ-Driven Cancer: Insights from a Rare Fusion Sarcoma. Cancers. 2018; 10(7):229. https://doi.org/10.3390/cancers10070229
Chicago/Turabian StyleLamar, John M., Vijeyaluxmy Motilal Nehru, and Guy Weinberg. 2018. "Epithelioid Hemangioendothelioma as a Model of YAP/TAZ-Driven Cancer: Insights from a Rare Fusion Sarcoma" Cancers 10, no. 7: 229. https://doi.org/10.3390/cancers10070229