Downregulation of TRAIL-Receptor 1 Increases TGFβ Type II Receptor Expression and TGFβ Signalling Via MicroRNA-370-3p in Pancreatic Cancer Cells

 ,
,  and
and 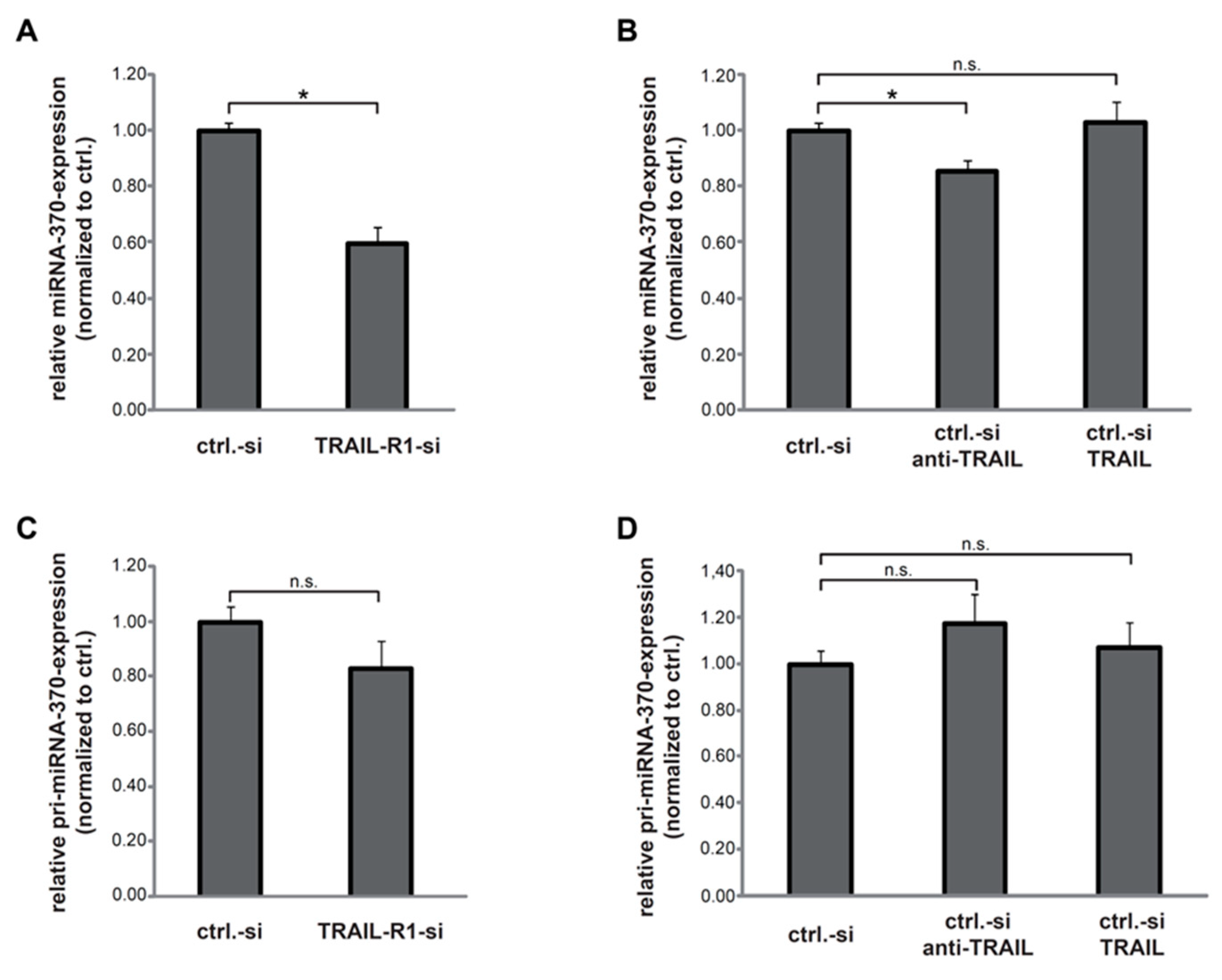{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. TRAIL-R1 Regulates the Expression of miR-370
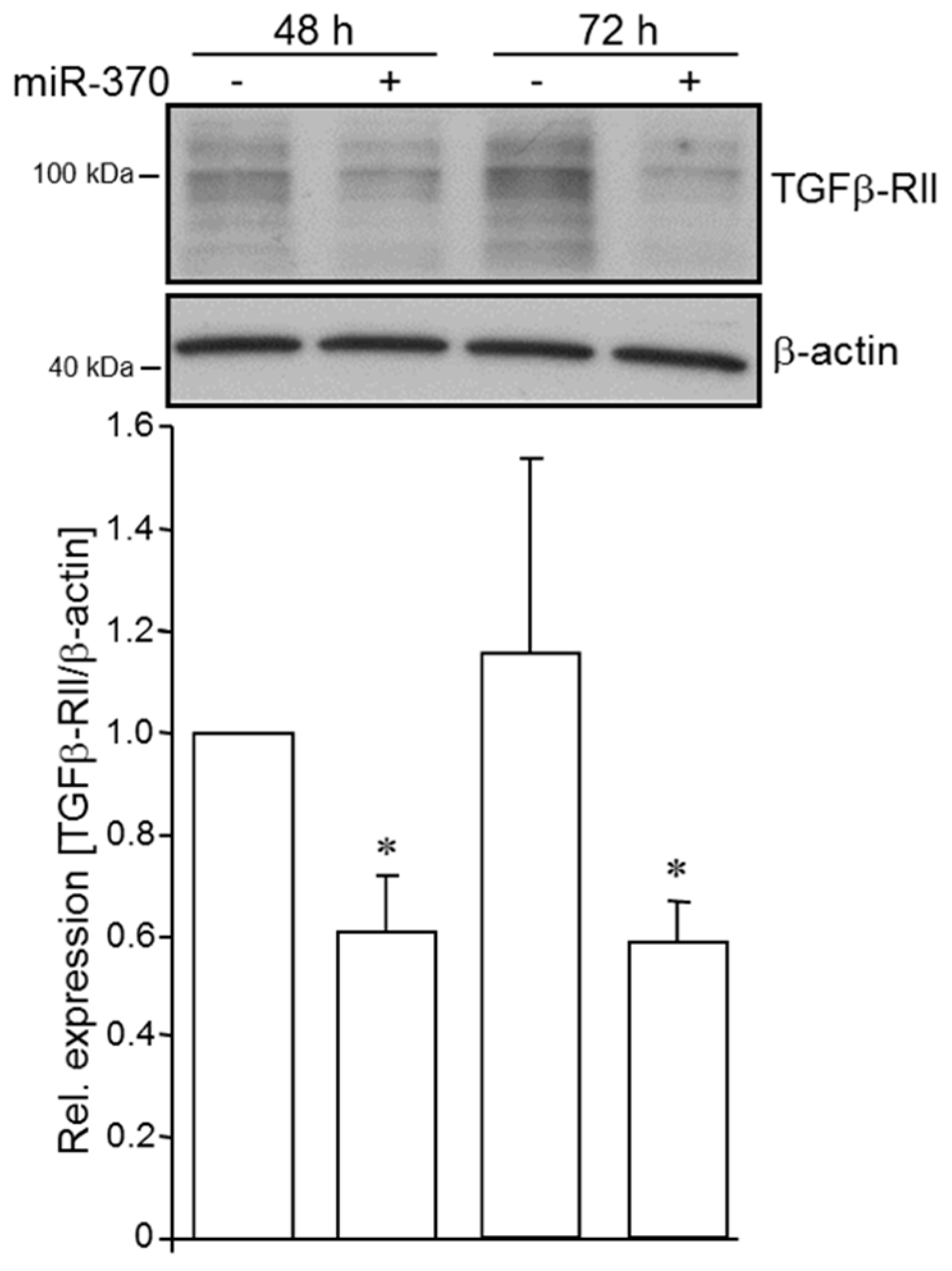
2.2. MiR-370-3p Negatively Controls TGFβ-RII in PDAC Cells
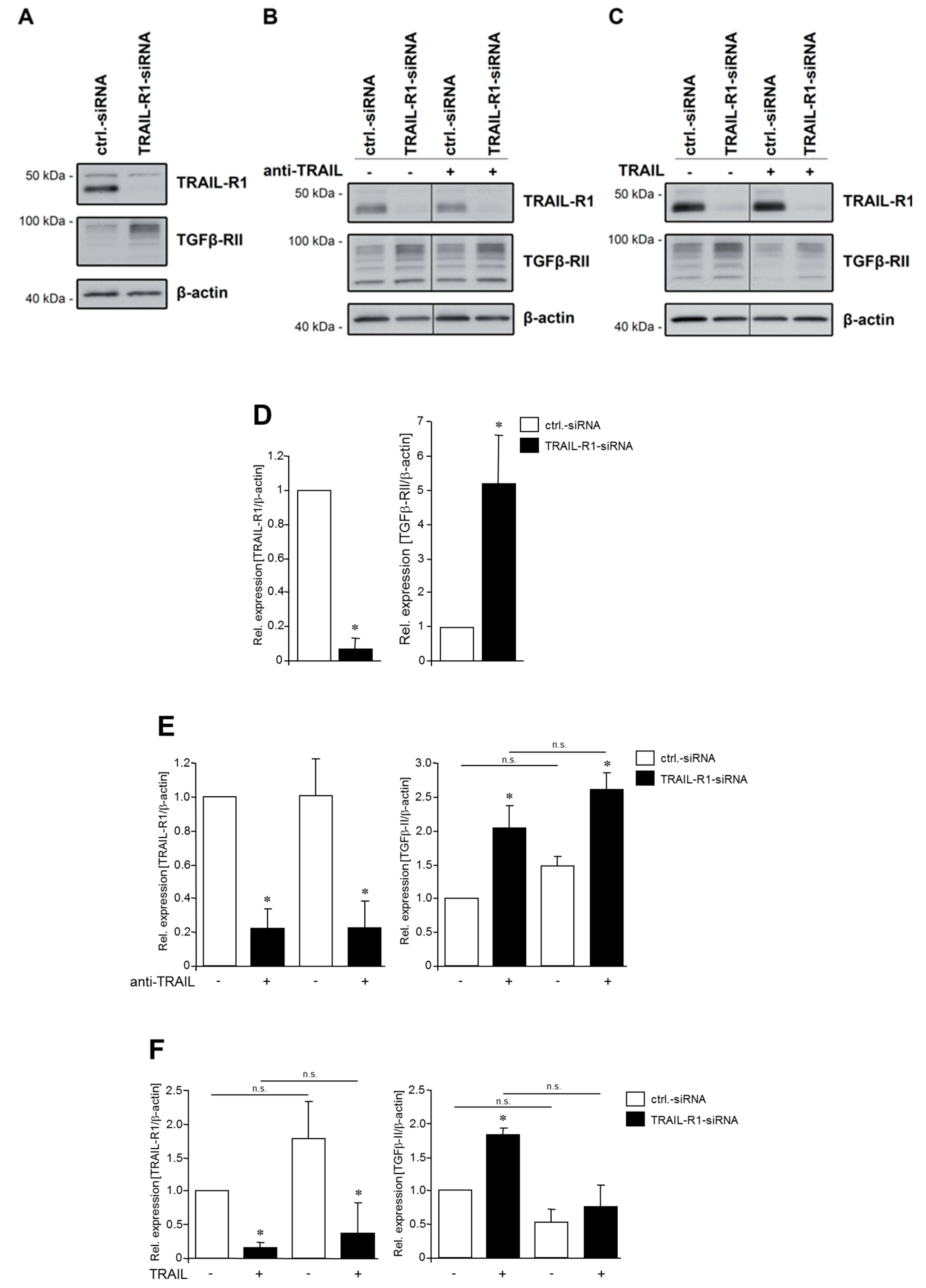
2.3. TRAIL-R1 Knockdown Increases the Abundance of TGFβ-RII
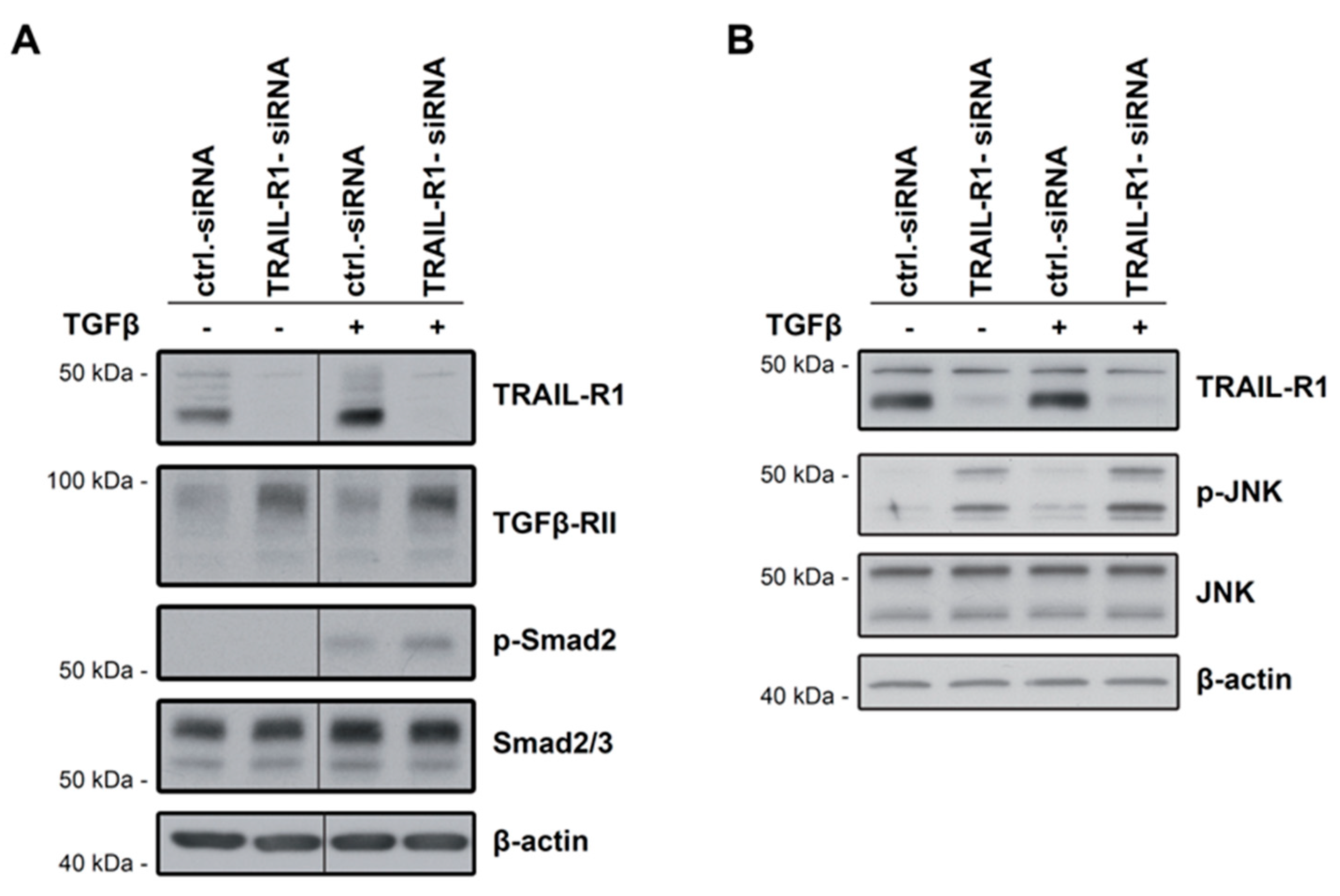
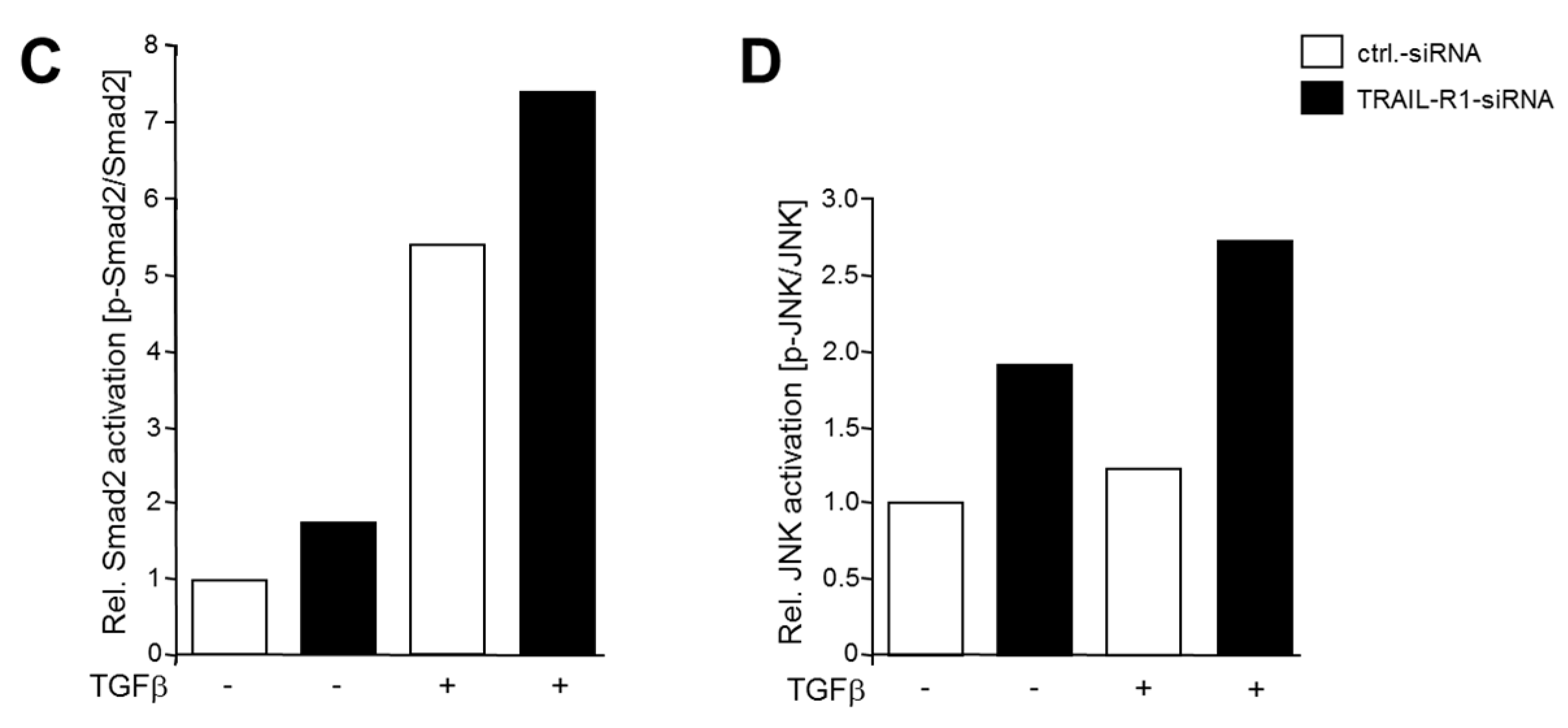
2.4. Knockdown of TRAIL-R1 Enhances Activation of Smad and Non-Smad Pathways after TGFβ Stimulation
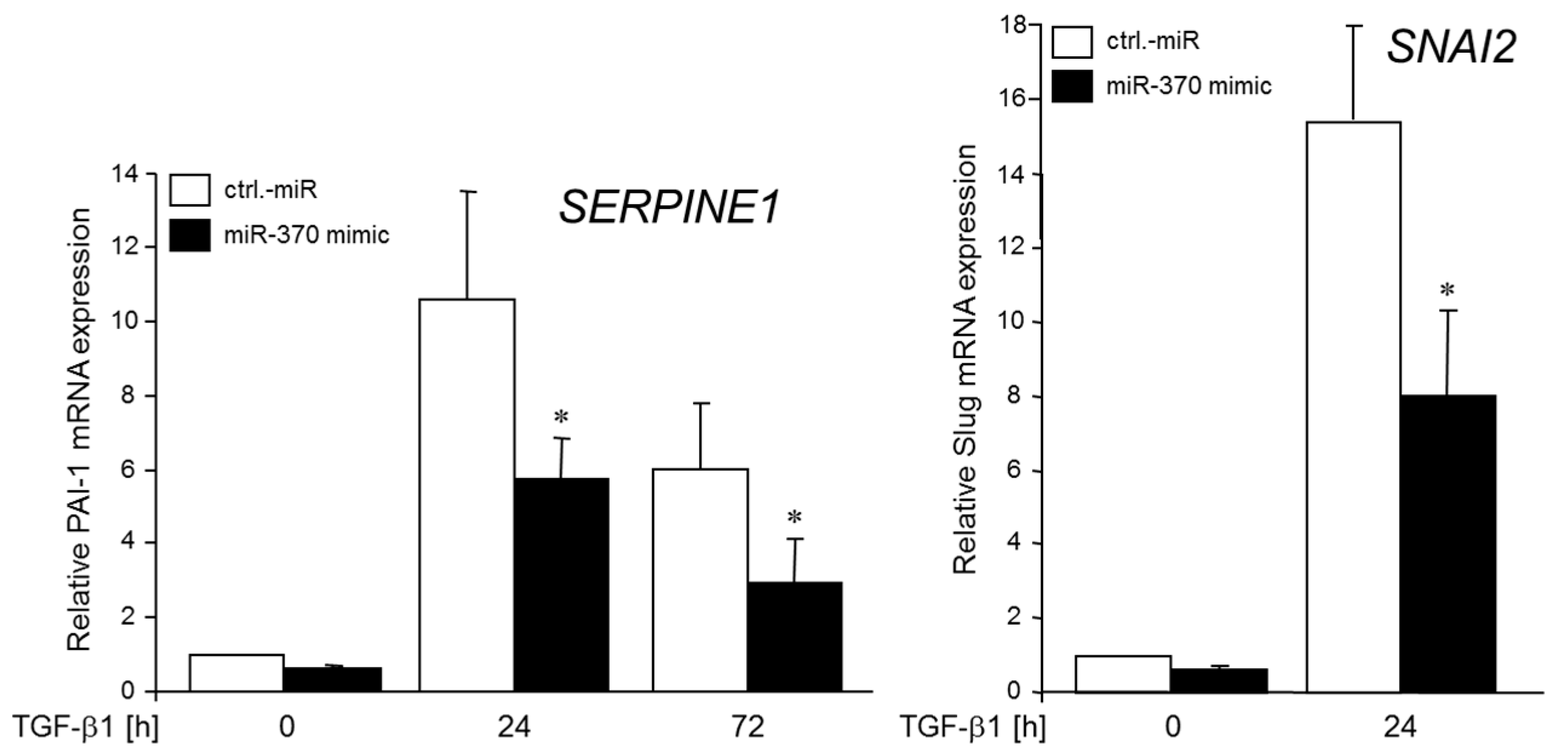
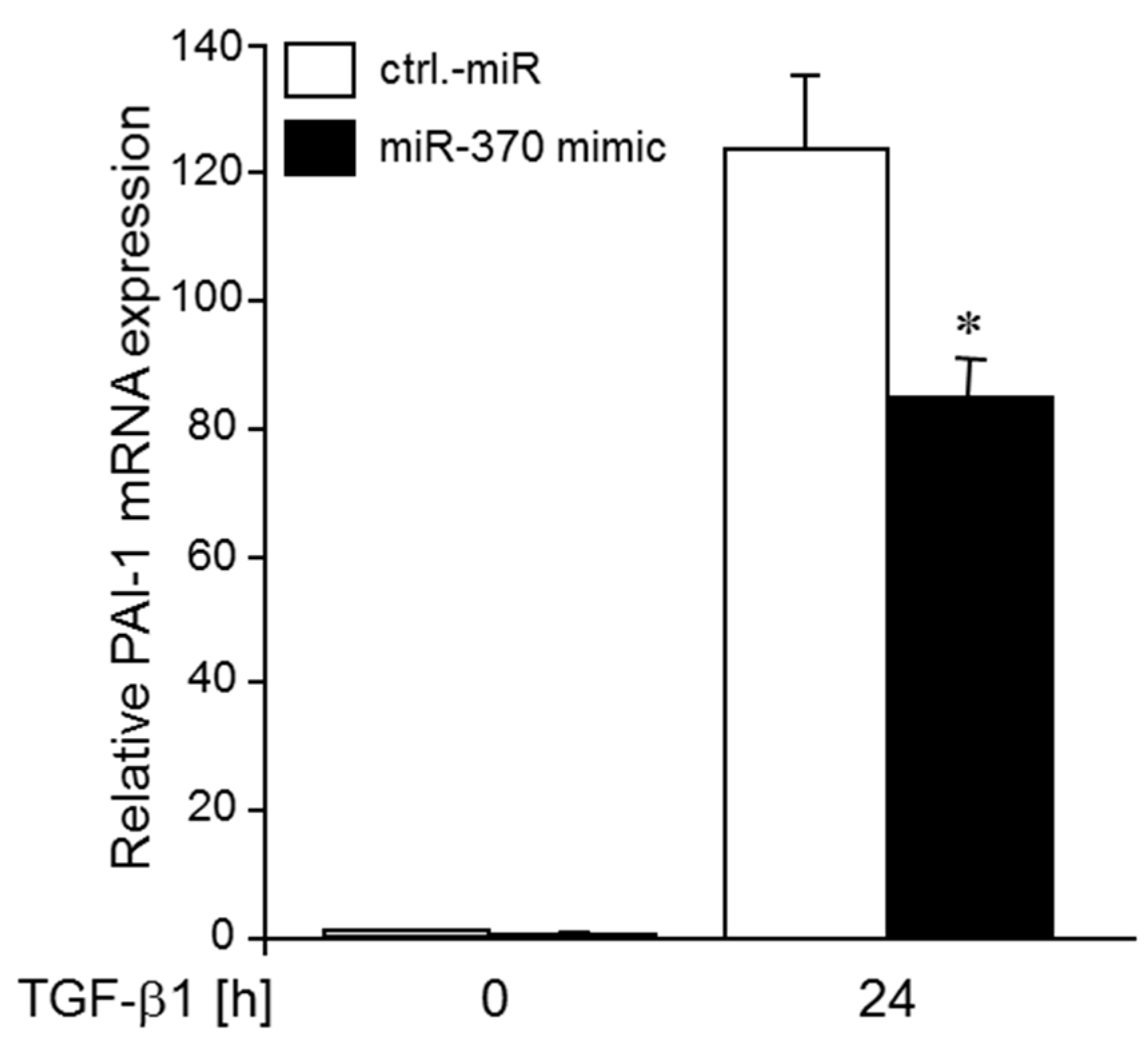
2.5. MiR-370-3p Reduces TGFβ-induced Expression of PAI-1 and SLUG
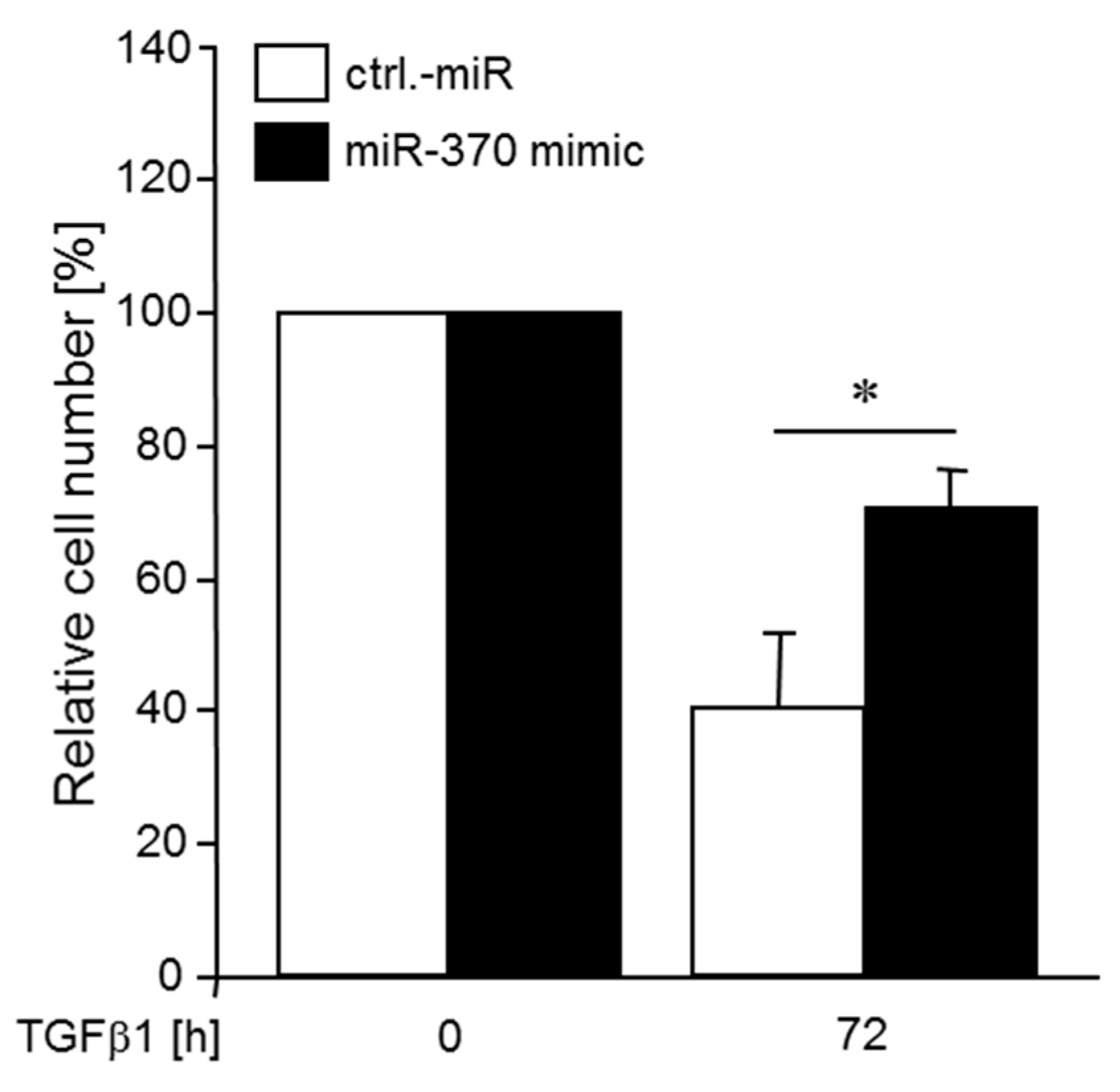
2.6. MiR-370 Reduces TGFβ-induced Growth Inhibition
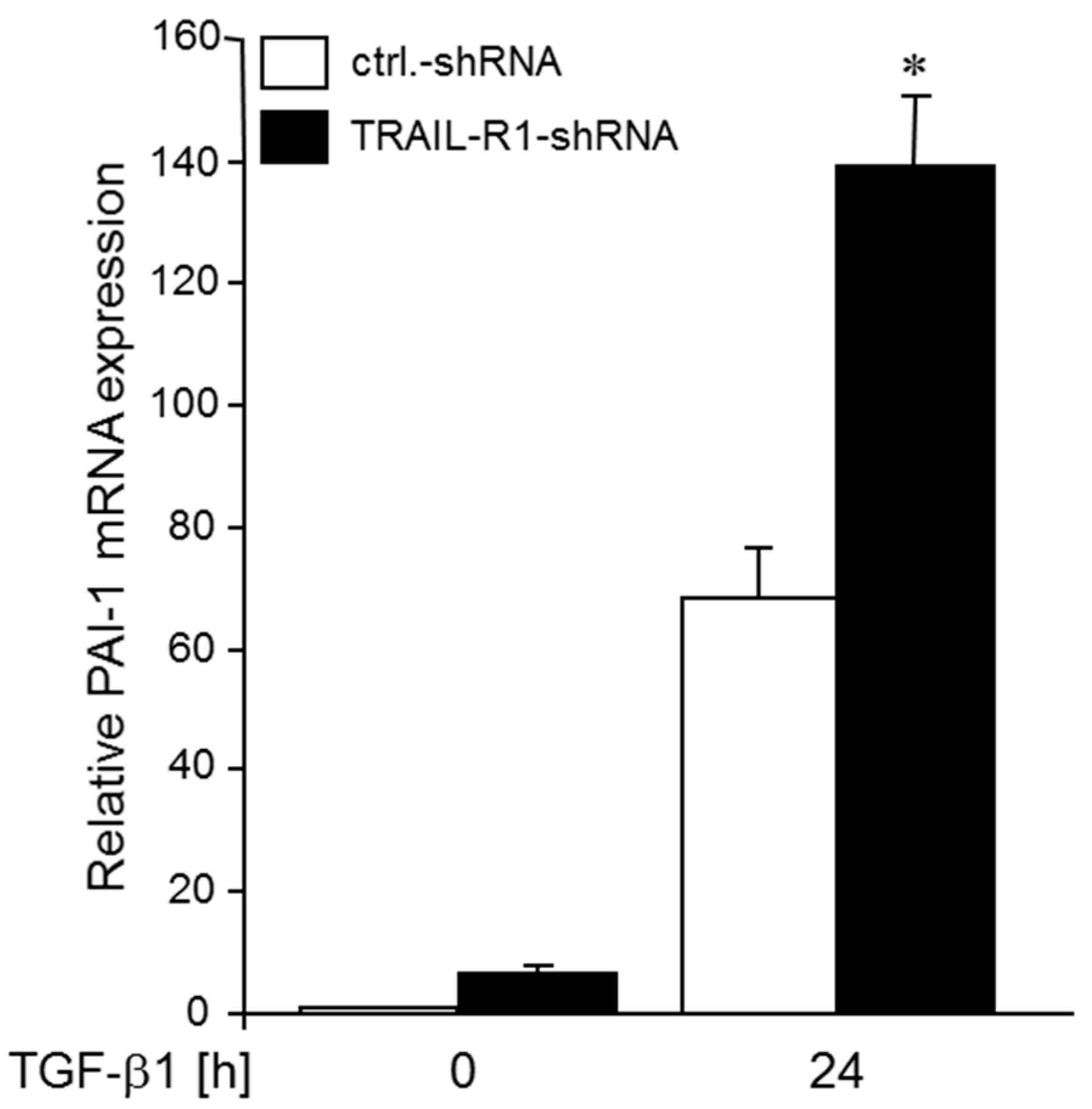
2.7. Knockdown of TRAIL-R1 Enhances TGFβ-Induced PAI-1 Expression
2.8. Ectopic Expression of a miR-370-3p Mimic Partially Reverses the TRAIL-R1 shRNA-induced TGFβ Hyperstimulation of PAI-1 Expression
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Culture Conditions, Cell Stimulation and Counting
5.2. Transfection of siRNA, shRNA and miR-370
5.3. Western Blot Analysis
5.4. RNA Isolation, Reverse Transcription and Polymerase Chain Reaction
5.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Kretz, A.L.; von Karstedt, S.; Hillenbrand, A.; Henne-Bruns, D.; Knippschild, U.; Trauzold, A.; Lemke, J. Should We Keep Walking along the Trail for Pancreatic Cancer Treatment? Revisiting TNF-Related Apoptosis-Inducing Ligand for Anticancer Therapy. Cancers (Basel) 2018, 3, e77. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolk, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef] [PubMed]
- Screaton, G.R.; Mongkolsapaya, J.; Xu, X.N.; Cowper, A.E.; McMichael, A.J.; Bell, J.I. TRICK2, a new alternatively spliced receptor that transduces the cytotoxic signal from TRAIL. Curr. Biol. 1997, 7, 693–696. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Smolak, P.J.; Walczak, H.; Waugh, J.; Huang, C.P.; DuBose, R.F. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signalling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumour necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer cell-autonomous TRAIL-R signalling promotes KRAS-driven cancer progression, invasion and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.J.; de Jong, S.; Kruyt, F.A. Non-canonical kinase signalling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ. 2013, 20, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Ehrenschwender, M.; Siegmund, D.; Wicovsky, A.; Kracht, M.; Dittrich-Breiholz, O.; Spindler, V.; Waschke, J.; Kalthoff, H.; Trauzold, A.; Wajant, H. Mutant PIK3CA licenses TRAIL and CD95L to induce non-apoptotic caspase-8-mediated ROCK activation. Cell Death Differ. 2010, 17, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, D.; Klose, S.; Zhou, D.; Baumann, B.; Röder, C.; Kalthoff, H.; Wajant, H.; Trauzold, A. Role of caspases in CD95L- and TRAIL-induced non-apoptotic signalling in pancreatic tumour cells. Cell Signal. 2007, 19, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Siegmund, D.; Schniewind, B.; Sipos, B.; Egberts, J.; Zorenkov, D.; Emme, D.; Röder, C.; Kalthoff, H.; Wajant, H. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006, 25, 7434–7439. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, U.; Röder, C.; Kalthoff, H.; Trauzold, A. Compartmentalization of TNF-related apoptosis-inducing ligand (TRAIL) death receptor functions: Emerging role of nuclear TRAIL-R2. Cell Death Dis. 2014, 5, e1390. [Google Scholar] [CrossRef] [PubMed]
- Haselmann, V.; Kurz, A.; Bertsch, U.; Hübner, S.; Olempska-Müller, M.; Fritsch, J.; Häsler, R.; Pickl, A.; Fritsche, H.; Annewanter, F.; et al. Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumour cells. Gastroenterology 2014, 146, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Shen, H.C.; Rivera Rosado, L.A.; Zhang, Y.; Di, X.; Zhang, B. Mislocalization of death receptors correlates with cellular resistance to their cognate ligands in human breast cancer cells. Oncotarget 2012, 3, 833–842. [Google Scholar] [PubMed]
- Kojima, Y.; Nakayama, M.; Nishina, T.; Nakano, H.; Koyanagi, M.; Takeda, K.; Okumura, K.; Yagita, H. Importin β1 protein-mediated nuclear localization of death receptor 5 (DR5) limits DR5/tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced cell death of human tumour cells. J. Biol. Chem. 2011, 286, 43383–43393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Franco, A.V.; Nguyen, T.; Gray, C.P.; Hersey, P. Differential localization and regulation of death and decoy receptors for TNF-related apoptosis-inducing ligand (TRAIL) in human melanoma cells. J. Immunol. 2000, 164, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, H.; Heilmann, T.; Tower, R.J.; Hauser, C.; von Au, A.; El-Sheikh, D.; Campbell, G.M.; Alp, G.; Schewe, D.; Hübner, S.; et al. TRAIL-R2 promotes skeletal metastasis in a breast cancer xenograft mouse model. Oncotarget 2015, 6, 9502–9516. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Agami, R. MicroRNAs, RNA binding proteins and cancer. Eur. J. Clin. Investig. 2010, 40, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Transcription and processing of human microRNA precursors. Mol. Cell 2004, 16, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.; Zamore, P.D. Rethinking the microprocessor. Cell 2006, 125, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Redfern, A.D.; Colley, S.M.; Beveridge, D.J.; Ikeda, N.; Epis, M.R.; Li, X.; Foulds, C.E.; Stuart, L.M.; Barker, A.; Russell, V.J.; et al. RNA-induced silencing complex (RISC) Proteins PACT, TRBP and Dicer are SRA binding nuclear receptor coregulators. Proc. Natl. Acad. Sci. USA 2013, 110, 6536–6541. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kunej, T.; Godnic, I.; Horvat, S.; Zorc, M.; Calin, G.A. Cross talk between microRNA and coding cancer genes. Cancer J. 2012, 18, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Hatzimichael, E.; Londin, E.; Vartholomatos, G.; Loher, P.; Rigoutsos, I.; Briasoulis, E. The microRNAs within the DLK1-DIO3 genomic region: Involvement in disease pathogenesis. Cell. Mol. Life Sci. 2013, 70, 795–814. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Bock, C.; Paulsen, M. Structural conservation versus functional divergence of maternally expressed microRNAs in the Dlk1/Gtl2 imprinting region. BMC Genomics 2008, 9, e346. [Google Scholar] [CrossRef] [PubMed][Green Version]
- An, F.; Yamanaka, S.; Allen, S.; Roberts, L.R.; Gores, G.J.; Pawlik, T.M.; Xie, Q.; Ishida, M.; Mezey, E.; Ferguson-Smith, A.C.; et al. Silencing of miR-370 in human cholangiocarcinoma by allelic loss and interleukin-6 induced maternal to paternal epigenotype switch. PLoS ONE 2012, 7, e45606. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, A.; Hong, S.M.; Hruban, R.H.; Goggins, M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin. Cancer Res. 2012, 18, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sun, H.; Zeng, W.; He, J.; Mao, X. Upregulation of MircoRNA-370 induces proliferation in human prostate cancer cells by downregulating the transcription factor FOXO1. PLoS ONE 2012, 7, e45825. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Zhang, C.; Lin, C.; Zhang, J.; Zhang, W.; Zhang, W.; Lu, Y.; Zheng, L.; Li, X. Circular RNA circITGA7 inhibits colorectal cancer growth and metastasis by modulating the Ras pathway and upregulating transcription of its host gene ITGA7. J. Pathol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2013. Ann. Oncol. 2013, 24, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Moore, M.J. Advanced pancreatic carcinoma: Current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010, 7, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Goggins, M.; Shekher, M.; Turnacioglu, K.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 1998, 58, 5329–5332. [Google Scholar] [PubMed]
- Xu, P.; Liu, J.; Derynck, R. Post-translational regulation of TGF-β receptor and Smad signalling. FEBS Lett. 2012, 586, 1871–1884. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.S.; Hung, P.S.; Chen, J.H.; Tu, H.F.; Fang, W.L.; Chen, C.Y.; Chen, W.T.; Gong, N.R.; Wu, C.W. Overexpression of miR-370 and downregulation of its novel target TGFβ-RII contribute to the progression of gastric carcinoma. Oncogene 2012, 31, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.P.; Yi, M.; Li, Q.Q.; Zhou, W.P.; Cong, W.M.; Yang, Y.; Ning, B.F.; Yin, C.; Huang, Z.W.; Wang, J.; et al. Perturbation of MicroRNA-370/Lin-28 homolog A/nuclear factor kappa B regulatory circuit contributes to the development of hepatocellular carcinoma. Hepatology 2013, 58, 1977–1991. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wu, T.; Li, Y.; Xu, Z.; Zhang, S.; Liu, B.; Chen, Q.; Tian, D. MicroRNA-370-3p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting β-catenin. Brain Res. 2016, 1644, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.T.; Chen, X.B.; Liu, H.L. Up-regulation of miR-370-3p restores glioblastoma multiforme sensitivity to temozolomide by influencing MGMT expression. Sci. Rep. 2016, 6, e32972. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; von Heydebreck, A.; Zhang, J.D.; Gunawan, B.; Langer, C.; Ramadori, G.; Wiemann, S.; Sahin, O. Localization- and mutation-dependent microRNA (miRNA) expression signatures in gastrointestinal stromal tumours (GISTs), with a cluster of co-expressed miRNAs located at 14q32.31. J. Pathol. 2010, 220, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Chiyomaru, T.; Enokida, H.; Kawakami, K.; Tatarano, S.; Nishiyama, K.; Nohata, N.; Seki, N.; Nakagawa, M. The tumour-suppressive function of miR-1 and miR-133a targeting TAGLN2 in bladder cancer. Br. J. Cancer 2011, 104, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Z.; Ander, B.P.; Tian, Y.; Stamova, B.; Jickling, G.C.; Davis, R.R.; Sharp, F.R. Integrated analysis of mRNA and microRNA expression in mature neurons, neural progenitor cells and neuroblastoma cells. Gene 2012, 495, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Mühlenbeck, F.; Schneider, P.; Bodmer, J.L.; Schwenzer, R.; Hauser, A.; Schubert, G.; Scheurich, P.; Moosmayer, D.; Tschopp, J.; Wajant, H. The tumour necrosis factor-related apoptosis-inducing ligand receptors TRAIL-R1 and TRAIL-R2 have distinct cross-linking requirements for initiation of apoptosis and are non-redundant in JNK activation. J. Biol. Chem. 2000, 275, 32208–32213. [Google Scholar] [CrossRef] [PubMed]
- Radke, D.I.; Ungefroren, H.; Helm, O.; Voigt, S.; Alp, G.; Braun, H.; Hübner, S.; Dilchert, J.; Sebens, S.; Adam, D.; et al. Negative control of TRAIL-R1 signaling by transforming growth factor β1 in pancreatic tumor cells involves Smad-dependent down regulation of TRAIL-R1. Cell Signal 2016, 28, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Gundlach, J.P.; Hauser, C.; Schlegel, F.M.; Böger, C.; Röder, C.; Röcken, C.; Becker, T.; Egberts, J.H.; Kalthoff, H.; Trauzold, A. Cytoplasmic TRAIL-R1 is a positive prognostic marker in PDAC. BMC Cancer. 2018, 18, e777. [Google Scholar] [CrossRef] [PubMed]
- Gallmeier, E.; Bader, D.C.; Kriegl, L.; Berezowska, S.; Seeliger, H.; Göke, B.; Kirchner, T.; Bruns, C.; De Toni, E.N. Loss of TRAIL-receptors is a recurrent feature in pancreatic cancer and determines the prognosis of patients with no nodal metastasis after surgery. PLoS ONE 2013, 8, e56760. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ge, Q.; Liu, J.; Zhang, Q.; Wang, C.; Cui, K.; Chen, Z. Effects of miR-1236-3p and miR-370-5p on activation of p21 in various tumors and its inhibition on the growth of lung cancer cells. Tumour Biol. 2017, 39, e1010428317710824. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, Y.; Deng, X.; Luo, M.; Wang, X.; Hu, H.; Liu, C.; Zhong, M. Long noncoding RNA H19 promotes transforming growth factor-β-induced epithelial-mesenchymal transition by acting as a competing endogenous RNA of miR-370-3p in ovarian cancer cells. Onco. Targets Ther. 2018, 11, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of TGF-β signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar] [PubMed]
- Chitkara, D.; Mittal, A.; Mahato, R.I. MiRNAs in pancreatic cancer: Therapeutic potential, delivery challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Ganju, A.; Khan, S.; Hafeez, B.B.; Behrman, S.W.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. MiRNA nanotherapeutics for cancer. Drug Discov. Today 2017, 22, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Fransvea, E.; Dituri, F.; Lupo, L.; Antonaci, S.; Giannelli, G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010, 51, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Fransvea, E.; Angelotti, U.; Antonaci, S.; Giannelli, G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008, 47, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, R.S.; Dumont, N.; Ritter, C.A.; Dugger, T.C.; Brantley, D.M.; Chen, J.; Easterly, E.; Roebuck, L.R.; Ryan, S.; Gotwals, P.J.; et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration and metastases. J. Clin. Investig. 2002, 109, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Wermann, H.; Arlt, A.; Schütze, S.; Schäfer, H.; Oestern, S.; Röder, C.; Ungefroren, H.; Lampe, E.; Heinrich, M.; et al. CD95 and TRAIL receptor-mediated activation of protein kinase C and NF-kappaB contributes to apoptosis resistance in ductal pancreatic adenocarcinoma cells. Oncogene 2001, 20, 4258–4269. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radke, D.I.; Ling, Q.; Häsler, R.; Alp, G.; Ungefroren, H.; Trauzold, A. Downregulation of TRAIL-Receptor 1 Increases TGFβ Type II Receptor Expression and TGFβ Signalling Via MicroRNA-370-3p in Pancreatic Cancer Cells. Cancers 2018, 10, 399. https://doi.org/10.3390/cancers10110399
Radke DI, Ling Q, Häsler R, Alp G, Ungefroren H, Trauzold A. Downregulation of TRAIL-Receptor 1 Increases TGFβ Type II Receptor Expression and TGFβ Signalling Via MicroRNA-370-3p in Pancreatic Cancer Cells. Cancers. 2018; 10(11):399. https://doi.org/10.3390/cancers10110399
Chicago/Turabian StyleRadke, David I., Qi Ling, Robert Häsler, Gökhan Alp, Hendrik Ungefroren, and Anna Trauzold. 2018. "Downregulation of TRAIL-Receptor 1 Increases TGFβ Type II Receptor Expression and TGFβ Signalling Via MicroRNA-370-3p in Pancreatic Cancer Cells" Cancers 10, no. 11: 399. https://doi.org/10.3390/cancers10110399
APA StyleRadke, D. I., Ling, Q., Häsler, R., Alp, G., Ungefroren, H., & Trauzold, A. (2018). Downregulation of TRAIL-Receptor 1 Increases TGFβ Type II Receptor Expression and TGFβ Signalling Via MicroRNA-370-3p in Pancreatic Cancer Cells. Cancers, 10(11), 399. https://doi.org/10.3390/cancers10110399