Targeted Tumor Therapy Remixed—An Update on the Use of Small-Molecule Drugs in Combination Therapies


Department of Dermatology and Allergic Diseases, University of Ulm, 89081 Ulm, Germany
Cancers 2018, 10(6), 155; https://doi.org/10.3390/cancers10060155
Submission received: 23 April 2018
/
Revised: 18 May 2018
/
Accepted: 22 May 2018
/
Published: 24 May 2018
Abstract
:Over the last decade, the treatment of tumor patients has been revolutionized by the highly successful introduction of novel targeted therapies, in particular small-molecule kinase inhibitors and monoclonal antibodies, as well as by immunotherapies. Depending on the mutational status, BRAF and MEK inhibitor combinations or immune checkpoint inhibitors are current first-line treatments for metastatic melanoma. However, despite great improvements of survival rates limitations due to tumor heterogeneity, primary and acquired therapy resistance, immune evasion, and economical considerations will need to be overcome. Accordingly, ongoing clinical trials explore the individualized use of small-molecule drugs in new targeted therapy combinations based on patient parameters and tumor biopsies. With focus on melanoma therapy this review aims at providing a comprehensive overview of such novel alternative and combinational therapy strategies currently emerging from basic research. The molecular principles and drug classes that may hold promise for improved tumor therapy combination regimens including kinase inhibition, induction of apoptosis, DNA-damage response inhibition, epigenetic reprogramming, telomerase inhibition, redox modulation, metabolic reprogramming, proteasome inhibition, cancer stem cell transdifferentiation, immune cell signaling modulation, and others, are explained in brief. In addition, relevant targeted therapy combinations in current clinical trials and individualized treatment strategies are highlighted.

1. Introduction
Cancer pathogenesis evolves like a drama with a series of acts—including tumor initiation, promotion, progression, invasion, immune evasion, metastasis, chemo-resistance, and further progression ([1,2,3] and others). As per the model of multi-step tumorigenesis, mutations in oncogenes and in tumor suppressor genes as well as further genetic and epigenetic changes mediate this transformation process [1,2]. In interaction with the tumor micro-environment tumor cell plasticity and clonal evolution decide about tumor cell survival and aggressiveness-leading to substantial heterogeneity within the tumor as major limitation to durable therapeutic responses [4]. Conventional cytotoxic drugs (“chemotherapy”) for the treatment of advanced tumors include DNA-alkylating agents, anti-metabolites, anthracyclines, vinca alkaloids, topoisomerase inhibitors, and others and commonly target cycling cells with substantial side-effects in normal proliferative tissues like hair loss, diarrhea, and infections [5]. For certain types of tumors derived from hormonally responsive tissues, i.e., breast, endometrium, prostate, testes, thyroid, and adrenal cortex, hormone modulation and hormone receptor antagonists constitute effective anti-cancer therapy options [6].
With improved sequencing techniques and deeper molecular understanding of cancer pathogenesis, new targeted therapies could be developed-with the advantage of greater specificity towards molecular pathways altered in cancer cells compared with “classical chemotherapy” (recently reviewed in [7]). Apart from approved antibodies and kinase inhibitors, targeting either growth factor receptor tyrosine kinases (EGFR, PDGFR, VEGFR, c-MET et al.), downstream kinases (BRAF, MEK, AKT, mTOR et al.), or fusion proteins (BCR-ABL) dysregulated in tumors, a number of alternative small-molecule drugs and natural compounds are available for tumor therapy. Most recently, novel immunotherapies, in particular checkpoint inhibition with anti-CTLA-4 and anti-PD-1/-PD-1L antibodies targeting negative regulatory receptors on T cells, have been successfully applied against a growing number of tumor types [8]. Because intrinsic and acquired therapy resistance to approved cytotoxic and targeted therapies, and in part also to immunotherapies, still represent a major hurdle to long-term progression-free survival (PFS) and lasting remissions in cancer patients [7,8,9], different classes of small-molecule drugs are currently under investigation in pre-clinical and clinical trials in alternative combination regimens and will be discussed in this review.
2. Approved Targeted Therapy Regimens against Kinase Pathways and Their Limitations
In melanoma, the most common driver mutation occurring in over 50% of patients affects the serine/threonine protein kinase BRAF (BRAFV600E), a downstream effector of growth factor receptor signaling, that can be therapeutically targeted by specific BRAFV600mut inhibitors (BRAFi) [10]. Because of the rapid development of acquired resistance during BRAFi monotherapy within months, combination therapy with BRAFi and MEK-inhibitors (MEKi) is currently standard in the treatment of BRAFV600mut-positive stage IV melanoma patients [11]. Combination treatment with BRAFi and MEKi has synergistic anti-tumor effects, reduced toxic side effects, and prolongs resistance development to a year on average. Depending on the presence of NRAS- or c-KIT-mutations, either MEKi, c-KIT inhibitors, immunotherapy with anti-PD-1 checkpoint blockers, or polychemotherapy are recommended for the treatment of advanced non-BRAFV600mut melanoma [12]. Different mechanisms of acquired therapy resistance to either monotherapy with BRAFi or combination treatment could be identified in melanoma [8,13,14,15]: In response to BRAFi, reactivation of the mitogen activated protein kinase (MAPK) pathway, e.g., due to switching among BRAF, ARAF and CRAF isoforms, and enhanced insulin like growth factor (IGF)-IR/PI3K signaling occurred [13]. Reduced cytotoxicity mediated by upregulation of BCL-2 family pro-survival factors through activation of the PI3K/AKT pathway downstream of IGF-R or in context with activation of the hepatocyte growth factor (HGF)/c-MET or fibroblast growth factor receptor (FGF) pathway or via melanocyte master regulator microphthalmia associated transcription factor (MITF) was also observed under BRAFi and MEKi combination therapy. In addition, BRAFi and MEKi treatment may interfere with immune responses to melanoma cells.
Similarly, the targeted therapy combination with BRAFi and MEKi has recently been approved for the treatment of a specific type of metastatic non-small cell lung carcinoma (NSCLC) by the US Food and Drug Administration (FDA) [16]. In other types of lung cancer with epidermal growth factor receptor (EGFR) mutations, commonly tyrosine kinase inhibitors (TKi) against the mutant EGFR or anti-EFGR antibodies are applied alone or in new combination therapy trials with other anti-cancer therapeutics [17]. Combination therapy is also key to the treatment of metastatic colorectal cancer (CRC)-most frequently bearing KRAS, NRAS, BRAF, EGFR, and MYC mutations, loss of functions, amplifications, and microsatellite instability (MSI)-as reflected by ongoing clinical trials (e.g., HERACLES trial, NCT03225937) [18]. In general, mutations in key oncogenic kinase signaling pathways-including the RAS-RAF-MEK-ERK/MAPK, the PI3K/PTEN-AKT-mTOR, the JAK-STAT-or the WNT/β-catenin/MYC pathways occur with high frequency in a variety of tumors [2,3]. Because of parallel activation and significant signaling cross talk between these pathways, successive and combination therapies targeting different components with established small-molecule inhibitors based on an individual assessment of tumor or liquid biopsies may have the potential to overcome, or at least delay, therapy resistance (e.g., my pathway trial, NCT02091141 [7,19]).
3. Alternative and Emerging Strategies for Improved Targeted Cancer Therapy
Apart from oncogenic kinases in growth factor receptor signaling, a variety of cellular pathways are altered in tumor cells and may provide alternative druggable targets for tumor therapy (summarized in Figure 1). The following sections will provide an overview of molecular pathways and functions commonly altered in tumor cells in context with emerging strategies how to therapeutically interfere with these processes with small-molecule agents (Table 1). Moreover, selected current approaches targeting the tumor micro-environment—extracellular matrix, angiogenesis, or immune cells—will be addressed in brief [20]. With focus on treatment options for advanced melanoma and therapy resistance management, information about small-molecule tumor therapies and potentially beneficial therapy combinations in ongoing clinical trials was extracted from listings in public databases [19] and published research articles and will be highlighted.
3.1. Kinase Inhibitor Combinations
Based on a proven principle the use of new kinase inhibitors and inhibitor combinations may enhance established first line medications-because additional mutations or activation of compensatory growth factor signaling pathways are frequently observed in response to cancer therapy. To overcome the acquisition of melanoma therapy resistance commonly occurring on average after 12 months in patients on BRAFi and MEKi combinations [7], the ongoing LOGIC-2 clinical trial explores the effects of a third agent for the treatment of advanced BRAFV600-positive melanoma. Based on the genetic assessment of a tumor biopsy taken at progression of disease (PD) patients will receive either an inhibitor of cyclin-dependent kinases CDK4/6 (ribociclib, LEE011), of class I PI3K (buparlisib, BKM120), of the c-MET receptor tyrosine kinase (capmatinib, INC280), or of fibroblast growth factor receptor (FGFR) kinase (BGJ398) in a triple therapy approach (NCT02159066). Whereas the rationale for use of PI3Ki, METi, or FGFRi lies in the potential parallel activation of respective growth factor signaling pathways, adding CDKi may be beneficial for the treatment of tumors with elevated CDK4/6 activity or genetic alterations in the Cyclin D-CDK4/6-p16INK4A-RB pathway. In addition, a triple combination of BRAFi and MEKi with a heat shock protein 90 (HSP90) inhibitor (onalespib) is being tested in an ongoing phase I trial (NCT02097225).
The efficacy of the combination of MEKi (binimetinib/MEK162) and CDK4/6 inhibitors is currently also assessed in patients with advanced KRAS-mutant non-small cell lung cancer (NSCLC) (palbociclib, NCT03170206)—and will be followed up on in NRAS-mutant melanoma based on promising phase Ib trial results (ribociclib, NCT01781572). In advanced squamous cell lung, pancreatic, head & neck and other solid tumors, combining the inhibition of CDK4/6 (palbociclib) and PI3K/mTOR (gedatolisib) may have advantage over the use of each drug alone or the Platinum-based chemotherapy (NCT03065062). For another type of NSCLC with activation of the c-MET pathway, the efficacy of MET inhibitors (crizotinib and capmatinib, INC280) is being assessed either as monotherapy (NCT02414139) or in combination with epidermal growth factor (EGF) tyrosine kinase inhibitor erlotinib (NCT01911507).
To target the signaling of angiogenic factors that control tumor neovascularization, including vascular epithelial growth factor (VEGF), PDGF, and FGF receptor tyrosine kinase signaling, oral multi-kinase inhibitors, such as lucitanib, have been tested in certain forms of NSCLC and breast cancer with genetic FGR alterations [21]. The combination of VEGF inhibitor nintedanib with paclitaxel chemotherapy may provide an alternative therapy regimen for patients with BRAFWT metastatic melanoma (NIPAWILMA, NCT02308553). Similarly, inhibitors of focal adhesion kinase (FAK), a key regulator of integrin signaling, target tumor cell proliferation, invasion, metastasis, and angiogenesis and are promising drugs for combination therapy—in melanoma with BRAFi and in other tumors such as CRC with activated stroma—because of a limitation of tumor cell escape mechanisms [22,23]. Due to supporting effects of FAK inhibition on CD8+ T cell adaptive immune responses with potential synergy with anti-PD1 therapy in preclinical studies, a current trial explores the safety and tolerability of the combination of FAKi defacitinib with anti-PD1 antibody pembrolizumab in advanced solid tumors (NCT02758587).
3.2. Apoptosis Induction and Autophagy Modulation
Most anti-cancer therapies directly or indirectly exploit “programmed cell death” (apoptosis) and other cell death pathways. However, during step-wise transformation, tumor cells acquire various genetic alterations to reduce their sensitivity to cell death and increase their survival under stress conditions—limiting the efficacy of “cell death drugs” at doses that will not harm healthy cells [1,2,3]. A number of emerging therapeutics therefore focus on “reactivating” cell death programs in tumor cells. In general, apoptosis, the so called “programmed cell death”, can be initiated by an extrinsic (death receptor) and an intrinsic (mitochondrial) death pathway—both leading to a common death executing program mediated by the Caspase family of proteases (reviewed in [24]). Accordingly, drugs that either trigger extrinsic death receptor signaling or enhance intrinsic mitochondrial pathways are currently tested for clinical use in tumor therapy. In addition, inhibition of autophagy may enhance tumor cell apoptosis.
Because death receptors (DR), including CD95 (Fas/APO1), DR3, DR6, TNF-R1, and TNF-related apoptosis-inducing factor (TRAIL)-R1/DR4 and TRAIL-R2/DR5, depending on their expression, have the ability to trigger apoptosis in most tumor cells, strategies to activate death signaling via DR agonists or agonistic antibodies have strong therapeutic potential against cancer. In melanoma, due to variable expression of TRAIL-R1/DR4, TRAIL-R2/DR5, and other DRs, the choice and specificity of the agonistic antibody as well as its ability to crosslink Fcγ receptors on myeloid cells turned out to be crucial for effective induction of apoptosis signaling and therapeutic efficacy [25]. An ongoing clinical study investigates the potential of a TRAIL/DR5 antibody (DS-8273a) to augment the clinical efficacy of PD1-blocker nivolumab in melanoma combination therapy (NCT02983006).
Different classes of small-molecule drugs enforce the intrinsic mitochondrial pathway: On the one hand, by mimicking the natural antagonists of BCL-2 family survival proteins (so called BH3-mimetics or BCL-2 family inhibitors) or of the inhibitors of apoptosis (IAPs) (so called SMAC-mimetics); on the other hand, by directly targeting BCL-2 expression with antisense oligonucleotides—all sensitizing tumor cells to death [26]. In melanoma and other tumors, resistance to kinase inhibitors like BRAFi and MEKi is often mediated by an abrogation of intrinsic apoptosis signaling pathways—namely by downregulation of BH3-only proteins or by induction and activation of BCL-2 family members like BCL-2, BCL-XL, BFL-1, and MCL-1 [27]. While melanoma and other solid tumor are mostly insensitive against mono-therapy with either BH3-or SMAC-mimetics, combination approaches of kinase inhibitors with apoptosis mimetics or epigenetic drugs inducing BH3-proteins (see Section 3.4) have the potential to override drug resistance. In melanoma resistant to BRAFi and MEKi, in particular BH3-mimetics targeting MCL-1 and manipulation of the MCL-1/NOXA-axis maybe beneficial and warrant clinical testing [28]. The BH3-mimetic venetoclax, selectively inhibiting BCL-2, often overexpressed in leukemia and lymphoma, has been approved for the treatment of refractory chronic lymphatic leukemia (CLL) [29]. Currently, this drug is being tested in combination with navitoclax and chemotherapy in acute lymphoid leukemia (ALL) (NCT03181126) and with various other targeted therapeutics in different types hematologic malignancies. In addition, a first Phase-Ib dose-finding study evaluates the SMAC-mimetic debio 1143 in combination with anti-PD-L1 antibody avelumab in patients with advanced solid malignancies, especially NSCLC after Platinum-based therapy (NCT03270176).
Another promising strategy to enhance tumor cell apoptosis involves the “re-activation” of tumor suppressor p53, a key player in apoptosis and cell cycle arrest that is frequently mutated and inactivated in various cancers [30]. Restoration of p53 function in melanoma, often expressing inactivated wildtype p53, and in other tumors can be archived by targeting its antagonists, E3 ligase Mdm2, Mdm4, Mdmx, and inhibitors of apoptosis stimulating protein of p53 (iASPPs) [31] or through direct p53 activators [32]. p53 reactivation may have synergistic effects with BRAFi potentially overcoming therapeutic resistances. In a current phase-I dose-escalation study, an MDM2 antagonist (RO6839921) is intravenously applied in patients with advanced malignancies, including acute myeloid leukemia (AML) (NCT02098967).
In addition, targeting autophagy, another homeostatic cellular process pathway involved in survival and proliferation of tumor cells, with established drugs, such as the anti-malaria agent hydroxychloroquine (HCQ), may induce tumor cell apoptosis and enhance the potency of many cancer therapies [33]. Hence, several ongoing clinical trials are conducted to evaluate the anti-tumor effects of autophagy inhibition in a variety of solid tumors in mono- or combination therapy. These include clinical studies assessing the safety and efficacy of the combination of hydroxychloroquine with histone deacetylase (HDAC) inhibitor vorinostat in refractory metastatic colorectal cancer (mCRC) (NCT02316340) and in other advanced solid tumors (NCT01023737, also see Section 3.4).
3.3. DNA Damage Response Inhibitors
In response to DNA insults caused by endogenous and exogenous stressors—including reactive oxygen species (ROS), stalled replication (RS), DNA-damaging drugs, or irradiation—normal cells and tumor cells activate so called DNA-damage response (DDR) pathways. Among the principle outcomes of DDRs are either transient cell cycle arrest and restoration of the damaged nucleotide-sequence of the DNA (DNA repair) or apoptosis and clearance of the damaged cells (reviewed in [34]). In a variety of tumors, functional inactivation of such DDR pathways contributes to increased genomic instability and mutational load, but also sensitizes cells towards standard genotoxic (DNA damaging) treatments—providing a rationale to explore the use of DDR inhibitors in novel combination therapy regimens.
Among the main therapeutic targets for DDR inhibitors, currently being tested in phase 0‒II clinical trials, are the phosphatidyilinositol-3-OH-kinase (PI3K) family kinases ataxia teleangiectasia mutated (ATM) and RAD3-related (ATR) and their downstream DDR kinases CHK1 and WEE1 [35]. In addition, inhibitors of poly(ADP-ribose) polymerase (PARP), an enzyme involved in base excision repair (BER) of single strand breaks (SSB), are clinically evaluated singly or in combination with chemotherapy [36]. In brief, ATM kinase is activated in response to double strand breaks (DBS), initially recognized by the so called MRN-complex (composed of MRE11, RAD50, and NBS1). ATM then phosphorylates histone H2AX on serine residue S139 close to the break (γH2AX) as an amplifier to recruit more MRN-molecules and additional DNA repair proteins like BRCA1 and 53BP1. This leads to the activation of the G1-S cell cycle checkpoint and p53-dependent induction of CDK-inhibitor p21 WAF1/CIP1 as well as apoptosis genes. In contrast, single stranded DNA (ssDNA), arising at either stalled replication forks or at resected DSB, causes activation of ATR kinase that mediates cell cycle arrest and DNA repair at the break via downstream kinase CHK1. In addition, PARP acts as “molecular sensor” to identify SSB and contributes DNA repair.
Despite some limitations in the use of DDR inhibitors due to lipophilicity and toxicity in proliferating normal cells, the concept of synthetic lethality based on mutations in DDR pathways in tumor cells can be exploited therapeutically. Based on the enhanced sensitivity of tumor cells with inactivated tumor suppressor genes BRCA 1 and 2 towards PARP-1 inhibition [37], the PARP inhibitors olaparib, rucaparib, and niraparib could recently receive FDA approval for the treatment of BRCA-mutant ovarian and refractory breast cancers. Similarly, due to mutations in the p53/RB-pathway, a deregulated G1 cell cycle checkpoint, and increased replication stress tumor cells often fully depend on the S/G2 checkpoint. This generates a therapeutic window for ATR and CHK1 inhibitors and increases the sensitivity to DNA damaging drugs [38]. ATM inhibitors have been shown to be effective in pre-clinical studies in brain and prostate cancers in combination with chemo-or radiotherapy with greater sensitivity of p53-mutant or PTEN-mutant cells, respectively [39]. Following up on the pre-clinical synthetic lethality of ATM loss-of-function in combination with drugs inhibiting either growth factor kinases MEK1/2 or PARP [40,41,42], a current phase-I clinical trial assesses the ATM inhibitor AZD01566 alone or in combination with PARPi olaparib or chemotherapy in patients with advanced solid tumors (AToM, NCT02588105). Similarly, the combination of CHK1 inhibitor prexasertib and PARP inhibitor olaparib in advanced solid tumors is tested based on their synergistic effect against cancer cells in vitro (NCT03057145). In melanoma, pre-clinical studies suggest that inhibition of PARP and DNA damage responses is a promising strategy for combination therapy [43]. In summary, enhanced effects of DDR inhibitors in cells with sensitizing mutations and synergistic effects of combinations of different checkpoint inhibitors warrant further clinical testing.
3.4. Epigenetic Drugs
Cellular transformation and tumor cell plasticity require specific epigenetic changes occurring in response to environmental and intrinsic stimuli. Often, epigenetic modifications in tumor cells are involved in the silencing of tumor suppressor genes—at different stages of cellular dedifferentiation, during epithelial-mesenchymal transition (EMT), and during drug resistance development (reviewed in [44]). Therefore, DNA-methylation patterns, histone modifications and altered microRNA expression not only represent valuable biomarkers, but in particular, DNA methylation via DNA methyltransferases (DNMTs) and histone deacetylation via HDACs have also been explored as therapeutic targets for combination therapy. More recently, additional histone modifying enzymes and histone code readers of the bromodomain-extraterminal (BET) family have come to the spotlight as potential weapons against tumor resistance to kinase inhibitors.
DNA hypomethylating agents such as 5-azacytidine and 5-aza-2-deoxy-cytidine (decitabine), both inhibitors of DNMT1, have been clinically approved for the treatment of hematological malignancies [45] and induce differentiation und apoptosis due to reactivation of cell cycle regulating genes. In BRAF-mutated metastatic melanoma, a phase I–II clinical study assess the effect of DNA methylation on tumor progression and resistance development under therapy with BRAFi and MEKi with or without decitabine (NCT01876641). Similarly, the drug may induce cancer stem cell differentiation and thereby aid in combination regimens (see Section 3.9).
Histone deacetylase inhibitors (HDACi) were also among the first anti-cancer drugs targeting epigenetic enzymes that received approval for the treatment of hematological malignancies. The first HDACi, vorinostat, approved as third line therapy for cutaneous T-cell lymphoma (CTL), is currently clinically tested as adjuvant treatment of CRC in combination with hydroxychloroquine (see Section 3.3) against TKi regorafenib (NCT02316340) and in advanced solid tumors (NCT01023737). In addition, the combination of vorinostat with immunotherapy using checkpoint inhibitor pembrolizumab is tested in patients with advanced NSCLC (NCT02638090). Other HDACi, including belinostat, panobinostat, and etinostat, are currently evaluated for the treatment of refractory solid malignancies, including mesothelioma, melanoma, lymphoma, and others–in part in combinations with other targeted drugs or with classical chemotherapy [20,46]. In a subset of drug-resistant melanoma, HDACi may restore BRAFi sensitivity via different mechanisms, including induction of pro-apoptotic pathways and decreased PI3K signaling [47]. In addition, histone-independent actions of HDACi, in particular the hyperacetylation of tumor suppressor p53, chaperone HSP90, and NF-κB subunit p65/RelA, may contribute to their effects on drug-resistant tumor cells and reflect the broad impact of HDAC/i on cancer in general [48]. Consequently, safety and efficacy of HDACi will currently be evaluated in particular in “problem” melanoma and tumors, such as resistant BRAFV600mut advanced melanoma (vorinostat, NCT02836548), high risk uveal melanoma (NCT03022565), and in combination therapies with checkpoint inhibitor immunotherapy (PEMDAC, NCT02697630, NCT02032810 and NCT02437136)—seeking to overcome therapy resistance.
Closer analyses of the critical roles of different histone modifying enzymes and histone readers in cancer progression and therapy resistance have triggered the development of additional inhibitors. Drugs targeting the polycomb repressive complex (PRC) 2 factor EZH2, catalyzing H3K27 trimethylation (EZH2i), or BET protein family epigenetic readers, affecting transcription of genes with super-enhancers (BETi), are already being tested in clinical studies in different malignancies [49]. Due to their potential to induce apoptosis in melanoma cells by increasing NOXA and AIF and decreasing MCL-1 levels, EZH2 inhibitors may enhance the sensitivity to MAPK inhibition in combination regimens with BRAFi and MEKi. In current early clinical trials, the EZH2 inhibitor tazemetostat is assessed in relapsed or refractory malignant mesothelioma with BAP1 loss of function in adults (NCT02860286). However, due to a case of secondary lymphoma, new enrollment of pediatric patients in current tazemetostat trials for synovial sarcoma (NCT02601937) or different relapsed or refractory advanced solid tumors with sensitizing mutations is currently suspended (NCT03213665). In principle, BET inhibitors (BETi) as well may override resistance to MAPK inhibitors in certain tumors (such as NRAS-mutant melanoma) by sensitizing them to apoptosis and have been shown to potently inhibit growth of solid and hematological tumor cells in vitro and in vivo in animal models [50]. According to dose findings studies, BETi OTX015 can be safely applied in patients with acute leukemia (NCT02698189) or selected advanced solid tumors including triple negative breast cancer (TNBR), NSCLC, and castration-resistance prostate cancer (CRPC) and pancreatic cancer (NCT02698189). In additional ongoing phase I dose-escalation studies, other BETi are tested in advanced or recurrent solid malignancies (NCT02630251, and NCT02419417) and in hematologic malignancies (NCT02543879), as well as in a phase I/IIa study as monotherapy or in combination with nivolumab immunotherapy (NCT02419417). In other regimes, combinations of BETi with inhibitors of cyclin dependent kinases (CDKi) or HDACi or ATMi or PARPi maybe tested -following up on promising synergistic anti-tumor effects in vitro. Moreover, in pre-clinical studies novel roles of enzymes, either affecting the ubiquitination of histone H2A K119, such as PCR1 factor BMI1, 2A-DUB/MYSM1, and RNF2 or the methylation of H3 K9, in progression and drug resistance of melanoma and other cancers are currently being investigated [51,52,53].
On the other hand, clinical testing of the safety of microRNA MRX3 in a dose-escalation study in patients with unresectable primary liver cancer, advanced or metastatic cancers, or hematological malignancies was recently terminated due to several serious immune related events (NCT01829971). Current clinical studies mainly focus on microRNAs as biomarkers [20].
3.5. Telomerase Inhibitors
Immortalization of tumor cells usually requires the induction and activation of telomerase to maintain telomere length and integrity and to prevent DNA damage responses [54]. Consequently, genes involved in telomere maintenance—including telomerase reverse transcriptase (hTERT, the catalytic subunit of telomerase), shelterin complex proteins (TRF1, TRF2, POT1, TIN2, TPP1, and Rap1), and telomerase-associated proteins (such as hTEP, p23, HSP90 and dyskerin)—are relatively frequently activated or mutated in tumors including both familial and acquired melanoma, often in context with BRAF mutations (recently reviewed in [55,56]).
Therapeutic inhibition of telomerase activity can be achieved on the one hand by targeting either the hTERT catalytic subunit—by nucleoside analoga such as 3′-azido-2′,3′-dideoxythymine (AZT)—or on the other hand, the telomerase RNA (hTR) component—via antisense oligonucleotides or the more specific telomerase template antagonists such as the 13-mer oligonucleotide N3′-P5′-thio-phosphoramidate (GRN163L, imetelstat) [55,57]. Telomerase enzyme inhibition and targeting of hTR in cancer cells generally result in progressive telomere shortening and reduced cellular viability. However, despite significant inhibition of tumor growth and metastasis, in line with off-target effects of telomerase inhibitors (Ti) in in vitro studies [58], a number of clinical trials had to be withdrawn due to unfavorable toxicity profiles. For instance, imetelstat inhibited primary tumor growth and metastases in in vitro and in vivo studies. But in early phase clinical studies in adults and children, dose-limiting higher grade hematologic cytotoxicity and cases of intratumoral hemorrhage secondary to thrombocytopenia were the causes of premature cancellation [59,60]. An alternative approach to target telomeres in cancer cells is the use of so called “T-oligos”—guanine-rich oligonucleotides homologous to the 3′ telomere overhang sequence. T-oligos, in particular a specific 11-base oligonucleotide (5′-dGTTAGGGTTAG-3′ or T11), have been shown to induce DNA damage responses (DDRs) such as senescence, apoptosis, and cell cycle arrest in numerous cancer cell types, including melanoma, with only minimal cytostatic effects in normal cells [61]. Clinical testing of T-oligos may therefore be warranted. In addition, peptides derived from TERT have been in focus in anti-cancer immunotherapy—either to raise CD8+ cytotoxic T cell responses against TERT-epitopes on tumor cells [62] or to monitor tumor-specific anti-telomerase-specific CD4+ T cell immunity to TERT-neoantigens before and after chemo-or immunotherapy (Telocap02, NCT02846103).
3.6. Redox Modulators
By definition reactive oxygen species (ROS) occurring as byproducts of cellular metabolism or due to exposure to external stressors are oxygen species with reactive properties (such as peroxides, superoxide, hydroxyl radicals and singlet oxygen)—that can damage other cellular molecules including proteins and DNA if insufficiently detoxified by cellular antioxidant systems. In tumor cells, in brief, ROS are “double-edged swords”: on the one hand, ROS have tumor-suppressive functions due to the induction of tumor cell apoptosis. On the other hand, ROS may promote tumorigenesis through induction of DNA damage and mutations, as well as epithelial-mesenchymal transition (EMT) and metastasis by regulating extracellular matrix and cytoskeleton remodeling [63]. Because oxidative stress responses interfere with a complex network of cellular mitochondrial detox enzymes, transcriptional regulators, and tumor suppressors—including NFκB, nuclear factor erythroid 2 like 2 (NRF2), and BRCA1—therapeutic strategies targeting with these processes with small-molecules or neutraceuticals and phytochemicals may hold promise for targeted tumor combination therapy and may reduce cytotoxicity of conventional therapies.
The redox modulator dimethyl fumarate (DMF), an approved treatment for autoimmune diseases such as multiple sclerosis and psoriasis, induces recycling of intracellular antioxidant glutathione (GSH) pools, inhibits NFκB p65/RelA and activates NRF2—thereby overall promoting anti-oxidant responses in different cell types [64]. Current clinical trials therefore assess the safety and suitable dose of DMF to reduce cytotoxicity in combination with radiotherapy (RT) and temozolomide (TMZ) in glioblastoma multiforme (GBM) (NCT02337426) and in other cancers [65]. In addition, DMF has been shown to induce apoptosis in T cell lymphoma cells [66] and is currently being tested in phase-I-clinical trials in refractory chronic lymphocytic leukemia (CLL) (NCT02784834). In melanoma, anti-proliferative and pro-apoptotic effects of DMF could be demonstrated leading to reduced growth and metastasis of melanoma in pre-clinical models [67,68]. Similarly, the selective GSK-3β inhibitor thiadiazolidinone (TDZD-8) has been shown to selectively cause death of stem cell marker expressing leukaemia cells through depletion of thiols with rapid accumulation of ROS—with only minimal toxicity to normal hematopoietic cells [69]. TDZD-8 may undergo clinical testing.
An example for the use of vitamins with antioxidant properties as alternative anticancer therapies is a phase II-clinical trial assessing the efficacy of high dose vitamin C (sodium ascorbate) infusions to synergistically act with chemotherapy to kill cancer cells and to reduce toxic side effects (NCT02655913) [70]. In addition, other vitamins and antioxidants, such as vitamin E, N-acetylcysteine (NAC), green tee catechins, and others, are being assessed in combination regimens. Because bioenergetics and cellular metabolism are connected, the drugs discussed in the next section also in part interfere with the redox modulation and energy requirements of tumor cells.
3.7. Metabolic Reprogramming Drugs and Enzymatic Inhibitors
To maintain high proliferation rates under nutrient- and oxygen-limited conditions, tumor cells need to adapt their nutrient transport, metabolism, and bioenergetics—a process known as “metabolic reprogramming” in cancer biology. On the one hand, oncogenic signaling pathways, such as the MAPK pathway, regulate the use of glucose and amino acids and the switch from oxidative phosphorylation (oxphos) to anaerobic glycolysis (known as “Warburg effect”) in tumor cells (reviewed in [71]). On the other hand, nutrient metabolism interferes with responsiveness to BRAF (kinase) inhibition in melanoma and other tumors [72]. Induction of anabolic pathways supporting tumorigenesis often results from sustained activation of the PI3K/Akt/mTOR pathway, mTOR complex 1 (mTORC1), and transcriptional networks involving HIF-1α/MYC/SREBP1. Therefore, strategies interfering with both, glucose and amino acid supplies, and the regulating anabolic signaling pathways, may be therapeutically effective against tumors in a relatively selective manner and are currently explored in pre-clinical and clinical settings.
One approach to specifically interfere with cancer cell glucose and energy metabolisms involves the application of the anti-diabetic biguanides metformin and phenformin that exhibit anti-cancer properties by interfering with mTOR signaling and inhibiting mitochondrial complex I [73]. In the presence of biguanides, tricarbocylic acid (TCA) cycle intermediates and mitochondrial ATP production are reduced leading to tumor cell death when glycolytic ATP levels decrease as a result of limited glucose availability to tumor cells. In melanoma cell cultures, phenformin reduced cellular viability and growth of both CSC and non-CSC and abrogated invasion more potently than metformin [74]. Current phase-I clinical trials therefore assess the safety and dose-escalation options for the use of phenformin in combination with BRAFi and MEKi therapy in metastatic melanoma (NCT03026517), as well the combination of metformin and temsirolimus, an mTOR inhibitor, in advanced cancers (NCT01529593). Another metabolic drug with anti-cancer activity is the pyruvate mimetic compound dichloroacetate (DCA), an established treatment for pediatric mitochondrial disorders. DCA stimulates mitochondrial function by inhibiting regulatory pyruvate dehydrogenase kinases (PDK) 1–4 at the expense of glycolysis to reverse the Warburg effect and block the growth advantage of tumor cells [75]. Ongoing clinical studies evaluate the effects of DCA vs. placebo in combination with cisplatin and radiation treatment in patients with stage III–IV squamous cell carcinoma of the head and neck (SCCHN) (NCT01386632). In pre-clinical studies, inhibition of glutamine transport and uptake were suggested as potential therapeutic strategies in multiple myeloma [76] and both, BRAFWT and BRAFV600mut, melanoma. Based on similar principles, the benefits of nutraceuticals such as vitamins, curcumin, and green tee polyphenols—interfering with metabolic and redox balances—are clinically explored in treatment and prevention of various cancers [77].
3.8. Proteasome Inhibitors (PI)
The ubiquitin-proteasome pathway, regulating the proteolytic degradation of proteins involved in cell cycle control und survival (including cyclins, p53, NF-κB, and others), is involved in many aspects of tumor cell transformation and tumor growth and also has important roles in normal cells (reviewed in [78]). Toxicities and acquired resistances have therefore so far limited the use of this drug class—with a few exceptions. Clinical evaluation of the first-generation reversible small molecular (26S) PI bortezomib, already approved as second line treatment for multiple myeloma and for mantel cell lymphoma [79], may hold promise for therapeutic use in combination with HDAC inhibitor vorinostat or chemotherapy in NSCLC or with purine nucleoside metabolic inhibitor clofarabine in other refractory solid tumors (NCT02211755). However, in metastatic melanoma, the combination of bortezomib with paclitaxel and carboplatin was associated with considerable cytotoxicity and had only limited clinical benefit [80]. Newer second-generation PIs with improved selectivity and novel combinations restoring apoptotic pathways may have better clinical efficacy.
3.9. Cancer Stem Cell Transdifferentiation
Tumor heterogeneity and cellular plasticity towards environmental changes confer critical survival advantages to a tumor cell population and limit lasting therapeutic success [81]. In particular, the slow-cycling population of cancer stem cells (CSC), also called “cancer-initiating” cells, has been implicated in driving relapses during targeted and conventional cytotoxic tumor therapy [82]. CSC may therefore constitute important targets for combination therapies or two-component drugs targeting both proliferating and SC tumor cells. Exploiting their stem cell properties, the so called “transdifferentiation” therapy aims at inducing terminal differentiation of CSC towards benign cells of the original lineage the tumor arose from or to related lineages through pharmacological manipulation of transcription factor balances.
One such differentiation strategy involves the use of retinoids: the retinoid acid receptor (RAR) agonist all trans-retinoid acid (ATRA) has been successfully applied in acute promyelocytic leukemia (APL) [83]. Moreover, the selective retinoid X receptor (R(X)R) agonist bexarotene may constitute a weapon against multidrug-resistance in breast cancer [84]. Similarly, retinoids may induce differentiation in other cancer stem cell types. In melanoma, the balance of master regulator microphthalmia-associated transcription factor (MITF) and tyrosine kinase AXL has been shown to function as rheostat determining the transition between proliferative and invasive state—with highest MITF levels promoting neuronal differentiation [85]. In addition, either high MIFT-levels or a low MIFT/AXL-ratio were shown to be associated with resistance of BRAF-and NRAS-mutant melanoma cells towards kinase inhibitors and other targeted therapies [86]. Because ATRA treatment of melanoma cells in culture induced differentiation and apoptosis in context with increased expression of MITF [87], ATRA may provide a combinational therapeutic tool against certain melanomas—to tackle both, melanoma SC and therapy resistance, via key regulator MITF and related gene expression. As a single agent, on the opposite, R(X)R agonist bexarotene was not efficient against metastatic melanoma in an earlier trial [88]. Biomarkers as well as effects on immune cells may have to be considered in future anti-cancer therapy approaches using retinoids.
Second, certain epigenetic drugs (see Section 3.4) have the potential to regulate tumor cell transdifferentation, in particular DNA demethylating agents and HDACi [89]. In this respect, the DNA hypomethylating drug 5-aza-(2-deoxy-)cytidine (decitabine) induced the neuronal marker gene microtubule associated protein 2 (MAP2), progressively methylated during melanoma progression, in metastatic melanoma cells. However, in a phase II clinical trial 5,6-dihydro-5-azacytidine (DHAC) demonstrated only limited benefit against malignant melanoma—without serious myelosuppression. Contrarily, decitabine plus high dose interleukin-2 induced regression of melanoma in 31% of patients, but was associated with significant occurrence of neutropenia. In addition, decitabine is tested in combination with BRAFi and MEKi to measure the time to progression under this combination (NCT01876641). Similarly, HDACi have been shown to promote MAP2 expression and induce benign neuron-like differentiation in a metastatic melanoma mouse cell line in vitro [90]. HDAC inhibitors also inhibit the growth of uveal melanoma cells both in vitro and in vivo, and other problem melanoma types (see Section 3.4).
A promising alternative transdifferentiation strategy is to induce adipocyte-like differentiation of tumor cells by application of unsaturated fatty acids—such as oleic, palmitoleic, or linoleic acid—or peroxisome proliferation-activated receptor gamma (PPARγ) agonists. This approach has successfully been used to enforce transdifferentiation in melanoma spheres and in many tumor cell lines in vitro [91] and will now need to be transferred to clinical applications. Although a growing number of in vitro studies could demonstrate significant effects, currently, there is no clinically approved fixed transdifferentiation therapy regimen for cancers yet.
3.10. Immunomodulatory Drugs
Apart from the inhibitory surface receptors CTLA-4 and PD-1 several cytoplasmic proteins, such as ubiquitin ligases and kinases, downstream of the T cell receptor (TCR) act as negative regulators of T cell activation and may limit anti-tumor immune responses. Cytoplasmic inhibitors of TCR signaling include Casitas B cell lymphoma (CBL) family E3 ligases (c-CBL, CBL-b, ITCH, GRAIL, DELTEX, and NEDD), adaptor proteins DOK-1/2 and STS1/2, as well as kinases such as DRAK2, and others (reviewed in [92]). Using siRNA and shRNA approaches to down-modulate CBL-b in T cells, increased T cell activity towards melanoma cells could be achieved in pre-clinical models [93]. Although no clinically approved inhibitors are currently available, targeting intracellular negative regulators of TCR signaling may represent a promising alternative anti-tumor strategy for combination immunotherapy as it simultaneously deactivates several adaptive immune “checkpoints” including TFG-β, CTLA-4, and PD-1 signaling [94,95]. According to pre-clinical studies, avidity-tuning through manipulation of negative TCR regulation may also enhance target cell selectivity in adoptive chimeric antigen receptor (CAR) T cell cancer therapy [96]. Interestingly, targeting CBL E3 ligases may also have synergistic effects in certain tumor cell types, such as melanoma, where c-CBL has recently been shown to promote tumor growth and mobility in part via the focal adhesion kinase (FAK)-GRB2-SRC nexus. Overall, immuno-stimulatory and growth-inhibitory effects therefore provide a rationale to further explore the therapeutic value of inhibiting these ubiquitination proteins in combination therapy of melanoma and other tumors [97].
An alternative strategy to limit negative regulation of T cell function and to break immunotolerance to tumor cells involves the modulation of L-tryptophan metabolism via the kynurenine pathway with the indoleamine 2,3-dioxygenase-1 (IDO1) inhibitor epacadostat [98]. However, despite initial excitement, the phase III-clinical trial of epacadostat with pembrolizumab for melanoma was recently halted because the combination therapy missed the first primary endpoint of improving PFS vs. pembrolizumab alone (ECHO-301/KEYNOTE-252, NCT02752074). Another ongoing clinical trial assesses the benefit of epacadostat administration in context of a multipeptide melanoma vaccine (NCT01961115). Moreover, HSP90 inhibitor therapy may work as indirect “immune adjuvant” leading to increased therapeutic T cell recruitment against EphA2 expressing tumors, such as BRAFi-resistant melanoma, due to transient proteasome-dependent degradation of HSP90 client protein EphA2 in absence of the functional chaperone [99,100].
3.11. Other Substances
A few other small-molecule anti-tumor therapeutics are currently being tested clinically and will not be discussed in more detail here. In brief, dose-finding studies are ongoing for WNT/β-catenin inhibitors (LGK974) as single agent or in combination with anti-PD1 antibody PDR001 (NCT01351103). In melanoma, altered expression and functions of WNT pathway genes including Frizzled receptors and their functions are ongoing areas of research. To follow up on an alternative strategy to destabilize survival proteins in cancer cells by interfering with cellular chaperones, a number of HSP90 inhibitors are tested in combination with BRAFi and MEKi in melanoma (NCT02721459), in NSCLC (NCT01784640), and in other cancers [101].
4. Towards Algorithms for Improved Combination Therapy and Individualized Approaches
In the light of the different targeted therapy strategies against cancers available, new possibilities arise to (1) overcome therapy resistance to current kinase inhibitor combinations by adding a third or fourth agent, (2) treat tumors not bearing the most common driver mutations (such as BRAFWT melanoma) and “problem” tumors (such as mucosal or uveal melanoma or brain metastases), (3) find alternative strategies for individual patient needs (such as elderly patients, children, or patients with immune or metabolic disorders), e.g., by reducing cytotoxicity to normal cells. (4) to enhance established immuno-and chemotherapy regimens.
According to ongoing clinical trials, in BRAFV600mut melanoma, therapy resistance could be addressed by either using triple combinations of kinase inhibitors (see Section 3.1), or combinations of BRAFi and MEKi with either apoptosis inducing drugs (death receptor agonists, BH3-or SMAC-mimetics, see Section 3.2.) or epigenetic drugs (DNMT1i, HDACi, EZH2i, BETi, see Section 3.4)—promoting tumor cell killing—or with metabolic drugs (phenformin, Section 3.7). Commonly, acquired resistance to kinase inhibitors may involve compensatory activation of other growth factor signaling pathways or additional kinase mutations, increased survival signaling, or altered cell cycle control [9]. To interfere with increased BCL-2 or MCL-1 in BRAFi-resistant melanoma and other cancer types, alternatively, different BH3-mimetics or epigenetic drugs may be applied—the later with potential additional benefit of impacting cell cycle regulation, EMT, and CSC transdifferentiation. To determine the most favorable individualized therapy regimens therefore, the current trend is towards a genetic assessment of tumor biopsies or “liquid biopsies” [102]. Based on patient parameters and molecular characterization of tumor biopsies, potentially from different metastases, or even single-cells [103], the design of individualized regimens will be optimized—as exemplified for the therapy of advanced melanoma (Figure 2).
In addition, alternative agents, such as DNA damage response inhibitors (PARPi, ATMi), newer telomer-based drugs (“T-oligos”), or anti-angiogenic strategies (VEGFi) could be applicable for combination therapies of BRAFWT melanoma. These drug classes might also be promising for combination therapies with kinase inhibitors in other advanced cancers. Importantly, several of the emerging small-molecule approaches reviewed here are not directed against tumor-specific mutant proteins, but instead rather target metabolic, gene expression, or epigenetic changes more commonly occurring across different tumor types.
Although increased toxicity of targeted therapy combinations is a concern, molecular analyses of tumor biopsies may identify aberrations rendering tumor cells particularly sensitive to a specific drug combination as well as guide the use of combinations with non-overlapping toxicity profiles. Accordingly, preliminary results of clinical studies of novel kinase inhibitor combinations containing CDKi suggest that presence of alterations in the Cyclin D-CDK4/6-p16INK4A-RB axis confers increased tumor cell responsiveness—which may lead to overall manageable safety profiles of these combinations and favorable efficacies [104]. Similarly, adding retinoids or certain epigenetic drugs in combination regimens may have synergistic anti-tumor effects without increasing toxicity for the patient because of non-overlapping side effects [105]. The awaited results of ongoing clinical trials assessing the safety and tolerability of novel triple combinations of small molecule drugs or of combinations with conventional or immune therapies will therefore guide future therapy decisions. Moreover, therapy strategies that were previously not successful against a broader number of cancer types as monotherapy due to toxic side effects (such as proteasome or telomerase inhibitors)—might be applicable at lower doses in combination regimens or in modified forms.
Importantly, newer small-molecule targeted therapies may also help to enhance immunogenicity of tumors for immunotherapies or to reduce side effects of established chemo- and radiotherapy regimens. Because the combination of immunotherapy using checkpoint inhibitors with kinase inhibitors such as BRAFi for metastatic BRAFV600mut melanoma may be effective, clinical trials assessing the best sequence, dose, and duration of treatments to archive the highest response rates are currently underway (NCT02818023, NCT02130466) [106].
5. Conclusions and Outlook
A variety of small-molecule targeted therapies are currently available for novel combinational and alternative therapy regimens based on individual patient and tumor parameters. Therefore, it is now important to establish additional biomarkers including gene mutation patterns, expression changes, epigenetic makers, liquid markers, and tumor micro-environmental markers—to improve the prediction of the best combination regimen for each patient, tumor, and therapeutic stage. In the treatment of advanced melanoma, both additional triple combinations with BRAFi and MEKi and successive application of targeted therapies, as well as combinations of small-molecules with immunotherapy, chemotherapy, and radiotherapy may open novel perspectives. Furthermore, identification of additional tumor biomarkers may also lead to improved prevention and adjuvant strategies.
Funding
This work was supported by a grant from the German Research Foundation, grant number [DFG: GA-2052].
Acknowledgments
Martina V. Gatzka worked as research group leader in the Department of Experimental Dermatology at the University of Ulm. Her research was supported by a grant from the German Research Foundation (DFG, GA-2052). The author thanks Karin Scharffetter-Kochanek for the departmental support and all past and present members of the Exp. Dermatology laboratories for helpful discussion, in particular Adelheid Hainzl for her continued expert technical support and friendship. In addition, all authors of articles and studies related to targeted tumor therapies that could not be mentioned here due to space limitations are acknowledged at this point and apologized to—in many instances, only the initiating report or the latest review could be cited. The references and trials cited here represent a selection based on information accessible to the author at the time of writing.
Conflicts of Interest
The author declares no conflict of interest.
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Analysis of FDA approved anticancer drugs reveals the future of cancer therapy. Cell. Cycle 2004, 3, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.E.; Feigelson, H.S. Hormonal carcinogenesis. Carcinogenesis 2000, 21, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol. Rev. 2017, 276, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, F.H.; Bernards, R. Drug resistance to targeted therapies: Déjà vu all over again. Mol. Oncol. 2014, 8, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.J.; Ribas, A. Targeted therapy for melanoma. Cancer Treat. Res. 2016, 167, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Peris, K.; Haushild, A.; Saiag, P.; Middleton, M.; Bastholt, L.; Grob, J.J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.J.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2016. Eur. J. Cancer 2016, 63, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabris, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediate by a RAF kinase switch in melanoma can be overcome by co-targeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [PubMed]
- aaijmakers, M.I.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.A.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co- existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicenter phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef]
- Chan, B.A.; Hughes, B.G.M. Targeted therapy for non-small cell lung cancer. Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.M.; Coyle, V.M.; Kennedy, R.D.; Wilson, R.H. Molecular subtypes and personalized therapy in metastatic colorectal cancer. Curr. Colorectal Cancer Rep. 2016, 12, 141–150. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. NIH. Available online: https://clinicaltrials.gov (accessed on 15 April 2018).
- Li, H.; Fan, X.; Houghton, M. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; DeBraud, F.; Bahleda, R.; Adamo, B.; Andre, F.; Dienstmann, R.; Delmonte, A.; Dereda, R.; Isaacson, J.; Litten, J.; et al. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann. Oncol. 2014, 25, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinae inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin b1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Gatzka, M.; Walsh, C.M. Apoptotic signal transduction and T cell tolerance. Autoimmunity 2007, 40, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Kurbanov, B.M.; Geilen, C.C.; Fecker, L.F.; Orfanos, C.E.; Eberle, J. Efficient TRAIL-R1/DR4-mediated apoptosis in melanoma cells by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Investig. Dermatol. 2005, 125, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Mohana-Kumaran, N.; Hill, D.S.; Allen, J.D.; Haass, N.K. Targeting the intrinsic apoptosis pathway as a strategy for melanoma therapy. Pigment. Cell Melanoma Res. 2014, 27, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Aplin, A.E. BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ. 2012, 19, 2029–2039. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xie, M.; Song, T.; Shen, H.; Yu, X.; Zhang, Z. A novel BH3 mimetic efficiently induces apoptosis in melanoma cells through direct binding to anti-apoptotic Bcl-2 family protein, including phosphorylated Mcl-1. Pigment Cell Melanoma Res. 2015, 28, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Chen, L.; Lanning, M.E.; Fletcher, A. Expanding the cancer arsenal with targeted therapies: Disarmament of the antiapoptotic Bcl-2 proteins by small-molecules. J. Med. Chem. 2017, 60, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Jochemsen, A.G. Reactivation of p53 as therapeutic intervention for malignant melanoma. Curr. Opin. Oncol. 2014, 26, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Breyssens, H.; Salter, V.; Zhong, S.; Hu, Y.; Baer, C.; Ratnayaka, I.; Sullivan, A.; Brown, N.R.; Endicott, J.; et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell 2013, 23, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. p53 reactivation by PRIMA-1(Met) (APR-246) sensitizes (V600E/K)BRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Mukhopadhyay, S.; Das, D.N.; Sinha, D.; Naik, P.P.; Bhutia, S.K. Mechanism of autophagic regulation in carcinogenesis and cancer therapeutics. Semin. Cell Dev. Biol. 2015, 39, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Damia, G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat. Rev. 2017, 60, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Beumer, J.H.; Hamieh, L.; Tawbi, H. Role of PARP inhibitors in cancer biology and therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Macurek, L. Targeting the checkpoint to kill cancers cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Hanna, C.; Walker, S.M.; Gonda, D.; Li, J.; Wikstrom, K.; Savage, K.I.; Butterworth, K.T.; Chen, C.; Harkin, D.P.; et al. Mechanistic rationale to target PTEN-deficient tumor cells with inhibitors of the DNA damage response kinase ATM. Cancer Res. 2015, 75, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Smida, M.; Fece de la Cruz, F.; Kerzendorfer, C.; Uras, I.Z.; Mair, B.; Mazouzi, A.; Suchankova, T.; Konopka, T.; Katz, A.M.; Paz, K.; et al. MEK inhibitors block growth of lung tumours with mutations in ataxia telangiectasia mutated. Nat. Commun. 2016, 7, 13701. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient colorectal cancer cells are sensitive to the PARP inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.W.-S.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Chevanne, M.; Zampieri, M.; Caldini, R.; Rizzo, A.; Ciccarone, F.; Catizone, A.; D’Angelo, C.; Guastafierro, T.; Biroccio, A.; Reale, A.; et al. Inhibition of PARP activity by PJ-34 leads to growth impairment and cell death associated with aberrant mitotic pattern and nucleolar actin accumulation in M14 melanoma cell line. J. Cell. Physiol. 2010, 222, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Derissen, E.J.B.; Beijnen, J.H.; Schellens, J.H.M. Concise Drug Review: Azacitidine and Decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic postential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Gunatilake, D.; Beaumont, K.A.; Sharp, D.M.; Tiffen, J.C.; Heinemann, A.; Weninger, W.; Haass, N.K.; Wilmott, J.S.; Madore, J.; et al. HDAC inhibitors restore BRAF-inhibitor sensitivity by altering PI3K and survival signalling in a subset of melanoma. Int. J. Cancer 2018, 142, 1926–1937. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, N. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Tiffen, J.C.; Hersey, P. Histone modifications, modifiers, and readers in melanoma resistance to targeted and immune therapy. Cancers 2015, 7, 1959–1982. [Google Scholar] [CrossRef] [PubMed]
- Echevarria-Vargas, I.M.; Reyes-Uribe, P.I.; Guterres, A.N.; Yin, X.; Kossenkov, A.V.; Liu, Q.; Zhang, G.; Krepler, C.; Cheng, C.; Wei, Z.; et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, R.; Bhutkar, A.; McNamara, M.C.; Lees, J.A. BMI1 induces an invasive signature in melanoma that promotes metastasis and chemoresistance. Genes Dev. 2016, 30, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Wilms, C.; Kroeger, C.M.; Hainzl, A.V.; Banik, I.; Bruno, C.; Krikki, I.; Farsam, V.; Wlaschek, M.; Gatzka, M.V. MYSM1/2A-DUB is an epigenetic regulator in human melanoma and contributes to tumor cell growth. Oncotarget 2017, 8, 67287–67299. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Akdemir, K.C.; Kwong, L.N.; Fiziev, P.; Wu, C.J.; Keung, E.Z.; Sharma, S.; Samant, N.S.; Williams, M.; Axelrad, J.B.; et al. Dual Roles of RNF2 in melanoma progression. Cancer Discov. 2015, 5, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [PubMed]
- Martinez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.E.; Truong, A.; Meyer, L.J. Genetic predisposition to melanoma. Semin. Oncol. 2016, 43, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Jäger, K.; Walter, M. Therapeutic targeting of telomerase. Genes 2016, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Mender, I.; Senturk, S.; Ozgunes, N.; Akcali, K.C.; Kletsas, D.; Gryaznov, S.; Can, A.; Shaym, J.W.; Dikmen, Z.G. Imetelstat (a telomerase antagonist) exerts off-target effects on the cytoskeleton. Int. J. Oncol. 2013, 42, 1709–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, P.A.; Drissi, R.; Muscal, J.A.; Panditharatna, E.; Fouladi, M.; Ingle, A.M.; Ahern, C.H.; Reid, J.M.; Lin, T.; Weigel, B.J.; et al. A phase I trial of imetelstat in children with refractory or recurrent solid tumors: A Children’s Oncology Group Phase I Consortium Study (ADVL1112). Clin. Cancer Res. 2013, 19, 6578–6584. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Kolevaska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, G.; Wojdyla, L.; Frakes, M.; Schrank, Z.; Leviskas, B.; Ivancich, M.; Vinay, P.; Ganapathy, R.; Ramirez, B.E.; Puri, N. Mechanism of action of G-quadruplex forming oligonucleotide homologous to the telomere overhang in melanoma. J. Investig. Dermatol. 2018, 138, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Pankhong, P.; Shin, T.H.; Obeng-Adjei, N.; Morrow, M.P.; Walters, J.N.; Khan, A.S.; Sardesai, N.Y.; Weiner, D.B. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunol. Res. 2013, 1, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaderi, Z.; Maghazachi, A.A. Utilization of dimethyl fumarate and related molecules for treatment of multiple sclerosis, cancer, and other diseases. Front. Immunol. 2016, 7, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Shafer, D.A.; Chen, Z.J.; Harris, T.; Tombes, M.B.; Shrader, E.; Strickler, K.; Ryan, A.A.; Dent, P.; Gordon, M. Phase I trial of dimethyl fumarate, temozolomide, and radiation therapy in glioblastoma multiforme. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Nicolay, J.P.; Müller-Decker, K.; Schroeder, A.; Brechmann, M.; Möbs, M.; Géraud, C.; Assaf, C.; Goerdt, S.; Krammer, P.H.; Gülow, K. Dimethyl fumarate restores apoptosis sensitivity and inhibits tumor growth and metastasis in CTCL by targeting NF-κB. Blood 2016, 128, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Loewe, R.; Valero, T.; Kremling, S.; Pratscher, B.; Kunstfeld, R.; Pehamberger, H.; Petzelbauer, P. Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res. 2006, 66, 11888–11896. [Google Scholar] [CrossRef] [PubMed]
- Kaluzki, I.; Hrgovic, I.; Hailemariam-Jahn, T.; Doll, M.; Kleemann, J.; Valesky, E.M.; Kippenberger, S.; Kaufmann, R.; Zoeller, N.; Meissner, M. Dimethylfumarate inhibits melanoma cell proliferation via p21 and p53 induction and BCL-2 and cyclin B1 downregulation. Tumour Biol. 2016, 37, 13627–13635. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.L.; Li, X.; Corbett, C.A.; Rossi, R.M.; Bushnell, T.; Liesveld, J.L.; Hébert, J.; Young, F.; Jordan, C.T. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8). Blood 2007, 110, 4436–4444. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, D.; Pelosi, E.; Castelli, G.; Lo-Coco, F.; Testa, U. Mechanisms of anti-cancer effects of ascorbate: Cytotoxic activity and epigenetic modulation. Blood Cells Mol. Dis. 2018, 69, 57–64. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin enhances the therapeutic benefit of BRAFV600E inhibition in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrachi, T.; Romagnani, A.; Albini, A.; Longo, C.; Argenziano, G.; Grisendi, G.; Dominici, M.; Ciarrocchi, A.; Dallaglio, K. Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget 2017, 8, 6914–6928. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Roudebush, P.; Davenport, D.J.; Novotny, B.J. The use of nutraceuticals in cancer therapy. Vet. Clin. North Am. Small Anim. Pract. 2004, 34, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. Bench-to-bedside translation of targeted therapies in multiple myeloma. J. Clin. Oncol. 2012, 30, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Croghan, G.A.; Suman, V.J.; Maples, W.J.; Albertini, M.; Linette, G.; Flaherty, L.; Eckardt, J.; Ma, C.; Markovic, S.N.; Erlichman, C. A study of paclitaxel, carboplatin, and bortezomib in the treatment of metastatic malignant melanoma: A phase 2 consortium study. Cancer 2010, 116, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Dragu, D.L.; Necula, L.G.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Therapies targeting cancer stem cells: Current trends and future challenges. World J. Stem Cells 2015, 7, 1185–1201. [Google Scholar] [CrossRef] [PubMed]
- Kakizuka, A.; Miller, W.H., Jr.; Umesono, K.; Warrell, R.P., Jr.; Frankel, S.R.; Murty, V.V.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663–674. [Google Scholar] [CrossRef]
- Yen, W.C.; Lamph, W. The selective retinoid X receptor agonist bexarotene (LGD1069, Targretin) prevents and overcomes multidrug resistance in advanced breast carcinoma. Mol. Cancer Ther. 2005, 4, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Arozarena, I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment. Cell Melanoma Res. 2005, 28, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Krijgsman, P.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Watabe, H.; Soma, Y.; Ito, M.; Kawa, Y.; Mizoguchi, M. All-trans retinoic acid induces differentiation and apoptosis of murine melanocyte precursors with induction of the microphthalmia-associated transcription factor. J. Investig. Dermatol. 2002, 118, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bedikian, A.Y.; Plager, C.; Papadopoulos, N.; Ellerhorst, J.; Smith, T.; Benjamin, R.S. A phase II evaluation of bexarotene (Targretin) capsules in patients with metastatic melanoma. Oncol. Rep. 2000, 7, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Shekhani, M.T.; Jayanthy, A.-S.; Moddodi, N.; Setaluri, V. Cancer stem cells and tumor transdifferentiation: Implications for novel therapeutic strategies. Am. J. Stem Cells 2013, 2, 52–61. [Google Scholar] [PubMed]
- Kuwajima, A.; Sakai, M.; Iwashita, J.; Abe, T. Differentiation of B16-BL6 melanoma cells into microtubule associated protein-2 positive cells after treatment with histone deacetylase inhibitors butyrate and trichostatin A. J. Health Sci. 2009, 55, 138–142. [Google Scholar] [CrossRef]
- Giampietri, C.; Petrungaro, S.; Cordella, M.; Tabolacci, C.; Tomaipitinca, L.; Facchiano, A.; Eramo, A.; Filippini, A.; Facchiano, F.; Ziparo, E. Lipid storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 2017, 18, 1271. [Google Scholar] [CrossRef] [PubMed]
- Gatzka, M.; Walsh, C.M. Negative regulation of TCR signaling in immunological tolerance: Taming good and evil. Curr. Immunol. Rev. 2008, 4, 190–198. [Google Scholar] [CrossRef]
- Fujiwara, M.; Anstadt, E.J.; Clark, R.B. Cbl-b deficiency mediates resistance to programmed death-ligand 1/programmed death-1 regulation. Front. Immunol. 2017, 26, 42. [Google Scholar] [CrossRef]
- Chiang, J.Y.; Jang, I.K.; Hodes, R.; Gu, H. Ablation of CBL-b provides protection against transplanted and spontaneous tumors. J. Clin. Investig. 2007, 117, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Loeser, S.; Loser, K.; Bijker, M.S.; Rangachari, M.; van der Burg, S.H.; Wada, T.; Beissert, S.; Melief, C.J.; Penninger, J.M. Spontaneous tumor rejection by CBL-b-deficient CD8+ T cells. J. Exp. Med. 2007, 204, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Sim, S.J.; Kim, S.H.; Singh, R.; Hwang, S.; Kim, Y.I.; Park, S.H.; Kim, K.H.; Lee, D.G.; Oh, H.S.; et al. Desensitized chimeric antigen receptor T cells selectively recognize target cells with enhanced antigen expression. Nat. Commun. 2018, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, N.; Wood, G.S. c-CBL regulates melanoma proliferation, migration, invasion and the FAK-SRC-GRB2 nexus. Oncotarget 2016, 7, 53869–53880. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Taylor, J.L.; Chi-Sabins, N.; Kawabe, M.; Gooding, W.E.; Storkus, W.J. Combination therapy with HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve recruitment of therapeutic T cells. Cancer Res. 2012, 72, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.; Ji, Z.; Tan, L.; Taylor, M.; Zhang, J.; Choi, H.G.; Frederick, D.T.; Kumar, R.; Wargo, J.A.; Flaherty, K.T.; et al. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015, 5, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Yuno, A.; Lee, M.J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical evaluation and biomarker profiling of Hsp90 inhibitors. Methods Mol. Biol. 2018, 1709, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Ryska, A. Molecular pathology in real time. Cancer Metastasis Rev. 2016, 35, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Tsoucas, D.; Yuan, G.C. Recent progress in single-cell cancer genomics. Curr. Opin. Genet. Dev. 2017, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.H.; Ascierto, P.A.; De Vos, F.Y.F.L.; Postow, A.M.; van Herpen, C.M.L.; Carlino, M.S. Phase 1b/2 trial of ribociclib+binimetinib in metastatic NRAS-mutant melanoma: Safety, efficacy, and recommended phase 2 dose (RP2D). J. Clin. Oncol. 2017, 35, 9519. [Google Scholar]
- Sato, T.; Cesaroni, M.; Chung, W.; Panjarian, S.; Tran, A.; Madzo, J.; Okamato, Y.; Zhang, H.; Chen, X.; Jelinek, J.; et al. Transcriptional Selectivity of Epigenetic Therapy in Cancer. Cancer Res. 2017, 77, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Aya, F.; Fernandez-Martinez, A.; Gaba, L.; Victoria, I.; Tosca, M.; Pineda, E.; Gascon, P.; Prat, A.; Arance, A. Sequential treatment with immunotherapy and BRAF inhibitors in BRAF-mutant advanced melanoma. Clin. Transl. Oncol. 2017, 19, 119–124. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The principles of targeted cancer therapy. A number of physiological processes are potentially altered in a cancer cell (light blue) and can be targeted by small-molecules and other drugs (1–9, strongly simplified). In addition, immune cells (purple) can be targeted (10) as well as other cells of the tumor micro-environment (not shown). A more detailed description of each cellular process and the therapeutic ways to interfere with it is provided in the text.
Figure 1.
The principles of targeted cancer therapy. A number of physiological processes are potentially altered in a cancer cell (light blue) and can be targeted by small-molecules and other drugs (1–9, strongly simplified). In addition, immune cells (purple) can be targeted (10) as well as other cells of the tumor micro-environment (not shown). A more detailed description of each cellular process and the therapeutic ways to interfere with it is provided in the text.
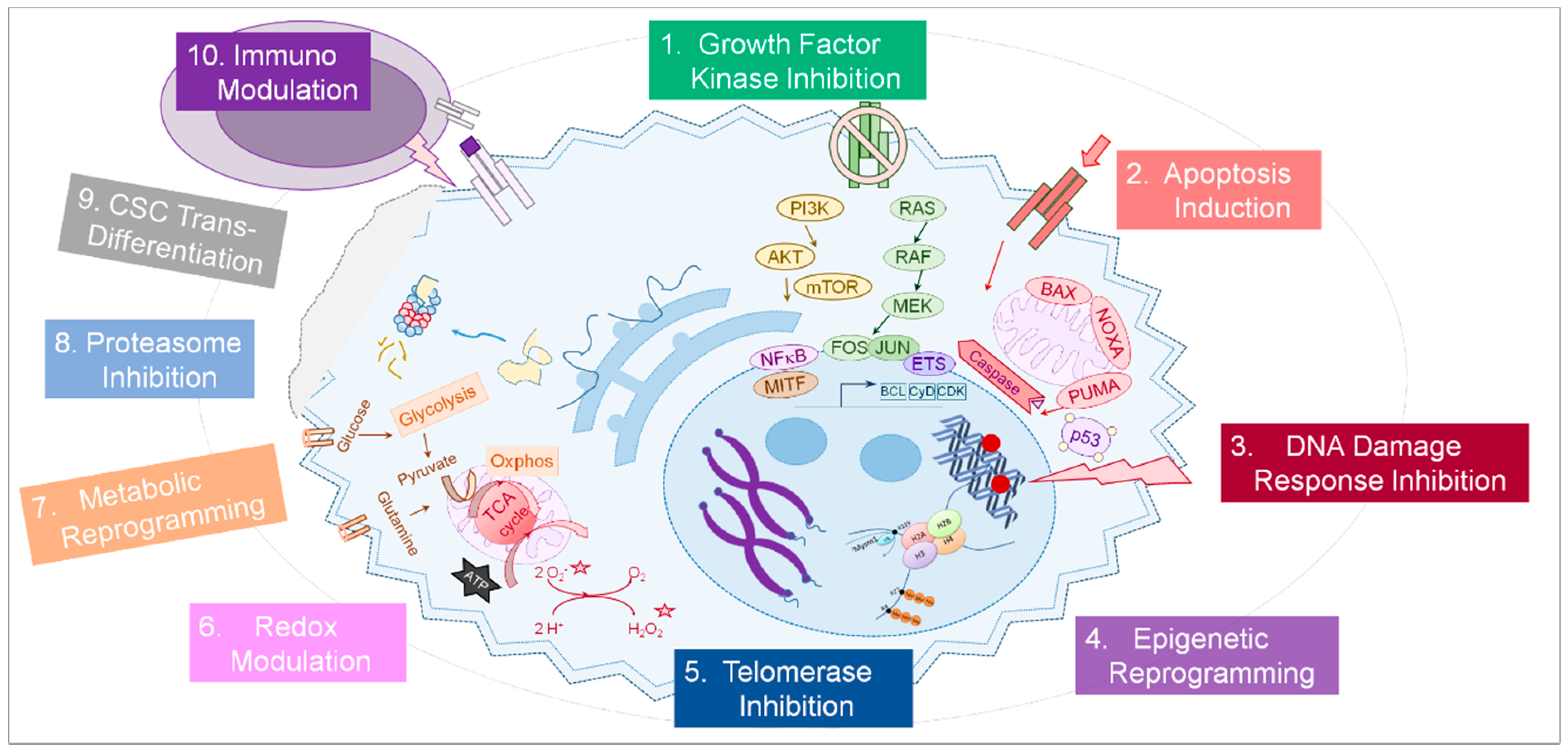
Figure 2.
Algorithm suggestion for therapy decisions based on the assessment of the tumor genetic and immunologic status and patient parameters for the example of metastatic melanoma.
Figure 2.
Algorithm suggestion for therapy decisions based on the assessment of the tumor genetic and immunologic status and patient parameters for the example of metastatic melanoma.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Targeted tumor therapy strategies, selected agents, and their molecular targets in selected ongoing clinical trials.
Table 1.
Targeted tumor therapy strategies, selected agents, and their molecular targets in selected ongoing clinical trials.
| Principle/Substance Group | Molecular Target | Cancer Type/Ongoing Trials (Selection) |
|---|---|---|
| 1. Signaling/Kinase inhibitors | ||
| - RAF-MEK-MAPK pathway inhibitors | BRAFV600mut, MEK | MM (LOGIC-2), NSCLC, CRC |
| - PI3K pathway inhibitors | PI3K, AKT, mTOR, GSK3 | MM (LOGIC-2), advanced solid tumors |
| - Cell cycle kinase inhibitors | Cyclin-dependent kinases (CDK4/6) | MM (LOGIC-2), NSCLC, solid tumors |
| - Growth factor signaling inhibitors | Oncogenic receptor kinases | Advanced cancers (multikinase inhibitors) |
| - FGFR | MM (LOGIC-2), TNBC, NSCLC | |
| - MET kinase | MM (LOGIC-2), RC, NSCLC, lymphoma | |
| - EGFR | NSCLC, CRC | |
| - VEGFR, PDGFR, others | MM (NIPAWILMA) | |
| - JAK-STAT pathway inhibitors | Janus kinases (JAKs) | advanced solid tumors, NSCLC, lymphoma |
| - FAK inhibitors (FAKi) | Focal adhesion kinase (FAK) | CRC, NSCLC, combination ImT |
| 2. Apoptosis modulators | ||
| - BH3 mimetics, BCL-2/XL-inhibitors | Intrinsic cell death (NOXA, PUMA, BIM) | ALL (Venetoclax), n/f 1 (pre-clinical) |
| - SMAC mimetics | Inhibitor of apoptosis proteins (IAPs) | advanced solid tumors, NSCLC |
| - BCL-2 antisense oligonucleotides | Anti-apoptotic BCL-2 members | HM, MM (Oblimersen, withdrawn) |
| - p53 reactivation | Tumor suppressor p53, MDM2/4 | Advanced cancers, lymphoma, AML |
| - Death receptor activation | Death receptors (FAS, TRAIL/APO-2) | MM (DS-8273a), NSCLC, CRC |
| - Autophagy inhibitors | Autophagy pathways | CRC, HCC, advanced solid tumors |
| 3. DNA damage response inhibitors | ||
| - PI3K/PIKK (ATM/ATR) inhibitors | ATM and ATR kinases | Advanced solid tumors, lymphoma, CNS |
| - Checkpoint kinase inhibitors | CHK1 kinase, WEE kinase | tumors (phase 0–II), ChT/RT combinations |
| - PARP inhibitors | poly(ADP-ribose) polymerase (PARP) | OC, combination therapies |
| 4. Epigenetic drugs | ||
| - 5-Aza-(2-deoxy-)cytidine | DNA-methyl-transferases (DNMT1-3) | HM (MDS), MM |
| - HDAC inhibitors (HDACi) | Histone deacetylases (HDAC1-10) | HM, lymphoma, N/SCLC, MM |
| - EZH2 inhibitors | Polycomb repressive complex (PRC) 2 | Sarcoma, lymphoma, mesothelioma |
| - BET inhibitors | BET family of histone readers | Refractory HM (MDS, ALL), solid tumors |
| - DUB inhibitors | (Histone) deubiquitinases | n/f 1 |
| - si/miRNAs | microRNAs, lncRNAs, tumor mRNAs | withdrawn, only biomarker studies |
| 5. Telomerase inhibitors | ||
| - hTR antagonists/competitors | hTR nucleotide (TERC) | solid tumors, withdrawn (Imetelstat) |
| - hTERT inhibitors (Ti) | Telomerase reverse transcriptase (TERT) | withdrawn |
| 6. Redox drugs | ||
| - Dimethylfumarate (DMF) | BCL2, PARP1, NFκB-NRF2-KEAP1-axis | Lymphoma (CTL), CNS (pre-clinical) |
| - Antioxidants (Vitamins, NAC, others) | EMT, cytotoxicity reduction | NSCLC, CRC, solid tumors, lymphoma |
| 7. Metabolic drugs | ||
| - Metformin, Phenformin | mTOR, TCA cycle | MM, BC, OC, PC, advanced solid tumors |
| - PDK inhibitors (Dichloroacetate) | Pyruvate dehydrogenase kinase, Glycolysis | CNS tumors (completed), SCC |
| 8. Proteasome inhibitors | ||
| - Bortezomib, PS-341 | (Ubiquitin)-Proteasome pathway | Refractory solid tumors, HM |
| 9. Transdifferentiation inducers | ||
| - Retinoids (ATRA, Bexarotene) | Nuclear RAR and RXR receptors | HM, CNS, MM (pre-clinical) |
| - Unsaturated fatty acids | PPARγ, adipogenic differentiation pathways | CNS, MM (pre-clinical) |
| 10. Immunomodulators | ||
| - T cell receptor signaling enhancers | Ubiquitin ligases (CBL, ITCH, GRAIL, et al.) | n/f 1 |
| Adaptor proteins, kinases | ||
| - Kynurenine pathway inhibitors | Indoleamine 2,3-dioxygenase (IDO1) | MM (KEYNOTE-252/ECHO-301, halted) |
| 11. Others | ||
| - Matrix metalloprotease inhibitors | MMP, COL3 | Advanced solid tumors (completed) |
| - WNT/β-Catenin inhibitors | WNT ligand | WNT-driven tumors (MM, CRC, others) |
1 n/f indicates that no ongoing clinical trial was found in public databases such as https://clinicaltrials.gov [19]. Abbreviations: acute lymphoblastic leukemia (ALL), central nervous system tumors (CNS), colorectal cancer (CRC), cutaneous T-cell lymphoma (CTL), hematological malignancies (HM), hepatocellular carcinoma (HCC), metastatic melanoma (MM), non-/small cell lung cancer (N/SCLC), ovarian carcinoma (OC), prostate carcinoma (PC), renal carcinoma (RC), squamous cell carcinoma (SCC), triple-negative/breast cancer (TN/BC), chemotherapy (ChT), immunotherapy (ImT), radiotherapy (RT).
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gatzka, M.V. Targeted Tumor Therapy Remixed—An Update on the Use of Small-Molecule Drugs in Combination Therapies. Cancers 2018, 10, 155. https://doi.org/10.3390/cancers10060155
AMA Style
Gatzka MV. Targeted Tumor Therapy Remixed—An Update on the Use of Small-Molecule Drugs in Combination Therapies. Cancers. 2018; 10(6):155. https://doi.org/10.3390/cancers10060155
Chicago/Turabian StyleGatzka, Martina V. 2018. "Targeted Tumor Therapy Remixed—An Update on the Use of Small-Molecule Drugs in Combination Therapies" Cancers 10, no. 6: 155. https://doi.org/10.3390/cancers10060155
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.